

Abstract
Purpose
Angiotensin-(1-7) [Ang-(1-7)] is an endogenous peptide hormone of the reninangiotensin system with antiproliferative and antiangiogenic properties. The primary objective of this study was to establish the recommended phase II dose of Ang-(1-7) for treating patients with advanced cancer. Secondary objectives were to assess toxicities, pharmacokinetics, clinical activity, and plasma biomarkers.
Experimental Design
Patients with advanced solid tumors refractory to standard therapy were treated with escalating doses of Ang-(1-7) in cohorts of three patients. Ang-(1-7) was administered by s.c. injection once daily for 5 days on a 3-week cycle. Tumor measurements were done every two cycles and treatment was continued until disease progression or unacceptable toxicity.
Results
Eighteen patients were enrolled. Dose-limiting toxicities encountered at the 700 μg/kg dose included stroke (grade 4) and reversible cranial neuropathy (grade 3). Other toxicities were generally mild. One patient developed a 19% reduction in tumor measurements. Three additional patients showed clinical benefit with stabilization of disease lasting more than 3 months. On day 1, Ang-(1-7) administration led to a decrease in plasma placental growth factor (PlGF) levels in patients with clinical benefit (P = 0.04) but not in patients without clinical benefit (P = 0.25). On day 5, PlGF levels remained lower in patients with clinical benefit compared with patients without clinical benefit (P = 0.04).
Conclusions
Ang-(1-7) is a first-in-class antiangiogenic drug with activity for treating cancer that is linked to reduction of plasma PlGF levels. The recommended phase II dose is 400 μg/kg for this administration schedule.
The systemic renin-angiotensin system is an essential regulator in the vasculature, controlling blood pressure and fluid homeostasis. Local tissue renin-angiotensin systems also exist and are involved in a variety of autocrine, intracrine, and paracrine functions (1, 2). The eight–amino acid peptide angiotensin II (Ang II), a major effector hormone of the system, is a potent vasoconstrictor and mitogen, whereas angiotensin-(1-7) [Ang-(1-7)] produces unique physiologic responses that often oppose Ang II actions (1-3). Ang-(1-7) is present in the circulation and tissues at concentrations similar to Ang II and functions as a vasodilator with antiproliferative and antiangiogenic properties (4).
Ang II is generated from angiotensin I following cleavage by the peptidase angiotensin-converting enzyme (3, 4). Ang-(1-7) is also formed from angiotensin I following cleavage by other peptidases, including neprilysin. Ang-(1-7) is alternatively generated following cleavage of Ang II by angiotensin-converting enzyme 2, as shown in Fig. 1. Both Ang II and Ang-(1-7) mediate their biological effects through interaction with distinct, high-affinity angiotensin receptors to activate molecular signaling pathways. Ang II is an agonist for the Ang II type 1 and type 2 receptors, whereas Ang-(1-7) activates the unique G protein–coupled receptor mas (1-5).
Fig. 1.
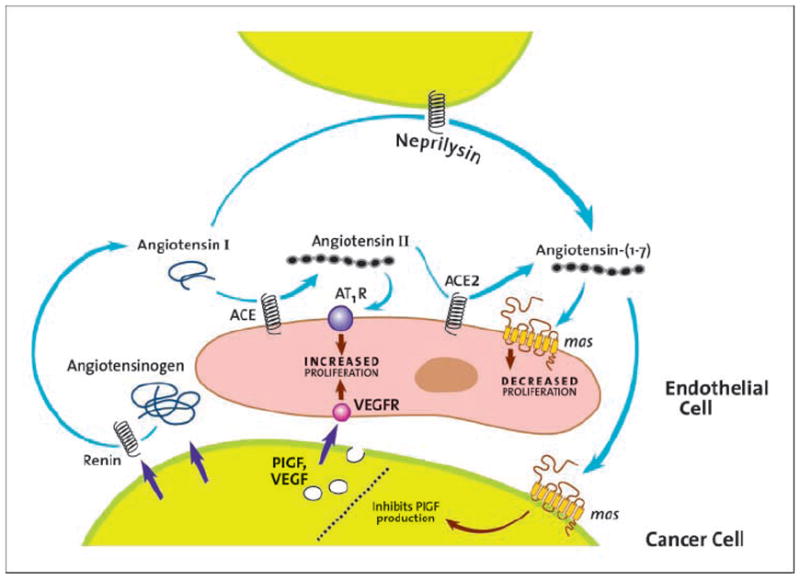
The renin-angiotensin system in cancer and antiangiogenic mechanisms targeted by Ang-(1-7).
