

Abstract
We describe a solution-phase sensor of lipid-protein binding based on localized surface plasmon resonance (LSPR) of silver nanocubes. When silica-coated nanocubes are mixed into a suspension of lipid vesicles, supported membranes spontaneously assemble on their surfaces. Using a standard laboratory spectrophotometer, we calibrate the LSPR peak shift due to protein binding to the membrane surface and then characterize the lipid-binding specificity of a pleckstrin-homology domain protein.
Introduction
The intracellular environment is dominated by membrane surfaces, and a significant fraction of biochemical processes involves membranes1. Analytical methods for membrane analysis based on chemical labeling have many drawbacks, and hence there is substantial demand for quantitative label-free detection. Techniques, such as backscattering interferometry2, colloidal assembly3, nanowire arrays4, microcantilevers5, acoustic sensing6, and surface plasmon resonance7 have all been reported, but most are impractical for widespread adoption in biological laboratories. More promising for protein-lipid interactions is localized surface plasmon resonances (LSPR), in which binding causes measurable changes in refractive index8–11. However, conventional LSPR techniques typically rely on analyte capture onto nanofabricated surfaces and often necessitate sophisticated instrumentation. The need for quantitative label-free detection methods that are simple, robustly reproducible, and accessible to scientists using generic laboratory equipment remains unmet.
Here, we report a platform that enables label-free measurements of protein binding to membrane surfaces on a standard laboratory spectrophotometer. We have previously described label-free detection using the LSPR of thiolated silver nanocubes immobilization on flat substrates.9 This configuration required multiple reactions, a customized detection system, and ultimately proved similarly impractical as the other methods mentioned above. A substantial improvement in utility is achieved here by modifying the system to allow measurements to be performed entirely in the solution phase. Highly monodisperse Ag nanocubes were prepared by an established synthetic protocol12 (Supplementary Fig. 1). In order to create a favorable surface for membrane assembly and suspension in solution, an ultra-thin layer of silica was then grown using Stöber synthesis (Methods). Transmission electron microscopy (TEM) micrographs revealed a uniform silica shell covering the Ag surface with average thickness 3.9 ± 0.2 nm (n = 5, mean ± s.d.) and corners with curvature radius of 19 nm (Fig. 1a and 1b). Elemental maps acquired by high-angle annular dark field scanning TEM show that the silicon and oxygen intensities were strongest on the edges of Ag@SiO2 core-shell nanocube particles (silver core @ silica shell), indicating the shell is conformal and uniform (Fig. 1c–1f, and Supplementary Fig. 2). Additionally, the SiO2 coating provides a shelf life in excess of one year by slowing silver oxidation.
Figure 1.
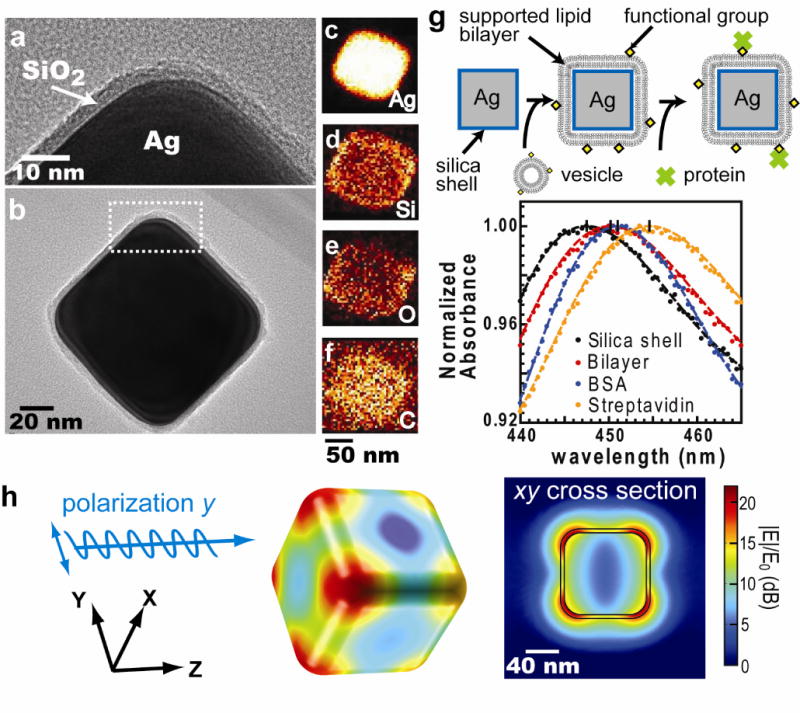
