

Abstract
In our functional dissection of the CD33 Alzheimer’s disease susceptibility locus, we find that the rs3865444C risk allele is associated with greater cell surface expression of CD33 in monocytes (t50 = 10.06, pjoint=1.3×10–13) of young and older individuals. It is also associated with (1) diminished internalization of Aβ42) (2) accumulation of neuritic amyloid pathology and fibrillar amyloid on in vivo imaging and (3), increased numbers of activated human microglia.
Recent genome-wide studies have identified nine AD susceptibility loci, including CD33 and MS4A4/MS4A6E, which implicate the immune system in AD susceptibility1–7. Currently, the most strongly associated marker in the CD33 locus is rs38654445, and higher CD33 expression levels in the brain have been associated with more advanced cognitive decline and AD.8, 9 CD33, also known as Siglec-3, is a 67-KDa transmembrane glycoprotein expressed on the surface of myeloid progenitor cells, mature monocytes and macrophages. It functions as a lectin, a carbohydrate-binding protein, and it contains putative immunoreceptor tyrosine-based inhibitory motifs (ITIMs) that are typically inhibitors of cellular activity. In support of this putative function, CD33 has been shown to constitutively repress monocyte-derived pro-inflammatory cytokines10.
To explore the functional consequences of the CD33 locus, we accessed genotyped, archived samples from the Brigham and Women’s Hospital PhenoGenetic Project (Supplemental Table 1a). In these 32 young subjects, we found a seven-fold increase in CD33 cell surface expression of circulating monocytes in relation to the rs3865444C risk allele (t31 = 8.48, p=1.4×10−9), but the frequency of CD33+ monocytes does not vary in relation to rs3865444 (Supplemental Fig. 1). rs3865444C explains 72.8% of the variance in CD33 cell surface expression level. We replicated this result in an independent set of 19 young subjects from the PhenoGenetic Project (t18 = 6.29, p=6.3×10−6), and a joint analysis of both sets of subjects is highly significant (t50 = 10.06, pjoint=1.3×10−13) (Fig. 1a). Given that AD manifests primarily in older age and that immune system function changes significantly with age, we performed an additional replication analysis using 175 older subjects, both with and without a diagnosis of AD, from three prospective cohort studies of aging: the Memory and Aging Project (MAP) (n=60)11, the Religious Orders Study (ROS)12 (n=91), and the Chicago Health and Aging Project (CHAP) (n=24)13 (Supplementary Table 1b). We once again replicated the association of higher CD33 expression with rs3865444C (z = 10.13, p=.4.1 × 10−24), after adjusting for age, sex and batch effect (Fig. 1b); here, rs3865444C explained 70.4% of the variance in CD33 cell surface expression. We found no significant interaction by AD diagnosis in the ROS/MAP subjects (z = −0.80, p=0.43) in whom this phenotype is available. Thus, the relation of genotype and CD33 expression appears to hold throughout the life course, and the effect of this susceptibility allele may therefore play a role in the earliest stages of AD. In our data, we do not find an effect of age on CD33 surface expression.
Figure 1. The CD33 risk allele is associated with increased CD33 expression and decreased uptake.

The MFI of CD33 protein expression on circulating monocytes of young healthy adults is increased with the rs3865444C risk allele (a). There is a sevenfold increase in CD33 expression in rs3865444CC vs. rs3865444AA subjects. Monocytes from older adults also have differential CD33 expression based on genotype (b). The MFI of FITC-labeled dextran (c) and Aβ(42) (d) uptake by monocytes from young healthy adults is decreased with the rs3865444C risk allele. Data shown is the change in MFI between samples incubated at 4°C and 37°C. Each circle represents an individual. The mean of the distribution is denoted by a horizontal line.
Since myeloid cells such as infiltrating macrophages or microglia are thought to play a role in AD by phagocytosis of amyloid β that accumulates in neuritic amyloid plaques (a key neuropathologic feature of AD14), we tested the hypothesis that monocytes from subjects with the rs3865444C CD33 risk allele have a reduced state of activation and therefore reduced phagocytic ability compared to subjects with the protective allele. We find that circulating monocytes bearing the rs3865444C risk allele demonstrate reduced uptake of both fluorescently-labeled dextran (t29 = 3.33, p=0.003) (Fig. 1c) and amyloid Aβ (Aβ (42)) (t31 = 3.64, p=0.001)(Fig. 1d). We replicated this observation in an independent set of subjects (t128 = 2.18, p= 0.03)(Supplementary Fig. 2). These results suggest that the alteration in uptake may be relevant to the context of AD pathology where amyloid toxicity caused by the fibrillogenic Aβ(42) form of amyloid plays an important role. Interestingly, we observed that, while the effect of rs3865444C is additive for CD33 expression, it may be dominant for functional traits (such as substrate uptake) (Supplementary Table 2).