The antimitogenic effects of Ang-(1-7) were initially shown in vitro and in vivo in vascular smooth muscle cells. Ang-(1-7) inhibited the proliferation of vascular smooth muscle cells (6) and reduced neointimal formation in the carotid artery following vascular injury (7) and in the abdominal aorta following stent implantation (8). Further, Ang-(1-7) significantly reduced lung cancer cell proliferation in a receptor-mediated process (9) and reduced lung tumor growth in a xenograft model with a concomitant reduction in cyclooxygenase-2 (10). Ang-(1-7) treatment also decreased microvessel density, which was associated with a reduction in vascular endothelial growth factor (VEGF) and placental growth factor (PlGF) in lung and breast tumor xenografts (11, 12). These results suggest that Ang-(1-7) may inhibit tumor growth by reducing proangiogenic factors to attenuate angiogenesis.
These observations led us to the hypothesis that Ang-(1-7) might be useful as a novel antiangiogenic therapy. A prior phase I study examining Ang-(1-7) as a myeloprotective agent failed to reach maximum tolerated dose, and the highest administered dose was used as the starting dose for this study (13). To our knowledge, no other drugs targeting the mas receptor have been developed for treating cancer.
This phase I trial was undertaken to establish a phase II dose of Ang-(1-7) for treating patients with advanced cancer. Plasma levels of proangiogenic hormones were measured to investigate whether changes in circulating levels of these paracrine hormones are associated with clinical outcomes.
Materials and Methods
Patient eligibility
Patients were required to have advanced solid tumors refractory to standard therapy. Patients also were required to have a pathologically documented malignancy and an Eastern Cooperative Oncology Group performance status of 0 to 2. Patients were ineligible if they were taking angiotensin-converting enzyme inhibitors or Ang II receptor blockers, had brain metastases, were pregnant or breast-feeding, or were receiving therapeutic anticoagulation. Required laboratory criteria at study entry included an absolute neutrophil count of ≥1,500/μL, platelet count of ≥100,000/μL, estimated creatinine clearance of >30 mL/min, total bilirubin of <2 mg/dL, and aspartate aminotransferase and alanine aminotransferase <3 times the upper limit of normal. Prior therapies (including chemotherapy, surgery, and radiation) had to be completed at least 4 wk before enrollment.
Study design
Ang-(1-7) was produced by Bachem AG under good manufacturing protocol conditions. Compounding was done to produce vials of Ang-(1-7) at concentrations of 10 and 50 mg/mL. Vials were stored frozen, and after thawing, vials were maintained at refrigerated temperatures for no more than 2 wk.
Ang-(1-7) was administered by s.c. injection once daily for 5 consecutive days every 21 d (one cycle = 21 d). Toxicities were assessed weekly during the first cycle and on the first day of each subsequent cycle. Tumor measurements were done every two cycles. In the absence of unacceptable treatment-related toxicity, patients continued treatment until disease progression. Intrapatient dose escalation was not allowed.
This study was approved by the Institutional Review Board of Wake Forest University and the Food and Drug Administration. It was registered in the National Cancer Institute PDQ Database and ClinicalTrials. gov as NCT00471562. All patients were required to give written informed consent.
Dose escalation
Planned dose levels were 100, 200, 400, 700, and 1,000 μg/kg. Serious adverse events (SAE) were defined as grade 3 or greater toxicities. Dose-limiting toxicities (DLT) were defined as SAEs at least possibly related to Ang-(1-7) treatment.
Patients were enrolled in cohorts of three patients starting at the 100 μg/kg dose. Escalation to the next level occurred if the maximum tolerated dose was not reached and a sufficient number of patients in a cohort were treated for at least 3 wk. If one of the first three patients enrolled in a cohort experienced a DLT, the cohort was expanded to include three additional patients. If none of these three additional patients experienced DLT, escalation to the next cohort occurred. If one or more of the additional patients experienced DLT, dose escalation was stopped, and this dose was designated the highest administered dose. A total of six patients were required to be treated at the maximum tolerated dose, which was defined as the highest dose level at which no more than one of six patients experienced DLT.
Patient assessment
Physical examination and routine laboratory tests were done weekly during the first cycle and on day 1 of subsequent cycles. Toxicities were graded using the Common Terminology Criteria for Adverse Events, version 3.0. Blood pressure was monitored before treatment for each dose and for 6 h after treatment on days 1 and 5 of the first cycle. Tumor measurements were done before initiation of treatment and then every two cycles until disease progression or unacceptable toxicity.
Measurement of drug and angiogenic hormone levels
Blood samples were obtained immediately before treatment as well as 1, 2, 3, 4, and 6 h after Ang-(1-7) administration on days 1 and 5 of the first cycle using a dedicated i.v. access. Blood samples were immediately placed on ice and processed within 30 min of collection. Plasma samples were stored at -80°C. Ang-(1-7) concentrations were measured by the Hypertension Core Facility of Wake Forest University using an established RIA method (14).