The physical properties of Ag@SiO2 core-shell nanocube. (a) & (b) TEM images of Ag@SiO2 nanocube. (a) is the close-up image of figure (b). (c)~(f) The elemental maps obtained by high-angle annular dark field scanning TEM (HAADF-STEM) with energy dispersive x-ray spectroscopy (EDS). (c) to (f) represent silver, silicon, oxygen, and carbon, respectively. (g) Top: Detection procedure of nanocube sensors. Supported lipid bilayers are formed by vesicle fusion onto the silica surface, and protein binding is monitored by shifts in the LSPR extinction spectrum. Bottom: Typical spectra of membrane coverage and protein binding to the membrane surfaces. Sequential addition of lipid vesicles, BSA, and streptavidin causes LSPR red shifts. (h) Electric field norm (|E|/E0) in decibel (dB) of a nanocube at resonance (n = 1.33303, λ0 = 474 nm) computed using finite-element analysis.
Ag@SiO2 nanocubes exhibit a sharp quadrupolar LSPR scattering peak (Fig. 1g). This is easily observed in the extinction spectrum of a suspension of nanocubes using standard laboratory tools such as a transmission ultraviolet-visible (UV-vis) spectrophotometer, micro-volume spectrometer (e.g. NanoDrop), dark-field microscopy (Supplementary Fig. 3), or light scattering spectrophotometer (Supplementary Fig. 4). Electromagnetic simulations based on the actual particle geometry confirm the time-averaged electric field norms exhibit quadrupole resonance with the highest near-field enhancement near the nanocube corners (Fig. 1h). At quadrupole resonance, |E|IE0 decays to 50% of its value at the silica-media interface over about 10 nm distance. The silica layer is sufficiently thin that the LSPR field still penetrates a lipid bilayer of 3–5 nm thickness (Supplementary Fig. 5). A widely used figure of merit (FOM) for LSPR is the peak shift per refractive index unit (nm / RIU) normalized to the linewidth of the LSPR peak (details in Method section). The FOM for Ag@SiO2 nanocubes is 1.7 versus 2.4 for bare silver nanocubes.
Supported lipid bilayers form spontaneously upon mixing Ag@SiO2 nanocubes into a lipid vesicle suspension (Fig. 1g). Supported membrane formation was confirmed using fluorescence recovery after photobleaching (FRAP) experiments to test the lateral fluidity and connectivity of membranes covering substrate-adsorbed nanocubes9 (Supplementary Fig. 6). The nanocubes were first immobilized on planar glass substrates and then exposed to lipid vesicle suspensions so that a supported lipid bilayer formed on top of both the glass substrate and nanocubes. Bilayers on Ag@SiO2 nanocube-covered substrates exhibited almost identical recovery behavior to bilayers on bare glass (Supplementary Fig. 6). This result indicates that the supported bilayers on Ag@SiO2 nanocubes are fluid and connected to the bilayer on surrounding glass. The magnitude of fluorescence recovery also confirms that the majority of nanocubes are covered with lipid membrane9. In contrast, bilayers on a bare Ag nanocube-covered substrates exhibited similar recovery times but only 60% of the recovery on bare glass, which illustrates that lipids absorbed on bare nanocubes did not form a fluid and continuous bilayer with the surrounding fluid bilayer. Although it has been suggested that supported lipid bilayer cannot form on a highly curved surfaces (11 nm radius of curvature) due to high elastic energy13, we did not observe any such limitation on the Ag@SiO2 nanocubes (19 nm radius of curvature over corner).