To investigate whether these functional consequences of the CD33 risk allele could lead to alterations of amyloid biology associated with AD, we examined two collections of older subjects (Supplementary Table 3a) that have undergone positron emission tomography imaging with Pittsburgh Compound B (PiB), which measures an individual’s burden of pathogenic fibrillar amyloid. Subjects are categorized into PiB positive (PiB+) and negative subsets (Fig. 2a,b), and the PiB+ subset is more likely to develop cognitive impairment and AD15. In a joint analysis of the two collections (n=218 total), we find that the rs3865444C risk allele is associated with a greater likelihood of being PiB+ (Fig. 2c,d), and as with the internalization phenotype, the effect of rs3865444C appears to fit a dominant model (χ2(1) = 5.52, p=0.02) better than an additive model (χ2(1) = 2.78, p=0.1). The association holds if we use the PiB burden as a quantitative trait (dominant model z = 2.35, p=0.02) in secondary analyses. Importantly, the magnitude and direction of the association linking rs3865444C with being PiB+ remains when the analysis is limited to asymptomatic older subjects, however the results are no longer significant, possibly because of the reduced sample size (Supplementary Table 3b). Overall, these data support a role for the rs3865444C susceptibility allele in in vivo amyloid accumulation in the presymptomatic phase of AD.
Figure 2. The susceptiblity allele of rs3865444 is associated with an increase in Pittsburgh Compound B (PiB) imaging.
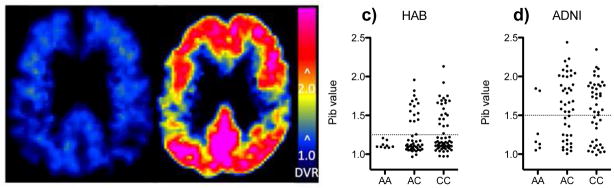
PiB-PET images from a PiB− rs3865444AA subject (a) and a PiB+ rs3865444CC subject (b) demonstrating a typical pattern of binding of PiB to amyloid plaque in the PiB positive (DVR > 2.0) subjects and lack of binding (DVR ~= 1.0) in the PiB negative subjects. The PiB values from the HAB (c) and ADNI (d) cohorts demonstrate an increased frequency of PiB+ subjects with the rs3865444C risk allele. PiB+ and PiB− values are separated by a dotted line.
Next, we assessed the role of rs3865444C in the accumulation of neuritic amyloid plaques measured at autopsy. In ROS/MAP subjects, we see that rs3865444C is also associated with greater neuritic amyloid plaque burden (z = 2.47, p=0.01). Further, we found that rs3865444C is associated with a greater likelihood of a pathologic diagnosis of AD (χ2(1) = 6.54, p=0.01). rs3865444C is not associated with the burden of neurofibrillary tangles (z = 1.40, p=0.16) (Supplementary Table 4). Given that these analyses were performed in individuals with measurements of CD33 expression in peripheral monocytes frozen prior to death, we assessed whether the level of CD33 expression in monocytes mediated some of the effect of the risk allele on neuropathologic traits. We found that the effect size of the relation of rs3865444C to neuritic amyloid plaque burden is diminished by 10% and is no longer significant (z = 1.74, p=0.08) when we add a covariate for the level of CD33 expression to the model. Thus, these two prospectively collected autopsy cohorts support a role for monocytes in statistically mediating some of the functional consequences of the CD33 risk allele that ultimately result in the accumulation of amyloid pathology and AD.
To further explore the possible role of CD33 in the brain of older subjects, we examined tissue sections from MAP subjects and report that CD33 is expressed in cells that have the morphologic attributes of microglia/macrophages (Fig. 3a–c). The CD33-expressing cells are found in the vicinity of neuritic plaques (Fig. 3a).
Figure 3. Immunohistochemistry reveals CD33 expression in the brain and an increase frequency of stage III microglia/macrophages associated with rs3865444C.

(a) CD33+ microglia/macrophages (brown) are seen in sections of the dorsolateral prefrontal cortex from a subject with pathologically confirmed AD; they are found in the vicinity of amyloid deposits (red). (b) Adjacent sections labeled with anti-CR3/43 (top panel), a marker for microglia/macrophage, and anti-CD33 (bottom panel) demonstrate that the cells staining with anti-CD33 have the morphologic attributes of microglia/macrophage. (c) CD33 is expressed at all 3 stages of activation in microglia/macrophages in the brain. The density of stage 3 CR3/43+ microglia is increased in the inferior temporal region (d) and posterior putamen region (e) with the rs3865444C risk allele. Each circle represents a subject. The line represents the mean.