Aliquots of plasma from the same time points were assayed by Pierce Biotechnology using Searchlight ELISA technology to quantify circulating VEGF, PlGF, and basic fibroblast growth factor. Samples were blinded before shipping. Aliquots of plasma from each time point were assayed at three dilutions (1:2, 1:50, and 1:1,000), and two replicates were done for each dilution. The Searchlight ELISA measurements were done using proprietary antibodies specific to VEGF, PlGF, and basic fibroblast growth factor (Pierce Biotechnology). A standard curve of known concentrations for each peptide was done at the same time using the same dilution scheme. For experimental samples, the value from the highest sample concentration within the range for the standard curve was reported as the peptide concentration for each time point.
Pharmacology and biomarker evaluation
Pharmacokinetic parameters were estimated using a one-compartment model. Biomarkers were analyzed using random coefficient modeling estimated by maximum likelihood. Biomarker levels were modeled after log transformation, consideration of quadratic effects of time (after centering), and adjustment for plasma drug levels. Hemolysis was assessed in all plasma samples and five samples from three patients were excluded from biomarker modeling due to marked hemolysis.
Statistical methods
Frequency and severity of treatment-related toxicities were examined by cohort. All analyses were two sided, and a P value of <0.05 was considered statistically significant. Analyses were done using Statistical Analysis System v9.1.3 (SAS Institute) and Stata v10.1 (StataCorp) and were done by the Core Biostatistics Facility of the Comprehensive Cancer of Wake Forest University.
Results
Patients
Eighteen patients were enrolled between April 2007 and October 2008. All patients were evaluable for toxicity. Fifteen patients were evaluable for response. The patient characteristics are displayed in Table 1.
Table 1.
Patient characteristics
| Parameter | n (%) or mean ± SD (min-max) |
|---|---|
| Age (y) | 61.3 ± 12.0 (40-82) |
| Gender | |
| Male | 12 (67) |
| Female | 6 (33) |
| Race | |
| Caucasian | 15 (83) |
| African-American | 3 (17) |
| Ethnicity | |
| Hispanic | 0 |
| Non-Hispanic | 18 (100) |
| Cancer diagnosis | |
| Colorectal | 6 (33) |
| Lung | 1 (6) |
| Pancreatic | 2 (11) |
| Prostate | 3 (17) |
| Sarcoma | 3 (17) |
| Other | 3 (17) |
| Prior therapy | |
| Chemotherapy | 18 (100) |
| Radiation therapy | 14 (78) |
| No prior therapy | 0 |
| No. subjects per cohort | |
| 100 μg/kg | 3 (17) |
| 200 μg/kg | 3 (17) |
| 400 μg/kg | 6 (33) |
| 700 μg/kg | 6 (33) |
Dose escalation and toxicity
No hematologic toxicity was observed. Nonhematologic toxicity possibly related to treatment is summarized in Table 2. Treatment was generally well tolerated with mild muscle cramps occurring in three patients. One patient developed gingival regression, which was attributed to Ang-(1-7) because no other etiology could be identified by a consulting dentist. There were no treatment-related deaths.
Table 2.
Adverse events at least possibly related to treatment
| Toxicity | Grade
|
|||
|---|---|---|---|---|
| 2 | 3 | 4 | Total | |
| Cerebrovascular ischemia | 0 | 0 | 1 | 1 |
| Fatigue | 2 | 0 | 0 | 2 |
| Gingival regression | 1 | 0 | 0 | 1 |
| Hypocalcemia | 1 | 0 | 0 | 1 |
| Orthostasis | 1 | 0 | 0 | 1 |
| Muscle weakness | 1 | 0 | 0 | 1 |
| Neuropathy, cranial | 0 | 1 | 0 | 1 |
| Musculoskeletal pain | 2 | 0 | 0 | 2 |
| Thrombosis/thrombus/embolism | 0 | 1 | 0 | 1 |
| Total | 10 | 2 | 1 | 12 |
Three SAEs possibly related to Ang-(1-7) were observed during the study. One patient with metastatic pancreatic cancer treated at 400 μg/kg developed a grade 3 deep vein thrombosis possibly related to Ang-(1-7) during the first cycle of therapy. The patient reported calf pain that started the evening after the first dose of treatment and worsened during the week of treatment. An ultrasound confirmed the presence of a deep vein thrombosis, and the patient was started on low–molecular weight heparin. A total of six patients were treated at the 400 μg/kg dose level, and no other treatment-related SAEs were observed.
One patient with metastatic lung cancer treated at 700 μg/kg developed multiple strokes (grade 4). On day 6 of the third cycle, the patient became acutely confused. Magnetic resonance imaging of the brain revealed multiple bilateral small strokes with a vasculitic pattern. A workup for an embolic source was negative. A consulting neurologist indicated that the event was possibly related to Ang-(1-7) administration. After discontinuing treatment, the patient’s neurologic symptoms improved but did not resolve completely. The patient eventually died from a pathologically confirmed malignant pleural effusion for which she declined chest tube placement 31 days after discontinuing treatment.