The LSPR response of the system is calibrated by monitoring the essentially irreversible binding of streptavidin to biotinylated lipids in the nanocube supported membrane (Supplementary Fig. 7). We employed three different approaches to control the surface density of membrane-bound streptavidin: (i) titrating biotinyl-cap-PE in bilayer; (ii) titrating streptavidin in solution; and (iii) measuring unbound fluorescent streptavidin (details in Supplementary Discussion). LSPR shifts were measured at different known surface densities of streptavidin and exhibited a linear relation with protein density (Fig. 2a). Consistent LSPR responses of 0.191 ± 0.025 ng mm−2 nm−1 (n = 3, mean ± s.d.) were determined by three independent approaches (Supplementary Table 1)
Figure 2.
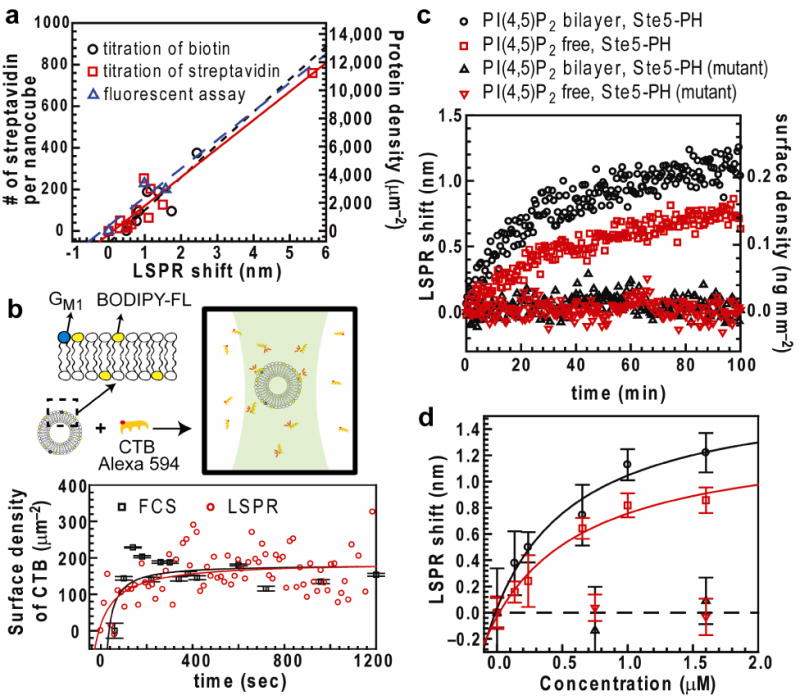
Calibration of the nanocube assay. (a) Relation between LSPR shift and number of streptavidin per nanocube (left vertical axis) and surface density (right axis) measured by titration of biotinyl-cap-PE, titration of streptavidin, and fluorescence measurement of streptavidin concentration. Linear fit slopes are reported in Supplementary Table 1. (b) Top: Concentrations of bound and unbound CTB are detected by multi-component FCS. Alexa 594-CTB binds to vesicles (average diameter 120 nm) containing 0.5% GM1 and 0.5% BODIPY-FL-DHPE lipids. BODIPY-FL-DHPE was used to determine the average number of vesicles diffusing within the excitation spot. Bottom: Binding kinetics measured by multi-component FCS and nanocube assay. (Error bar of FCS, n = 20, mean ± s.d.) CTB surface density was respectively calculated from known vesicle size and LSPR response to protein mass change in streptavidin-biotin systems (0.191 ng mm−2 nm−1). (c) Binding kinetics of wild-type and R407S K411S mutant of GST-Ste5 PH to different membrane surfaces. Concentrations of GST-Ste5 PH = 1.6 μM; GST-Ste5 PH mutant = 1.6 μM) (d) Equilibrium binding curves of GST-Ste5 PH to bilayers Kd = 0.49 ± 0.33 μM (PI(4,5)P2 bilayer) and 1.6 ± 0.45 μM (PI(4,5)P2-free bilayer) (n = 3, mean ± s.e.m.) Error limits of Kd are derived from the statistical error of curve fitting.