In older age, activated myeloid cells such as those that express CD33 are seen in the brain of cognitively non-impaired and demented individuals16. In ROS/MAP subjects, we find that the mean proportion of terminally activated microglia/macrophage (stage III microglia or macrophage) is significantly elevated relative to the rs3865444C risk allele (z = 3.06, p=0.003) in the inferior temporal lobe (which is an early target of AD-related amyloid neuropathology) (Fig. 3d), and there is a suggestion that it is elevated in the posterior putamen region (z = 2.42, p=0.017), after accounting for the testing of four hypotheses (Fig. 3e) (Supplemental Table 5a). These associations remain after adjusting for a diagnosis of AD (Supplemental Table 5b). This observation is consistent with a model for the AD association in which monocytes from subjects with the risk allele are less able to uptake Aβ(42), which could lead to an accumulation of activated but less functional microglia/macrophages in the brain, where they fail to clear amyloid β plaques.
We have begun to characterize the functional consequences of the CD33 AD susceptibility locus in early and late-life. These results are consistent with a recent report that decreasing CD33 expression in mice leads to enhanced Aβ(42) uptake by macrophage/microglia and decreased accumulation of brain amyloid in a mouse model.9 CD33 is thought to be an inhibitory lectin, and thus increased cell surface expression of CD33 due to the risk allele could lead to down-regulation of myeloid cell activity, such as the reduced uptake of Aβ(42). Such alterations of myeloid function throughout life could lead to the earlier accumulation of fibrillar amyloid as measured by PiB imaging in older brains, and, ultimately, the accumulation of more neuritic amyloid pathology and a greater likelihood of meeting a diagnosis of AD on neuropathologic examination.
The observation that the functional consequences of this variant are seen in younger and presymptomatic older subjects brings up the possibility that the CD33 risk allele and altered CD33 function could have a role in the very early presymptomatic phase of AD, in middle or younger age. Further, our data from young subjects with the risk allele suggest that AD-associated alterations in immune function that have previously been reported are not solely a feature of immune senescence in older age or a late reaction to the toxicity of amyloid and other AD-related pathologies. It is not yet clear how CD33-related dysfunction of innate immune cells relates to all manifestations of AD: it seems to contribute to known neuropathologic features of AD such as the accumulation of amyloid pathology but may well also be involved in inflammatory events that are associated with AD and remain to be further characterized.
The data reported here integrate well with an existing literature exploring the role of innate immune system dysfunction in AD14 and, critically, identifies a specific molecule, CD33, that links genetic susceptibility, altered myeloid function, and amyloid pathology to an early role in the pathogenesis of AD. These data therefore present (1) an intriguing novel target with which to manipulate amyloid pathology and (2) pertinent information that could be leveraged in algorithms with which to identify individuals at high risk of accumulating amyloid who are being targeted in clinical trials of presymptomatic AD.17, 18 More proximally, CD33 and its alteration of myeloid function may influence responses to the antibody-mediated therapeutic approaches that are currently being tested in AD19,20.
Online methods
Study Subjects
Informed consent was obtained on all human subjects. All blood draws, brain autopsies, experiments and data analysis were done in compliance with protocols approved by the Institutional Review Board of each Institution.
The Brigham & Women’s Hospital (BWH) PhenoGenetic Project
Peripheral venous blood was obtained from healthy control volunteers in compliance with protocols approved by the Institutional Review Board of Partners Healthcare. The PhenoGenetic Project is a living tissue bank that consists of healthy subjects that are re-contactable based on their genotype. 1741 healthy subjects >18 years old have been recruited from the general population of Boston, MA. They are free of chronic inflammatory, infectious and metabolic diseases. Their median age is 24, and 62.7% of subjects are women. For the CD33 expression experiments the median age was 26.5 (range=20.0–50.0). All of the PhenoGenetic subjects used in the uptake assay using dextran, and 25 of the subjects used in the Aβ(42) uptake assay were also used in the CD33 staining assay. 67% the subjects used in the Aβ(42) uptake assay were also used in the dextran uptake assay.