The 700 μg/kg cohort was expanded to include six patients, and one additional patient experienced a SAE possibly related to Ang-(1-7). The patient had metastatic prostate cancer and experienced a grade 3 cranial neuropathy. The patient reported sudden difficulty eating 2 weeks after receiving the first treatment. On exam, the patient was unable to deviate the tongue to one side. Magnetic resonance imaging of the brain was negative for stroke. Evaluation by a consulting neurologist indicated a peripheral neuropathy. Four weeks after discontinuing his Ang-(1-7) treatment, tongue function recovered completely.
SAEs not related to Ang-(1-7) included hospitalizations for the following reasons: pain due to cancer in one patient, ureteral obstruction due to cancer in one patient, lymphedema due to cancer in one patient, bradycardia due to medications in one patient, and gastroenteritis due to food poisoning in one patient.
With two of six patients experiencing SAEs at least possibly related to treatment at the 700 μg/kg dose, 400 μg/kg was determined to be the recommended phase II dose for this schedule. Six patients were treated at the 400 μg/kg dose and only one of six experienced a SAE (the patient with deep vein thrombosis discussed above).
Clinical activity
One patient with metastatic sarcoma that was progressing at the time of enrollment who was treated at the 700 μg/kg dose experienced a mixed response to therapy with an overall reduction in the sum of unidimensional measures of 19%. The patient continued on therapy for 10 months before eventually developing progressive disease (Table 3).
Table 3.
Number of cycles administered and best response for individual patients
| Dose level (μg/kg) | Cancer diagnosis | Cycles | DLT | Response |
|---|---|---|---|---|
| 100 | Urachal | 2 | No | PD |
| 100 | Prostate | 6 | No | SD |
| 100 | Sarcoma | 2 | No | PD |
| 200 | Sarcoma | 5 | No | SD |
| 200 | Pancreatic | 2 | No | PD |
| 200 | Colorectal | 2 | No | PD |
| 400 | Colorectal | 2 | No | PD |
| 400 | Pancreatic | 1 | Yes | n/a |
| 400 | Prostate | 2 | No | PD |
| 400 | Colorectal | 6 | No | SD |
| 400 | Colorectal | 1 | No | PD |
| 400 | Colorectal | 2 | No | PD |
| 700 | Colorectal | 2 | No | PD |
| 700 | Lung | 3 | Yes | n/a |
| 700 | Anal | 2 | No | PD |
| 700 | Sarcoma | 14 | No | MR |
| 700 | Prostate | 1 | Yes | n/a |
| 700 | Head and neck | 1 | No | PD |
Abbreviations: PD, progressive disease; SD, stable disease; MR, minor response; n/a, not available.
Three additional patients experienced stabilization of disease lasting more than 3 months. One of these patients had metastatic prostate cancer and showed a small consistent decrease in prostate-specific antigen on day 5 compared with day 1 of each cycle. One other patient with metastatic sarcoma developed cystic changes in a liver metastasis without a reduction in tumor size. This patient also experienced a 73% reduction in plasma PlGF levels after treatment on day 1.
Pharmacokinetic and biomarker analyses
Logarithmic plots of drug concentrations according to cohort are displayed in Fig. 2. Pretreatment concentrations of Ang-(1-7) were less than 35 pg/mL in all patients, similar to the values reported for healthy volunteers (15). The drug was rapidly bioavailable after s.c. injection with maximum drug levels achieved at 1 hour in almost all patients. Dose-dependent increases in drug exposure were observed only at the highest dose level. A single patient receiving the lowest drug dose had high drug levels and a prolonged drug half-life, which may explain the apparent lack of dose dependence among the lower dose levels. The mean half-life was similar across the three highest dose levels ranging from 0.42 to 0.61 hour (Supplementary Tables S1 and S2), in agreement with a prior phase I study that examined lower Ang-(1-7) doses (13).
Fig. 2.