To assess that bilayer-coated Ag@SiO2 nanocubes can quantify protein binding accurately, we compared the system with the established method of multi-component fluorescence correlation spectroscopy (multi-component FCS)14. Cholera toxin subunit B (CTB) binding to the membrane-associated receptor GM1 was used as a model system (Fig. 2b). In multi-component FCS measurements, lipid vesicles and CTB were labeled with different fluorophores and the concentrations of bound and unbound CTB were monitored. The average size of vesicles was determined independently by dynamic light scattering, which allowed determination of the surface density of vesicle-bound CTB (details in Supplementary discussion). Using the same materials and under the same experimental conditions, nanocube measurements were performed independently. LSPR response was converted to protein surface density using the LSPR response to protein mass change measured in the biotin-streptavidin system, 0.191 ng mm−2 nm−1 (Supplementary table 1). Kinetics measured by multi-component FCS and nanocube methods reached equilibrium state and the same surface density after 1000 sec (Fig. 2b). It is worth noting that unlike FCS, which only works at low concentration, the nanocube detection strategy has a much broader working range. (details in Supplementary Discussion)
Finally, we used the Ag@SiO2 nanocube assay to examine the heretofore unknown lipid binding specificity of a prototypic mitogen-activated protein kinase (MAPK) scaffold protein, Ste5. Ste5 contains a pleckstrin-homology domain (PH domain, residues 388–518) that is essential for its membrane recruitment and function, but the dependence of Ste5 binding on membrane composition is not well known15. We investigated the binding of Ste5 to membranes with and without PI(4,5)P2. GST-Ste5 PH domain fusion proteins (corresponding to Ste5 residue 369–517), with and without R407S and K411S mutations thought to abrogate lipid binding, were constructed, expressed, and purified from Entamoeba coli. To avoid interference of detergent with the membrane assay, we eliminated its use during protein purification (Supplementary discussion). Only wildtype GST-Ste5 PH domain bound to the membrane surface (Fig 2c). Although more Ste5 binding was observed on PI(4,5)P2 membranes, appreciable binding was also observed on membranes without PI(4,5)P2. This may be due to the presence of phosphatidic acid lipids, which have been observed to association with PH domains in other protein systems16. Binding curves were established to compute the binding affinity of GST-Ste5 on different compositions of membranes (Fig. 2d). At similar lipid compositions, we have previously reported rough estimates of Kd for Ste5-membrane binding using filter-immobilized lipids, liposome flotation assays, and surface plasmon resonance (SPR), that suggest a dissociation constant in the 5–10 μM range15. However, the lipid immobilization and tethering required for the filter and SPR assays are strongly disruptive of the membrane surface environment7 and liposome flotation assays are intrinsically error-prone. Thus, among all of the measurements, we consider the nanocube assay to be the most consistent and most accurate.
We report a core-shell Ag@SiO2 nanocube sensor that can measure protein binding to its membrane-coated surfaces. No complicated fabrication is necessary and these sensors can be prepared on the gram scale (> 1014) at minimal cost. Solution phase measurements readily integrate 1012 nanocubes in the illumination area of a standard spectrophotometer cuvette. This provides sensitivity of approximately 0.19 ng cm−2 based on 0.01nm standard error of 20 consecutive LSPR measurements (details in Supplementary discussion), in contrast to the immobilized format9 (109 nanocubes; sensitivity = 1.5 ng cm−2). This method is applicable to analytes that bind lipid membranes or membrane proteins, including proteins, peptides, nucleic acids, or even entire cells. The biggest advantage of this method is that simply adding Ag@SiO2 nanocubes to a vesicle suspension produces a system in which analytes binding to the membrane surface can be read out by standard spectral technique widely available in most labs, without labeling.