The Rush Religious Orders (ROS) Study and Memory and Aging Project (MAP), and Chicago Health and Aging Project
Study participants were free of known dementia at enrollment, agreed to annual clinical evaluations, and signed an Anatomic Gift Act donating their brains at death. ROS, started in 1994, enrolls Catholic priests, nuns and brothers, aged 53 or older from about 40 groups in 12 states. Since January 1994, more than 1,150 participants completed their baseline evaluation, of whom 87% are non-Hispanic white, and the follow-up rate of survivors and autopsy rate among the deceased both exceed 90%. MAP, started in 1997, enrolls men and women aged 55 or older and with no evidence of dementia at baseline from assisted living facilities in the Chicago area in compliance with protocols approved by the Institutional Review Board of RUSH University. Since October 1997, more than 1,550 participants completed their baseline evaluation, of which 87% were non-Hispanic white. The follow-up rate of survivors exceeds 90% and the autopsy rate exceeds 80%. More detailed descriptions of ROS, MAP and CHAP can be found in prior publications11, 12. The median age of subjects used in the CD33 expression experiments at sampling was 79.9, (range=65.8–94.8).
The Harvard Aging Brain Study
The Harvard Aging Brain Study is a longitudinal study of older individuals between the ages of 65 and 85 years old. Participants were evaluated with an extensive battery of functional and neuropsychological tests. All participants had a Clinical Dementia Rating of 021, performed no worse than 1.5 SD units below the age- and education-corrected norm on the Logical Memory IIa subtest of the Wechsler Memory Scale-Revised22, and scored 26 or above on the Mini-Mental State Examination23. Participants were excluded if previously diagnosed with a neurological or psychiatric condition or if they scored >11 on the Geriatric Depression Scale24. Participants provided informed consent in accordance with protocols approved by the Partners Healthcare Inc. Institutional Review Board.
The Alzheimer’s Disease Neuroimaging Initiative
The Alzheimer’s Disease Neuroimaging Initiative (ADNI) is a large multicenter, longitudinal, observational trial taking place across North America, in which subjects with normal cognition, amnestic MCI, and mild AD are followed up with periodic neuropsychological testing, multiple imaging techniques, and fluid biomarkers (www.loni.ucla.edu\ADNI). ADNI is a result of the efforts of many co-investigators from a broad range of academic institutions and private corporations, and subjects have been recruited from over 50 sites across the United States and Canada. Subjects (N= 96) with complete baseline clinical datasets were included in the current analysis. They were aged 55 to 89 years, in general good health or had stable medical problems at the time of screening, and had a study partner/caregiver able to provide an independent evaluation of the subject’s cognitive, behavioral, and functional status. Subjects did not have significant neurological conditions, significant active psychiatric disorders, or alcohol or substance abuse within 2 years of screening. All subjects underwent comprehensive neuropsychological testing (assessing memory, attention, executive function, language, and visuospatial function) and additional study partner questionnaires about behavior and daily functioning. All subjects had a global Clinical Dementia Rating scale21 (CDR) score = 0, 0.5 or 1.
Data used in the preparation of this article were obtained from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) database (adni.loni.ucla.edu). The ADNI was launched in 2003 by the National Institute on Aging (NIA), the National Institute of Biomedical Imaging and Bioengineering (NIBIB), the Food and Drug Administration (FDA), private pharmaceutical companies and non-profit organizations, as a $60 million, 5-year public-private partnership. The primary goal of ADNI has been to test whether serial magnetic resonance imaging (MRI), positron emission tomography (PET), other biological markers, and clinical and neuropsychological assessment can be combined to measure the progression of mild cognitive impairment (MCI) and early Alzheimer’s disease (AD). Determination of sensitive and specific markers of very early AD progression is intended to aid researchers and clinicians to develop new treatments and monitor their effectiveness, as well as lessen the time and cost of clinical trials.
The Principal Investigator of this initiative is Michael W. Weiner, MD, VA Medical Center and University of California – San Francisco. ADNI is the result of efforts of many co-investigators from a broad range of academic institutions and private corporations, and subjects have been recruited from over 50 sites across the U.S. and Canada. The initial goal of ADNI was to recruit 800 subjects but ADNI has been followed by ADNI-GO and ADNI-2. To date these three protocols have recruited over 1500 adults, ages 55 to 90, to participate in the research, consisting of cognitively normal older individuals, people with early or late MCI, and people with early AD. The follow up duration of each group is specified in the protocols for ADNI-1, ADNI-2 and ADNI-GO. Subjects originally recruited for ADNI-1 and ADNI-GO had the option to be followed in ADNI-2. For up-to-date information, see www.adni-info.org.
Genotypes
Subjects from the BWH PhenoGenetic Project and HAB were genotyped using DNA isolated from whole blood and the iPLEX™ Sequenom MassARRAY platform (genotype call rate > 95%, Hardy-Weinberg Equilibrium P value >0.001).