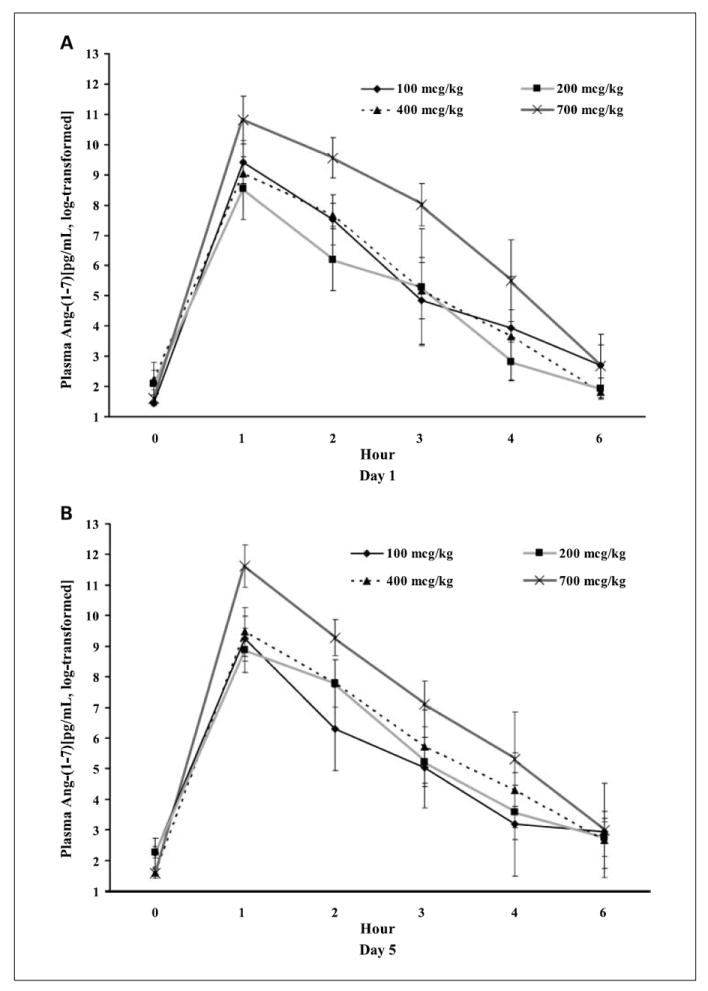
Plasma concentrations of Ang-(1-7) (A) on day 1 and (B) day 5 after log transformation.
Plasma levels of angiogenic biomarkers were examined over time after Ang-(1-7) administration. Circulating levels of basic fibroblast growth factor were below the limits of detection in numerous samples. Levels of PlGF and VEGF were compared in patients with clinical benefit and patients without clinical benefit as shown in Fig. 3. PlGF levels were not significantly different between the groups before treatment on day 1 (P = 0.32). After treatment on day 1, PlGF levels decreased over time in patients with clinical benefit (P = 0.04) but not in patients without clinical benefit (P = 0.25). On day 5, PlGF levels were lower in patients with clinical benefit compared with patients without clinical benefit (P = 0.04). This indicates that Ang-(1-7) administration resulted in a decrease in circulating levels of PlGF in some patients and links this effect to achieving clinical benefit.
Fig. 3.
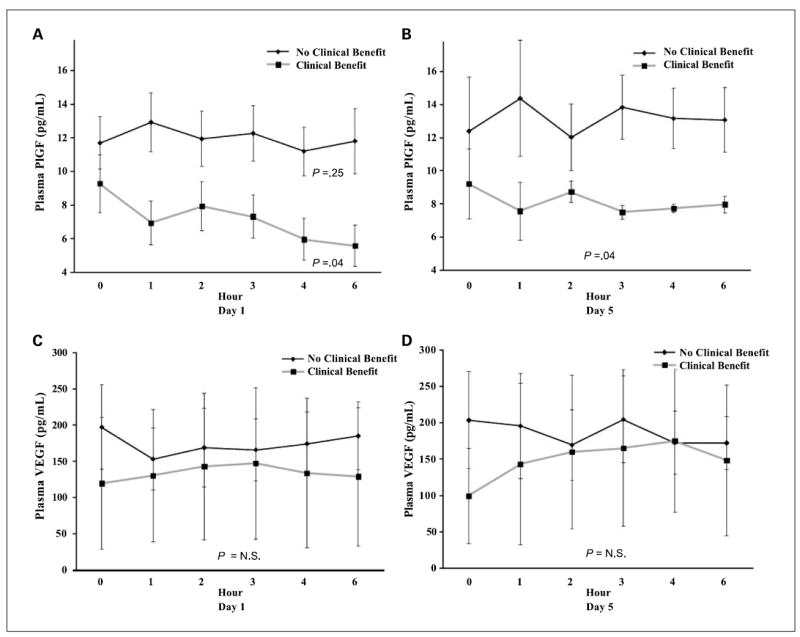
Comparison of plasma levels of PlGF (A and B) and VEGF (C and D) in patients who experienced clinical benefit versus those who did not experience clinical benefit. Statistical analyses evaluated changes over time on day 1 and differences between groups on day 5.
Discussion
This phase I study examined the toxicities and pharmacokinetics of Ang-(1-7) in patients with advanced cancer. The toxicities observed were generally mild aside from one case of multiple strokes and one case of cranial neuropathy. Arterial and venous thrombotic events such as those observed in this study seem to be a class effect of antiangiogenic drugs (16, 17). No bleeding complications or episodes of hypertension were observed. The recommended phase II dose of this drug is 400 μg/kg as a daily s.c. injection for 5 consecutive days on a 3-week cycle.