Methods
Materials
Lipids. The following lipids were purchased from Avanti Polar Lipids (Alabaster, AL): 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC), 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(cap-biotinyl) (Biotinyl-Cap-PE), Ganglioside GM1 (GM1), 1,2-dioleoyl-sn-glycero-3-phospho-L-serine (DOPS), 1,2-dioleoyl-sn-glycero-3-phosphate (DOPA), 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE), L-α-phosphatidylinositol (PI), and L-α-phosphatidylinositol-4,5-bisphosphate (PI(4,5)P2). The fluorescent lipid probes, Texas Red 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine (Texas red DPPE) and N-(4,4-difluoro-5,7-dimethyl-4-bora-3a,4a-diaza-s-indacene-3-propionyl)-1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine, triethylammonium salt (BODIPY-FL-DHPE), were purchased from Invitrogen.
Ethanol (200 proof), tetraethyl orthosilicate (TEOS), 28% ammonium hydroxide solution, unlabeled recombinant streptavidin, and bovine serum albumin were purchased from Sigma-Aldrich. The fluorescent proteins Alexa Fluor 647 streptavidin and cholera toxin subunit B (CTB) Alexa Fluor 594 were purchased from Invitrogen. Streptavidin and CTB binding experiments were performed in 1 × PBS buffer (Mediatech). GST-Ste5 binding measurements were performed in HKME buffer (20 mM HEPES-KOH at pH = 7.0, 160 mM KOAc, 1 mM MgCl2, 0.1 mM EGTA).
Silica-coated nanocube
Ag nanocubes are synthesized using the polyol method12,17,18 capped with poly(vinylpyrrolidone) (PVP), and stored in ethylene glycol before use. Silica shells were coated on Ag nanocubes using Stöber process.19 The concentration of ammonium hydroxide and reaction time affected the thickness and quality of the silica layer.20 Ag nanocubes were first washed extensively with ethanol. Silica layers were coated by mixing 7.5ml of Ag nanocube suspension in ethanol with 1950 μl of water, 600 μl of TEOS, and 300 μl of 0.28% ammonium hydroxide. The solution was sonicated during the entire reaction. After 40min reaction, the Ag@SiO2 nanocubes were washed with ethanol to remove the reagents and then washed extensively with water. The Ag@SiO2 nanocubes were stored in deionized water for future use.
LSPR measurement
Various approaches have been reported to collect nanoparticle extinction spectra21. We employed a general transmission ultraviolet-visible (UV-vis) spectrophotometer (Cary 100, Varian). Typically, spectral shifts were monitored by detecting the prominent quadrupolar LSPR peak λmax. These peaks were determined by fitting transmission spectra to a seventh-order polynomial (Fig. 1g). The dependence of LSPR peak shift on refractive index was measured in water-glycerol solutions of various ratios. To explore the effect of the silica shell, the refractive index sensitivity of Ag@SiO2 nanocubes was compared to Ag nanocubes using solutions of water and glycerol. (Supplementary Fig. 8) LSPR sensitivity was quantified using the widely reported figure of merit (FOM) calculated by dividing refractive index sensitivity by the line width of resonance spectrum (FOM = S/Δλ)22,23 The refractive index sensitivity (S) was evaluated from Supplementary Fig. 8 and represented as peak shift (reported in nm or eV) per refractive index unit (RIU). The line width of the resonance spectrum (Δλ) was obtained from the full width at half maximum (FWHM) of the LSPR peak (Fig. 1g).
To demonstrate the applicability of other detection schemes, scattering spectra were also measured by (1) dark field scattering microscopy using a dark field condenser and spectrometer (USB2000, Ocean Optics), and (2) a fluorescence spectrophotometer (Varian, Inc) configured for 90 degree scattering detection.
The nanocube concentrations were determined by counting deposited nanocubes on glass substrates. The silica-coated nanocube solutions were incubated in a sedimentation chamber for two days to create monolayers of nanocubes. Dark field microscopy was used to observe the nanocubes deposited on the bottom of each sedimentation chamber. A homemade image analysis program was developed to count the number of nanocubes in each imaging frame.