In the ROS, MAP, CHAP and ADNI subjects, imputed dosage values were used for analyses of the rs3865444C allele. In all four cohorts, DNA was extracted from whole blood or frozen post-mortem brain tissue. Genotype data was generated using the Affymetrix Genechip 6.0 platform at the Broad Institute’s Genetic Analysis Platform or the Translational Genomics Research Institute. Both sets of data underwent the same quality control (QC) analyses in parallel using the PLINK toolkit (http://pngu.mgh.harvard.edu/~purcell/plink/) and quality controlled genotypes were pooled. The QC process included a principal components analysis using default parameters in EIGENSTRAT25 to identify and remove population outliers. Imputation in ROS and MAP was performed using MACH software (version 1.0.16a) and HapMap release 22 CEU (build 36). The imputation quality score (INFO) for rs3865444 is 0.9963, suggesting that the SNP is imputed with a high level of confidence.
Cells and Flow Cytometry analysis
Peripheral blood mononuclear cells (PBMCs) were separated by Ficoll-Paque PLUS (GE Healthcare, Piscataway, NJ) gradient centrifugation. PBMCs were frozen at a concentration of 1–3×107 cells/ml in 10% DMSO (Sigma-Aldrich, St. Louis, MO)/90% FCS (Atlanta Biologicals, Lawrenceville, GA). After thawing, PBMCs were washed in 10 ml PBS. PBMCs were stained with anti-human CD33 (clone AC104.3E3; Miltenyi, Auburn, CA) or mouse IgG1 isotype (Miltenyi) in PBS plus 1% FCS. The monocyte gate was defined based on their distinct forward and side-scatter profile. The MFI was acquired on a FACSCalibur (BD Immunocytometry Systems, San Jose,CA) and analyzed with FlowJo software (Tree Star, Ashland, OR). An additive model was used in the analysis, adjusting for age and sex. Given the availability of genotyped samples in the phenogenetic cohort, the AA and CC genotype classes were over-sampled in figure 1a, while the subjects used in figure 1b were not selected by genotype and represent a random sample of subjects from the CHAP, MAP, and ROS cohorts, which are prospective cohorts of initially non-demented individuals.
Uptake assays
We tested monocyte uptake ability in circulating monocytes using two different substrates that have been deployed previously in this experimental paradigm26. After thawing, PBMCs were washed in 10 ml PBS. Either 100,000 PBMCs/well or 50,000/well purified monocytes were incubated with either 1 mg/ml FITC-labeled dextran (Sigma-Aldrich, Saint Louis, MO) or 15 ng/ml HiLyte Fluor 488-labeled Aβ(42) (AnaSpec, San Jose, CA) in 96 well polypropylene plates in HL-1 media plus 5% human serum for 1 hour at 37 °C or 4 °C. Cells were then washed 3 times with PBS and fixed in 4% PFA for 30 minutes. The monocyte gate was defined based on their distinct forward and side-scatter profile. The MFI was acquired on a FACSCalibur (BD Immunocytometry Systems, San Jose, CA) and analyzed with FlowJo software (Tree Star, Ashland, OR). Data shown is the change in MFI (delta MFI) between samples incubated at 4 °C (where uptake is blocked) and 37 °C (where uptake occurs). A dominant model was used in the analysis, adjusting for age and sex.
Pathology, Immunohistochemistry and Microglia quantification
Brains were removed in a standard fashion as previously described27–29. After weighing, each brain was cut into 1cm coronal slabs using a Plexiglas jig. Slabs from one hemisphere, and slabs from the other hemisphere not designated for rapid freezing, were fixed for at least 3 days in 4% paraformaldehyde. We used defined landmarks to obtain at least 2 tissue blocks from each of the following regions: dorsolateral prefrontal cortex, middle and inferior temporal cortex, inferior parietal, hippocampus CA1/subiculum, entorhinal cortex proper, ventromedial caudate, and posterior putamen. Tissue blocks were processed, embedded in paraffin, cut into either 6 micron or 20μm sections, and mounted on glass slides. Neuropathologic diagnoses were made by a board-certified neuropathologist blinded to age and clinical data. Bielschowsky silver stain 6 micron sections were used to visualize neuritic plaques, diffuse plaques, and neurofibrillary tangles in the frontal, temporal, parietal, entorhinal, and hippocampal cortices, as previously described, for the pathologic diagnosis of AD29. A neuropathologic diagnosis of “no AD,” “low likelihood AD,” “intermediate likelihood AD,” or “high likelihood AD” was given based on semiquantitative estimates of neuritic plaque density as recommended by CERAD and the neurofibrillary tangle stage by Braak and Braak as recommended by the National Institute on Aging (NIA) - Reagan criteria30. For analyses, the neuropathologic diagnosis of AD was considered absent if NIA-Reagan diagnosis was no or low and present if intermediate or high likelihood29. The density of neuritic plaques, diffuse plaques and neurofibrillary tangles was characterized using Bielschowsky silver stain for visualization and a graticule to count total number of each in a 1-mm2 area (100 magnification) of highest density. Counts for each marker were completed for each of five regions (midfrontal cortex, middle temporal cortex, inferior parietal cortex, entorhinal cortex, and hippocampus CA1/subiculum) and then converted to standardized scores using the mean of the entire deceased cohort for that region29.