Four patients on this study experienced clinical benefit with stabilization of disease lasting more than 3 months. Two of those showed some radiographic improvement and one other had a minor improvement in prostate-specific antigen. Patients with clinical benefit had a reduction in plasma PlGF, whereas no significant change in this proangiogenic hormone was observed in patients without clinical benefit. PlGF is emerging as a target for cancer therapy, and the findings of this study support the potential therapeutic value of targeting this hormone (18-20).
Resistance to VEGF-targeted drugs has been attributed to a process known as angiogenic escape (21), in which tumor angiogenesis is driven by alternative proangiogenic hormones, including PlGF, after VEGF signaling is blocked. Consistent with this theory, high levels of PlGF have been observed following treatment with existing VEGF targeting drugs (22, 23). The findings of this study suggest that Ang-(1-7) might enhance sensitivity to existing VEGF inhibitors by reducing PlGF levels and preventing angiogenic escape (18-23).
The findings of this study indicate that Ang-(1-7) treatment may reduce circulating levels of PlGF and that this effect may in turn lead to disease stabilization. It is possible, however, that these correlations do not represent causal relationships. It is possible that PlGF concentrations fall as a consequence of some other antitumor property of this drug but that the reduction in PlGF is not required to achieve clinical benefit. Further preclinical studies are needed to determine whether the reduction in PlGF is required for the antitumor effects of Ang-(1-7).
Clinical or molecular factors that predict who will achieve a reduction in PlGF following Ang-(1-7) treatment have not been determined. It is possible that expression of the mas receptor in cancer cells will be required for patients to benefit. Because the mas receptor is found on endothelial cells (24) and Ang-(1-7) directly inhibits endothelial cell tubule formation (12), receptor expression in cancer cells may not be required to effect tumor angiogenesis. Identification of predictive biomarkers may be useful for optimizing future clinical trials.
Antitumor activity was associated with reduction in VEGF and PlGF in human lung or breast tumor xenografts (11, 12). For the cohort of patients with clinical benefit, no significant change in levels of plasma VEGF was observed. Individual patient-level analyses of changes in plasma VEGF will be explored in future work.
This phase I trial validates the renin-angiotensin system as a target for cancer therapy (25-27). All of the necessary prohormones, peptidases, and receptors are found in cancers (27, 28), as shown in Fig. 1. The renin-angiotensin system also may prove to be an important target for cancer chemoprevention. Angiotensin-converting enzyme inhibitors are known to increase levels of Ang-(1-7) and have apparent chemopreventive activity observed in several large studies (29-33). Further preclinical and clinical studies of Ang-(1-7) and other drugs that target the renin-angiotensin system for cancer therapy and chemoprevention are needed.
Ang-(1-7) is a first-in-class drug targeting the mas receptor. Two of the three patients with sarcoma had clinical benefit and a phase II trial has been developed at the Wake Forest University to determine the activity of Ang-(1-7) for the treatment of this disease. Given that antiangiogenic drugs tend to have a broad spectrum of activity, phase II testing in other solid tumors is planned.
Supplementary Material
Translational Relevance.
Antiangiogenic drugs targeting mechanisms other than vascular endothelial growth factor (VEGF)-VEGF receptor pathway will likely play a vital role in future antiangiogenic combinations. This study found that angiotensin-(1-7) [Ang-(1-7)] has clinical activity and linked this activity to reduction in an emerging anti-angiogenic target, placental growth factor (PlGF). A phase II study of single-agent Ang-(1-7) in patients with sarcoma is ongoing, and strategies combining Ang-(1-7) with VEGF targeted drugs are in preclinical testing.
This study provides evidence of activity for a first-in-class antiangiogenic drug targeting the mas receptor. To our knowledge, this is also the first agent that has shown the ability to reduce PlGF levels in patients with cancer. Other drugs targeting PlGF are entering clinical trials. Our findings support development of Ang-(1-7) as well as these other drugs by validating PlGF as a target for cancer therapy.
Acknowledgments
We thank Shaunita A. Michael, R.N., B.S.N., and Christy S. Harris, Pharm.D., for their contributions to this study.
Grant support: Comprehensive Cancer Center of Wake Forest University grant NCI-P30-CA012197 (F.M. Torti), American Cancer Society grant ACS-MRSG-07-152-01-CNE (W.J. Petty), Cancer Challenge 2008, Farley-Hudson Foundation, and Royal Brown Cancer Research Fund (F.M. Torti).
Footnotes
Note: Supplementary data for this article are available at Clinical Cancer Research Online (http://clincancerres.aacrjournals.org/).
Disclosure of Potential Conflicts of Interest
If patented, E.A. Tallant and P.E. Gallagher have a potential financial interest in the use of the peptide for this project.