Bilayer preparation
Lipid vesicles. The desired composition of lipids was first mixed in chloroform. The mixture was then dried in a round bottom flask followed by desiccation under nitrogen for at least 30 minutes. Lipid films were then hydrated with 18.2 MΩ·cm deionized (DI) water. The resulting suspension was probe sonicated to clarity in an ice bath and ultracentrifuged at 4 °C for 45 min. The top small unilamellar vesicle (SUV) solution was extracted for use in experiments. For FCS and GST-Ste5 binding experiments, SUVs were prepared through an extrusion process. Instead of sonicating, the hydrated lipids were extruded through 100 nm polycarbonate pore filters (Whatman, UK) until the suspension reached clarity. The vesicle used in FCS measurement contains 0.5% GM1, 0.5% BODIPY-FL-DHPE and 99% DOPC lipids. The lipid membranes used in GST-Ste5 PH binding experiment contain: (1) 53% DOPC, 22% DOPE, 10% DOPS, 5% DOPA, 10% PI for PIP2-free bilayer and (2) 53% DOPC, 22% DOPE, 10% DOPS, 5% DOPA, 5% PI, 5% PI(4,5)P2 for PIP2 bilayer.
Supported lipid bilayers. Supported lipid bilayers were formed by adapting a standard vesicle fusion technique3. Bilayers were assembled by combining equal volumes of SUV suspension and the desired buffer in a small centrifuge tube, followed by vortex mixing. Excess vesicles and salt were removed by rinsing twice with the buffer using a benchtop centrifuge (minicentrifuge, VWR, maximum RCF = 2000 g). Membrane-coated particles were then diluted to the desired working concentration and introduced into the spectrophotometer cell.
Protein binding measurement
Bilayer-coated nanocubes were incubated with 0.05 mg ml−1 BSA solution to block nonspecific binding prior to adding desired proteins. Fifteen consecutive scans were performed to obtain the average λmax of the LSPR quadrupolar peak as a baseline. The desired amount of protein was directly cast into the spectrophotometer cell (400 μL sample volume) followed by pulse vortexing of the mixture. Spectra in the range of 430 nm to 480 nm were scanned immediately after mixing at 0.5 nm spectral resolution. The maximum attainable scanning rate was six seconds per spectrum, limited by the configuration of the UV-vis spectrophotometer. To minimize the use of protein in GST-Ste5 binding experiments, these measurements were performed with a sub-microvolume optical cuvette. Different volumes of protein (0.5–15μl) were incubated with 20μl of bilayer-coated Ag@SiO2 nanocube sensors for two hours. The average λmax of the LSPR quadrupolar peak were obtained from ten consecutive spectra. All experiments were performed at room temperature.
Fluorescent correlation spectroscopy
Fluorescence correlation spectroscopy (FCS) measurements were performed on a home-built FCS apparatus based on a Nikon TE2000 inverted fluorescence microscope as described previously24. Two laser beams, 488 nm and 568 nm, were coupled into an optical fiber and focused by a 100 × TIRF objective (Nikon) onto the sample to excite the fluorescent probes. The emitted light was filtered through notch filters and a confocal pinhole then separated by a 560 nm long-pass filter. Before focusing onto two avalanche photodiodes (APDs) (Perkin and Elmer), two color filters were used to minimize spectrum crosstalk. The photon arrival time was recorded and the auto-correlation functions of the two APD signals were calculated with a hardware correlator (Correlator.com) in real time. Using a double-labeled supported lipid bilayer as a sample, overlapping detection volumes were obtained by careful alignment of a collimator lens after the optical fiber and fine adjustment of the objective lens correction collar25. Measurements were made in eight-well chambered coverglass (Nunc) that were first soaked with 0.1 M NaOH for 20 min to clean the bottom surface. The supported lipid bilayers (100% DOPC) were formed on the bottom surface by vesicle fusion. The chamber was incubated with 0.1mg/ml BSA to prevent the protein and vesicle absorption. The size and the structure factor s of the excitation volume were calibrated using 200nM fluorescein in 1M NaOH solution with a known diffusion coefficient (D = 300 μm−2 s−1)26. All other measurements were performed at 24.5 °C in 1 × PBS buffer.