Immunohistochemistry for microglia was performed using an Automated Leica Bond immunostainer (Leica Microsystems Inc., Bannockborn IL) and anti-human HLA-DP, DQ, DR antibodies (clone CR3/43; DakoCytomation, Carpinteria CA; 1:100) using standard Bond epitope retrieval and detection. An investigator blinded to the clinical and pathologic data, outlined the cortical or subcortical gray region of interest on each slide using a Microbrightfield Stereology System. The Stereo Investigator 8.0 software program was used to place a 1000 × 750 μm sampling grid over the region and the program was engaged to sample 4.0% of the region with a 200 × 150 μm counting frame at 400x magnification at interval grid intersection points. Using separate tags for stage 1, 2 and 3 microglia, the operator marked the microglia at each intersection point. These counts were then upweighted by the stereology software to estimate total number of microglia (stage 1, 2 and 3) in the defined area. Different stages of activation from least (stage 1) to most (stage 3) activated can be defined morphologically; when these cells become activated, their long fine processes contract and thicken and the cell body adopts a larger more rounded cellular conformation. Data from the two adjacent blocks of tissue (0.5 to 1.0 cm apart) were averaged to obtain composite average densities of microglia in each region. For CD33 staining, an anti-human CD33 monoclonal antibody (clone PWS44; Leica Biosystems, Germany) was used with diaminobenzidene (DAB) as chromagen. The amyloid deposits were stained with the 10D5 antibody (courtesy of Elan pharmaceuticals) using alkaline phosphatase as the chromagen.
PiB imaging
Amyloid burden was measured with N-methyl-[11C]-2-(4-methylaminophenyl)-6-hydroxybenzothiazole (Pittsburgh Compound B; PiB), which binds to fibrillar amyloid, and was prepared at Massachusetts General Hospital as described previously31, 32. Participants underwent PiB PET imaging as described previously33, 34. Briefly, data were acquired using a Siemens/CTI ECAT HR scanner (3D mode; 63 image planes; 15.2 cm axial field of view; 5.6 mm transaxial resolution and 2.4 mm slice interval; 39 frames: 8×15s, 4×60s, 27×120s). After a transmission scan, 8.5 to 15mCi [11C]-PiB was injected as a bolus and followed immediately by a 60 min dynamic acquisition. PET data were reconstructed and attenuation corrected, and each frame was evaluated to verify adequate count statistics and absence of head motion (interframe head motion, if present, was corrected before further processing).
The dynamic PET data were reconstructed with scatter correction using commercially available routines for 3D PET data. PET data were parameterized by the distribution volume ratio (DVR) computed using the Logan graphical analysis technique35 applied to the frame data acquired 40 – 60 min after injection; this method has been fully validated for PiB imaging36. Time-activity curves were measured in each brain region under analysis (region of interest) and in a reference region in cerebellar cortex known to contain low levels of fibrillar amyloid. This approach has been applied to numerous PiB studies36–39 and yields data that are similar to arterial blood input methods37.
For each subject an index of PiB binding in cortical regions was calculated using the dynamic data via Logan graphical modeling within a large aggregate cortical region of interest consisting of frontal, lateral parietal and temporal, and retrosplenial cortices (the FLR region). PiB retention in the FLR region is substantial in patients with diagnosed AD and has been used as a summary measure of PiB retention in previous studies33, 34, 39. 1.25 is the value used to separate PiB+ and PiB− in the HAB cohort, and 1.5 is used to delineate between PiB+ and PiB− in the ADNI cohort.