References
- 1.Ferrario CM, Averill DB, Brosnihan KB, et al. Regulation of cardiovascular control mechanisms by angiotensin-(1-7) and angiotensin converting enzyme 2. In: Carey R, editor. Hypertension and hormonal mechanisms. 2007. pp. 43–59. [Google Scholar]
- 2.Bader M, Ganten D. Update on tissue renin-angiotensin systems. J Mol Med. 2008;86:615–21. doi: 10.1007/s00109-008-0336-0. [DOI] [PubMed] [Google Scholar]
- 3.Kumar R, Boim MA. Diversity of pathways for intracellular angiotensin II synthesis. Curr Opin Nephrol Hypertens. 2009;18:33–9. doi: 10.1097/MNH.0b013e32831a9e20. [DOI] [PubMed] [Google Scholar]
- 4.Santos RAS, Ferreira AJ, Silva ACS. Recent advances in the angiotensin-converting enzyme 2-angiotensin(1-7)-Mas axis. Exp Physio. 2008;93:519–27. doi: 10.1113/expphysiol.2008.042002. [DOI] [PubMed] [Google Scholar]
- 5.Santos RAS, Simoes E, Silva AC, et al. Angio-tensin-(1-7) is an endogenous ligand for the G-protein coupled receptor mas. Proc Natl Acad Sci U S A. 2003;100:8258–63. doi: 10.1073/pnas.1432869100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Freeman EJ, Chisolm GM, Ferrario CM, et al. Angiotensin-(1-7) inhibits vascular smooth muscle cell growth. Hypertension. 1996;28:104–8. doi: 10.1161/01.hyp.28.1.104. [DOI] [PubMed] [Google Scholar]
- 7.Strawn WB, Ferrario CM, Tallant EA. Angiotensin-(1-7) reduces smooth muscle growth after vascular injury. Hypertension. 1999;33:207–11. doi: 10.1161/01.hyp.33.1.207. [DOI] [PubMed] [Google Scholar]
- 8.Langeveld B, Van Gilst WH, Gio RA, et al. Angiotensin-(1-7) attenuates neointimal formation after stent implantation in the rat. Hypertension. 2005;45:138–41. doi: 10.1161/01.HYP.0000149382.83973.c2. [DOI] [PubMed] [Google Scholar]
- 9.Gallagher PE, Tallant EA. Inhibition of lung cancer cell growth by angiotensin-(1-7) Carcinogenesis. 2004;25:2045–52. doi: 10.1093/carcin/bgh236. [DOI] [PubMed] [Google Scholar]
- 10.Menon J, Soto-Pantoja DR, Callahan MF, et al. Angiotensin-(1-7) inhibits growth of human lung adenocarcinoma xenografts in nude mice through a reduction in cyclooxygenase-2. Cancer Res. 2007;67:2809–15. doi: 10.1158/0008-5472.CAN-06-3614. [DOI] [PubMed] [Google Scholar]
- 11.Soto-Pantoja DR, Menon J, Gallagher PE, et al. Angiotensin-(1-7) inhibits tumor angiogenesis in human lung cancer xenografts with a reduction in vascular endothelial growth factor. Mol Cancer Ther. 2009;8:1676–83. doi: 10.1158/1535-7163.MCT-09-0161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Soto-Pantoja DR, Petty WJ, Gallagher PE, et al. Angiotensin-(1-7) inhibits triple negative tumor growth through the inhibition of angiogenesis and a reduction in placental growth factor PIGF. 2008 [Google Scholar]
- 13.Rodgers KE, Oliver J, diZerega GS. Phase I/II dose escalation study of angiotensin 1-7 [A(1-7)] administered before and after chemotherapy in patients with newly diagnosed breast cancer. Cancer Chemother Pharmacol. 2006;57:559–68. doi: 10.1007/s00280-005-0078-4. [DOI] [PubMed] [Google Scholar]
- 14.Kohara K, Tabuchi Y, Senanayake P, et al. Reassessment of plasma angiotensins measurement: effects of protease inhibitors and sample handling procedures. Peptides. 1991;12:1135–41. doi: 10.1016/0196-9781(91)90070-6. [DOI] [PubMed] [Google Scholar]
- 15.Merrill DC, Karoly M, Chen K, et al. Angiotensin-(1-7) in normal and preeclamptic pregnancy. Endocrine. 2002;18:239–45. doi: 10.1385/ENDO:18:3:239. [DOI] [PubMed] [Google Scholar]
- 16.Scappaticci FA, Skillings JR, Holden SN, et al. Arterial thromboembolic events in patients with metastatic carcinoma treated with chemotherapy and bevacizumab. J Natl Cancer Inst. 2007;99:1232–9. doi: 10.1093/jnci/djm086. [DOI] [PubMed] [Google Scholar]