The model system, CTB binding to vesicles containing the membrane associated receptor monosialoganglioside GM1, was selected to directly compare FCS and nanocube measurements. To obtain a narrow size distribution of vesicles, SUVs were prepared by the standard extrusion method described above. Vesicles of 120 nm average diameter containing 0.5% GM1, 0.5% BODIPY-FL-DHPE and 99% DOPC lipids were measured by dynamic light scattering (Brookhaven Instruments Corp.). A detailed description of the multi-component FCS calculations is shown in the Supplementary Discussion.
TEM
Ag@SiO2 nanocubes were imaged using high-resolution transmission electron microscopy (JEOL 2100-F, 200 kV). The elemental x-ray analysis maps were generated using high-angle annular dark field scanning TEM (HAADF-STEM) with an energy dispersive x-ray spectroscopy (EDS) detector. TEM images revealed nanocube a lateral dimension of 98 nm, 19 nm radius of curvature at the edges, and silica shell thickness of 3.9 nm.
LSPR simulation
Finite element simulations using COMSOL were used to model the LSPR of silica-coated silver nanocubes. Free tetrahedral meshing of the geometry observed in TEM was performed in COMSOL, and further refined in the vicinity of the silica shell. The final mesh contained 359,000 tetrahedral elements, and convergence of absorption spectra within 0.1% error was confirmed by comparing results from a coarser mesh.
Frequency-domain scattered electric field solutions were computed using COMSOL’s RF module for a background oscillating field of arbitrary amplitude 1 V m−1. Real and imaginary refractive index dispersion was interpolated from literature tables for silver and silica27. The nanocube was simulated inside a sphere of diameter 400 nm, sufficiently large for all near-field effects to be negligible at the system boundary. A perfectly matched layer (PML) was additionally incorporated to cancel any reflection artifacts in the simulation. Field solutions were calculated for 50–100 different frequencies at a time.
GST-Ste5 Protein preparation
GST-Ste5 PH domain fusion proteins with and without R407S K411S mutations (corresponding to Ste5 residue 369–517) were constructed, expressed, and purified from Escherichia coli as described by Garrenton et al15. The use of Tween-20 detergent was omitted during protein purification to avoid the influence of detergent on lipid bilayers. Prior to binding experiments, GST-Ste5 proteins were treated with Amicon centrifuge filters (Millipore) for further purification and buffer exchange.
Supplementary Material
Acknowledgments
This work was supported by the Director, Office of Science, Office of Basic Energy Sciences, of the US Department of Energy under contract no. DE-AC02-05CH11231 (to J.T.G.) and by US National Institutes of Health Research Grant GM21841 (to J.T.). P.Y. would like to acknowledge the support from King Abdulaziz University.
Footnotes
Author Contributions
H.-J.W. and J.T.G. conceived the solution-phase nanocube sensor strategy. H.-J.W. implemented the experiments, J.H. synthesized nanocubes and performed TEM, W.-C.L. performed FCS measurements, C.R. performed LSPR simulations and Z.L. and E.S. prepared Ste5 proteins. H.-J.W., C.R. and J.T.G. wrote the manuscript. J.T.G., J.T. and P.Y. supervised the project. All authors discussed the results and commented on the manuscript at all stages.
Competing Financial Interests
The authors declare no competing financial interests.