Statistical analysis
Demographic information on all datasets was presented using counts and percentages for categorical variables and means and standard deviations or medians and ranges (minimum-maximum) for continuous variables. The rs3865444 single nucleotide polymorphism (SNP) was coded additively relative to the C allele in all analyses. Outcomes of interest were assessed for normality and a square root or natural log transformation was performed if necessary. For analysis of functional assays with small sample size all used Wilcoxon rank sum tests. The associations between rs3865444C and our outcomes of interest were assessed using linear regression models adjusted for experimental batch and study group where appropriate. We evaluated the impact of outliers and possible confounding variables (e.g. age, gender, education level), and adjusted for these variables in our statistical models if they changed the effect estimate associated with the predictor by >10%. In addition to include a covariate for experimental batch in our models where appropriate, to correct for potential batch effects we have also applied an Empirical Bayes priors distribution estimation framework (Combat v. 2.0)40, as implemented in Gene Pattern41 using a parametric method. To correct for multiple comparisons, we performed a standard Bonferroni correction by setting the threshold of significance equal to 0.05 divided by the number of tests performed under each a priori hypothesis. Statistical analyses were performed using SAS software, version 9.3 (SAS Institute, Cary, NC), the PLINK toolkit (http://pngu.mgh.harvard.edu/~purcell/plink/) and R, version 2.13 (www.r-project.org).
Supplementary Material
Acknowledgments
The authors are grateful to the participants in the ROS, MAP, CHAP, HAB, and ADNI studies. This work is supported by the NIH [R01 AG031553, R01 AG30146, R01 AG17917, R01 AG15819, P30 AG10161 and R01 AG11101], and the Illinois Department of Public Health. We also thank the participants of the Brigham & Women’s PhenoGenetic Project; this work was supported by R01 NS067305, RC2 GM093080, and R01 AG043617. EMB is also supported by the JDRF, ADA, BADERC and the Harvard NeuroDiscovery Center.
Footnotes
Author Contributions
E.M.B. and P.L.D. designed and implemented the study. E.M.B. developed the experimental methods utilizing PBMCs. E.M.B., L.O., A.T., L.L.R. and S.I. isolated PBMCs from the PhenoGenetic cohort and analyzed monocyte CD33 expression. E.M.B., A.T. and L.L.R. analyzed uptake ability of monocytes. B.T.K. and L.B.C. performed the statistical analyses and assisted with the interpretation and communication of results presented in the manuscript. A.V., M.L., and P.L.D. coordinated the collection of blood from the PhenoGenetic cohort. R.A.S., K.J., and ADNI provided the PiB imaging data and critically reviewed the manuscript. R.A.S. provided the HAB blood samples. M.C.M is the Principal Investigator of the study that contributed post-mortem data on brain microglia, and critically reviewed the manuscript. J.A.S. was responsible for the microglia and AD pathology data collection from the brains of deceased MAP participants and critical revision of the manuscript. D.A.B. is the principal investigator of the ROS-MAP cohort studies and contributed ante-mortem biospecimens, clinical and post-mortem data, and made critical revisions to the manuscript. P.L.D. is the principal investigator of the BWH PhenoGenetic project; he conceived the study, coordinated access to all of the cohorts, and wrote the manuscript with E.M.B.
References
- 1.Harold D, et al. Nature genetics. 2009;41:1088–1093. doi: 10.1038/ng.440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lambert JC, et al. Nature genetics. 2009;41:1094–1099. doi: 10.1038/ng.439. [DOI] [PubMed] [Google Scholar]
- 3.Seshadri S, et al. JAMA. 303:1832–1840. doi: 10.1001/jama.2010.574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hollingworth P, et al. Nature genetics. 2011;43:429–435. doi: 10.1038/ng.803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Naj AC, et al. Nature genetics. 2011;43:436–441. doi: 10.1038/ng.801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hollingworth P, Harold D, Jones L, Owen MJ, Williams J. Int J Geriatr Psychiatry. 26:793–802. doi: 10.1002/gps.2628. [DOI] [PubMed] [Google Scholar]