- 17.Feldman DR, Baum MS, Ginsberg MS, et al. Phase I trial of bevacizumab plus escalated doses of sunitinib in patients with metastatic renal cell carcinoma. J Clin Oncol. 2009;27:1432–9. doi: 10.1200/JCO.2008.19.0108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fischer C, Jonckx B, Mazzone M, et al. Anti-PlGF inhibits growth of VEGF(R)-inhibitor-resistant tumors without affecting healthy vessels. Cell. 2007;131:463–75. doi: 10.1016/j.cell.2007.08.038. [DOI] [PubMed] [Google Scholar]
- 19.Taylor AP, Goldenberg DM. Role of placenta growth factor in malignancy and evidence that an antagonistic PlGF/Flt-1 peptide inhibits the growth and metastasis of human breast cancer xenografts. Mol Cancer Ther. 2007;6:524–31. doi: 10.1158/1535-7163.MCT-06-0461. [DOI] [PubMed] [Google Scholar]
- 20.Loges S, Schmidt T, Carmeliet P. Antimyeloangiogenic therapy for cancer by inhibiting PlGF. Clin Cancer Res. 2009;15:3648–53. doi: 10.1158/1078-0432.CCR-08-2276. [DOI] [PubMed] [Google Scholar]
- 21.Casanovas O, Hicklin DJ, Bergers G, et al. Drug resistance by evasion of antiangiogenic targeting of VEGF signaling in late-stage pancreatic islet tumors. Cancer Cell. 2005;8:299–309. doi: 10.1016/j.ccr.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 22.Deprimo SE, Bello CL, Smeraglia J, et al. Circulating protein biomarkers of pharmacodynamic activity of sunitinib in patients with metastatic renal cell carcinoma: modulation of VEGF and VEGF-related proteins. J Transl Med. 2007;5:32. doi: 10.1186/1479-5876-5-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rosen LS, Kurzrock R, Mulay M, et al. Safety, pharmacokinetics, and efficacy of AMG 706, an oral multikinase inhibitor, in patients with advanced solid tumors. J Clin Oncol. 2007;25:2369–76. doi: 10.1200/JCO.2006.07.8170. [DOI] [PubMed] [Google Scholar]
- 24.Tallant EA, Lu X, Weiss RB, et al. Bovine aortic endothelial cells contain an angiotensin-(1-7) receptor. Hypertension. 1997;29:388–93. doi: 10.1161/01.hyp.29.1.388. [DOI] [PubMed] [Google Scholar]
- 25.Tallant EA, Menon J, Soto-Pantoja D, et al. Angiotensin peptides and cancer. In: Kastin AJ, editor. Handbook of Biologically Active Peptides. 2006. pp. 459–65. [Google Scholar]
- 26.Ager EI, Neo J, Christophi C. The renin-angiotensin system and malignancy. Carcinogenesis. 2008;29:1675–84. doi: 10.1093/carcin/bgn171. [DOI] [PubMed] [Google Scholar]
- 27.Khakoo AY, Sidman RL, Pasqualini R, et al. Does the renin-angiotensin system participate in regulation of human vasculogenesis and angiogenesis? Cancer Res. 2008;68:9112–5. doi: 10.1158/0008-5472.CAN-08-0851. [DOI] [PubMed] [Google Scholar]
- 28.Herr D, Rodewald M, Fraser HM, et al. Potential role of renin-angiotensin-system for tumor angiogenesis in receptor negative breast cancer. Gynecol Oncol. 2008;109:418–25. doi: 10.1016/j.ygyno.2008.02.019. [DOI] [PubMed] [Google Scholar]
- 29.Luque M, Martin P, Martell N, et al. Effects of captopril related to increased levels of prostacyclin and angiotensin-(1-7) in essential hypertension. J Hypertens. 1996;14:799–805. doi: 10.1097/00004872-199606000-00017. [DOI] [PubMed] [Google Scholar]
- 30.Lever AF, Hole DJ, Gillis CR, et al. Do inhibitors of angiotensin-I-converting enzyme protect against risk of cancer? Lancet. 1998;352:179–84. doi: 10.1016/S0140-6736(98)03228-0. [DOI] [PubMed] [Google Scholar]
- 31.Pahor M, Guralnik JM, Salive ME, et al. Do calcium channel blockers increase the risk of cancer? Am J Hypertens. 1996;9:695–9. doi: 10.1016/0895-7061(96)00186-0. [DOI] [PubMed] [Google Scholar]
- 32.Jick H, Jick S, Derby LE, et al. Calcium-channel blockers and risk of cancer. Lancet. 1997;349:525–8. doi: 10.1016/S0140-6736(97)80084-0. [DOI] [PubMed] [Google Scholar]
- 33.van der Knaap R, Siemes C, et al. Renin-angiotensin system inhibitors, angiotensin I-converting enzyme gene insertion/deletion polymorphism, and cancer: the Rotterdam Study. Cancer. 2008;112:748–57. doi: 10.1002/cncr.23215. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.