References
- 1.Kuriyan J, Groves JT. Nat Struct Mol Biol. 2010;17:659–665. doi: 10.1038/nsmb.1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baksh MM, Kussrow AK, Mileni M, Finn MG, Bornhop DJ. Nat Biotechnol. 2011;29:357–U173. doi: 10.1038/nbt.1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baksh MM, Jaros M, Groves JT. Nature. 2004;427:139–141. doi: 10.1038/nature02209. [DOI] [PubMed] [Google Scholar]
- 4.Zheng GF, Patolsky F, Cui Y, Wang WU, Lieber CM. Nat Biotechnol. 2005;23:1294–1301. doi: 10.1038/nbt1138. [DOI] [PubMed] [Google Scholar]
- 5.Braun T, et al. Nat Nanotechnol. 2009;4:179–185. doi: 10.1038/nnano.2008.398. [DOI] [PubMed] [Google Scholar]
- 6.Cooper MA. J Mol Recognit. 2004;17:286–315. doi: 10.1002/jmr.675. [DOI] [PubMed] [Google Scholar]
- 7.Beseničar M, Maček P, Lakey JH, Anderluh G. Chem Phys Lipids. 2006;141:169–178. doi: 10.1016/j.chemphyslip.2006.02.010. [DOI] [PubMed] [Google Scholar]
- 8.Dahlin A, et al. J Am Chem Soc. 2005;127:5043–5048. doi: 10.1021/ja043672o. [DOI] [PubMed] [Google Scholar]
- 9.Galush WJ, et al. Nano Lett. 2009;9:2077–2082. doi: 10.1021/nl900513k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jonsson MP, Jonsson P, Dahlin AB, Hook F. Nano Lett. 2007;7:3462–3468. doi: 10.1021/nl072006t. [DOI] [PubMed] [Google Scholar]
- 11.Baciu CL, Becker J, Janshoff A, Sonnichsen C. Nano Lett. 2008;8:1724–1728. doi: 10.1021/nl080805l. [DOI] [PubMed] [Google Scholar]
- 12.Tao A, Sinsermsuksakul P, Yang PD. Angew Chem Int Edit. 2006;45:4597–4601. doi: 10.1002/anie.200601277. [DOI] [PubMed] [Google Scholar]
- 13.Roiter Y, et al. Langmuir. 2009;25:6287–6299. doi: 10.1021/la900119a. [DOI] [PubMed] [Google Scholar]
- 14.Middleton ER, Rhoades E. Biophys J. 2010;99:2279–2288. doi: 10.1016/j.bpj.2010.07.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Garrenton LS, Young SL, Thorner J. Gene Dev. 2006;20:1946–1958. doi: 10.1101/gad.1413706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhao C, Du GW, Skowronek K, Frohman MA, Bar-Sagi D. Nat Cell Biol. 2007;9:706–U171. doi: 10.1038/ncb1594. [DOI] [PubMed] [Google Scholar]
- 17.Fievet F, Lagier JP, Blin B, Beaudoin B, Figlarz M. Solid State Ionics. 1989;32–33:198–205. [Google Scholar]
- 18.Sun YG, Xia YN. Science. 2002;298:2176–2179. doi: 10.1126/science.1077229. [DOI] [PubMed] [Google Scholar]
- 19.Stober W, Fink A, Bohn E. J Colloid Interface Sci. 1968;26:62. [Google Scholar]
- 20.Sioss JA, Stoermer RL, Sha MY, Keating CD. Langmuir. 2007;23:11334–11341. doi: 10.1021/la7019846. [DOI] [PubMed] [Google Scholar]
- 21.Willets KA, Van Duyne RP. Annu Rev Phys Chem. 2007;58:267–297. doi: 10.1146/annurev.physchem.58.032806.104607. [DOI] [PubMed] [Google Scholar]
- 22.Sherry LJ, et al. Nano Lett. 2005;5:2034–2038. doi: 10.1021/nl0515753. [DOI] [PubMed] [Google Scholar]
- 23.Mayer KM, Hafner JH. Chem Rev. 2011;111:3828–3857. doi: 10.1021/cr100313v. [DOI] [PubMed] [Google Scholar]
- 24.Forstner MB, Yee CK, Parikh AN, Groves JT. J Am Chem Soc. 2006;128:15221–15227. doi: 10.1021/ja064093h. [DOI] [PubMed] [Google Scholar]
- 25.Bacia K, Schwille P. Nat Protoc. 2007;2:2842–2856. doi: 10.1038/nprot.2007.410. [DOI] [PubMed] [Google Scholar]
- 26.Chen Y, Muller JD, Eid JS, Gratton E. In: In New Trends in Fluorescence Spectroscopy: Applications to Chemical and Life Sciences. Valeur B, Brochon JC, editors. Springer; Berlin: 2001. pp. 277–302. [Google Scholar]
- 27.Palik ED. Handbook of Optical Constants of Solids. Elsevier; Amsterdam: 1998. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.