- 7.Carrasquillo MM, et al. Molecular neurodegeneration. 2011;6:54. doi: 10.1186/1750-1326-6-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Karch CM, et al. PloS one. 2012;7:e50976. doi: 10.1371/journal.pone.0050976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Griciuc A, et al. Neuron. 2013 doi: 10.1016/j.neuron.2013.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lajaunias F, Dayer JM, Chizzolini C. Eur J Immunol. 2005;35:243–251. doi: 10.1002/eji.200425273. [DOI] [PubMed] [Google Scholar]
- 11.Bennett DA, et al. Curr Alzheimer Res [Google Scholar]
- 12.Bennett DA, Schneider JA, Arvanitakis Z, Wilson RS. Curr Alzheimer Res. doi: 10.2174/156720512801322573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bienias JL, Beckett LA, Bennett DA, Wilson RS, Evans DA. Journal of Alzheimer’s disease: JAD. 2003;5:349–355. doi: 10.3233/jad-2003-5501. [DOI] [PubMed] [Google Scholar]
- 14.Fiala M, Cribbs DH, Rosenthal M, Bernard G. Journal of Alzheimer’s disease: JAD. 2007;11:457–463. doi: 10.3233/jad-2007-11406. [DOI] [PubMed] [Google Scholar]
- 15.Okello A, et al. Neurology. 2009;73:754–760. doi: 10.1212/WNL.0b013e3181b23564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mackenzie IR. Neurology. 2000;55:132–134. doi: 10.1212/wnl.55.1.132. [DOI] [PubMed] [Google Scholar]
- 17.Sperling RA, et al. Alzheimer’s & dementia: the journal of the Alzheimer’s Association. 7:280–292. doi: 10.1016/j.jalz.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sperling RA, Jack CR, Jr, Aisen PS. Sci Transl Med. 3:111cm133. doi: 10.1126/scitranslmed.3002609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Salloway S, et al. Neurology. 2009;73:2061–2070. doi: 10.1212/WNL.0b013e3181c67808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Winblad B, et al. Lancet neurology. 11:597–604. doi: 10.1016/S1474-4422(12)70140-0. [DOI] [PubMed] [Google Scholar]
- 21.Morris JC. Neurology. 1993;43:2412–2414. doi: 10.1212/wnl.43.11.2412-a. [DOI] [PubMed] [Google Scholar]
- 22.Wechsler D. Wechsler Memory Scale–Revised (WMS–R) Psychologial Corporation; San Antonio, TX: 1987. [Google Scholar]
- 23.Folstein MF, Folstein SE, McHugh PR. Journal of psychiatric research. 1975;12:189–198. doi: 10.1016/0022-3956(75)90026-6. [DOI] [PubMed] [Google Scholar]
- 24.Yesavage JA, et al. Journal of psychiatric research. 1982;17:37–49. doi: 10.1016/0022-3956(82)90033-4. [DOI] [PubMed] [Google Scholar]
- 25.Price AL, et al. Nature genetics. 2006;38:904–909. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- 26.Ciaramella A, et al. Journal of Alzheimer’s disease: JAD. 19:559–572. doi: 10.3233/JAD-2010-1257. [DOI] [PubMed] [Google Scholar]
- 27.Bennett DA, et al. Neurology. 2006;66:1837–1844. doi: 10.1212/01.wnl.0000219668.47116.e6. [DOI] [PubMed] [Google Scholar]
- 28.Bennett DA, Schneider JA, Bienias JL, Evans DA, Wilson RS. Neurology. 2005;64:834–841. doi: 10.1212/01.WNL.0000152982.47274.9E. [DOI] [PubMed] [Google Scholar]
- 29.Schneider JA, Wilson RS, Bienias JL, Evans DA, Bennett DA. Neurology. 2004;62:1148–1155. doi: 10.1212/01.wnl.0000118211.78503.f5. [DOI] [PubMed] [Google Scholar]
- 30.Neurobiology of aging. 1997;18:S1–2. [PubMed] [Google Scholar]
- 31.Mathis CA, et al. Journal of medicinal chemistry. 2003;46:2740–2754. doi: 10.1021/jm030026b. [DOI] [PubMed] [Google Scholar]
- 32.Klunk WE, et al. Annals of neurology. 2004;55:306–319. doi: 10.1002/ana.20009. [DOI] [PubMed] [Google Scholar]
- 33.Gomperts SN, et al. Neurology. 2008;71:903–910. doi: 10.1212/01.wnl.0000326146.60732.d6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hedden T, et al. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2009;29:12686–12694. doi: 10.1523/JNEUROSCI.3189-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Logan J, et al. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 1990;10:740–747. doi: 10.1038/jcbfm.1990.127. [DOI] [PubMed] [Google Scholar]
- 36.Price JC, et al. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 2005;25:1528–1547. doi: 10.1038/sj.jcbfm.9600146. [DOI] [PubMed] [Google Scholar]
- 37.Lopresti BJ, et al. Journal of nuclear medicine: official publication, Society of Nuclear Medicine. 2005;46:1959–1972. [PubMed] [Google Scholar]
- 38.Fagan AM, et al. Annals of neurology. 2006;59:512–519. doi: 10.1002/ana.20730. [DOI] [PubMed] [Google Scholar]
- 39.Johnson KA, et al. Annals of neurology. 2007;62:229–234. doi: 10.1002/ana.21164. [DOI] [PubMed] [Google Scholar]
- 40.Johnson WE, Li C, Rabinovic A. Biostatistics. 2007;8:118–127. doi: 10.1093/biostatistics/kxj037. [DOI] [PubMed] [Google Scholar]
- 41.Reich M, et al. Nature genetics. 2006;38:500–501. doi: 10.1038/ng0506-500. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.