

Abstract
Mobilization of the T-cell response against cancer has the potential to achieve long-lasting cures. However, it is not known how to harness antigen-presenting cells optimally to achieve an effective antitumor T-cell response. In this study, we show that anti-CD47 antibody–mediated phagocytosis of cancer by macrophages can initiate an antitumor T-cell immune response. Using the ovalbumin model antigen system, anti-CD47 antibody–mediated phagocytosis of cancer cells by macrophages resulted in increased priming of OT-I T cells [cluster of differentiation 8-positive (CD8+)] but decreased priming of OT-II T cells (CD4+). The CD4+ T-cell response was characterized by a reduction in forkhead box P3-positive (Foxp3+) regulatory T cells. Macrophages following anti-CD47–mediated phagocytosis primed CD8+ T cells to exhibit cytotoxic function in vivo. This response protected animals from tumor challenge. We conclude that anti-CD47 antibody treatment not only enables macrophage phagocytosis of cancer but also can initiate an antitumor cytotoxic T-cell immune response.
Antigen presentation is the process by which innate immune cells such as macrophages and dendritic cells (antigen-presenting cells, APC) acquire antigens and present them to T cells to initiate the adaptive immune response. How APCs shape the immune response by both degrading antigens and preserving antigens for presentation to T cells has been a longstanding area of interest (1). Recently, the mechanism of antigen recognition by APCs has been shown to affect the preference of MHC I versus MHC II antigen-presentation pathways. For instance, mannose receptor-mediated endocytosis on dendritic cells has been associated with MHC I antigen presentation, whereas scavenger receptor-mediated endocytosis has been associated with MHC II presentation (2). Moreover, the functional outcomes of antigen presentation have been shown to be context dependent. For instance, targeting antigens to DEC-205 using monoclonal antibodies induced tolerance under noninflammatory conditions but mediated immunogenicity under activating conditions by cluster of differentiation 40 ligand (CD40L) (3). Harnessing APCs to enhance the antitumor T-cell response offers an exciting strategy for cancer immunotherapy. The ability of the T-cell immune response to be mobilized successfully against cancer has been demonstrated through preclinical and clinical studies of anti-CTLA4 antibody for T-cell activation (4).
Phagocytosis by macrophages relies on the cell’s recognition of prophagocytic (“eat me”) and antiphagocytic (“don’t eat me”) signals on target cells. Anti-CD47 blocking monoclonal antibodies (mAbs) induce macrophage phagocytosis of cancer cells by inhibiting an important antiphagocytic signal, allowing prophagocytic signals to dominate (5, 6). CD47 is highly expressed on cancer cells as compared with normal cells (5, 6) and interacts with the ligand signal regulatory protein α (SIRP-α) on macrophages (7). This interaction results in phosphorylation of immunoreceptor tyrosine-based inhibition (ITIM) motifs on SIRP-α’s cytoplasmic tail and the recruitment of Src homology phosphatase-1 (SHP-1) and SHP-2 phosphatases, which is thought to block phagocytosis by preventing myosin-IIA accumulation at the phagocytic synapse (8–12). We have demonstrated the therapeutic efficacy of anti-CD47 blocking mAbs against xenograft human cancers growing in immunodeficient mice, including cancers such as leukemia (5, 13), lymphoma (14), and multiple myeloma (15), solid tumors, including breast, colon, prostate, and bladder cancers, and sarcomas (6, 16). Whether the adaptive immune response also can be recruited against the cancer after anti-CD47 mAb treatment has not been tested, because the immunodeficient mice used to establish the xenograft models lack T, B, and NK cells. In this study, we tested the hypothesis that anti-CD47 antibody–mediated phagocytosis of cancer cells can facilitate an antitumor T-cell immune response.
Results
Macrophages Phagocytose Cancer Cells in the Presence of Anti-CD47 Blocking Antibody.
To follow the immune response to a model tumor antigen, the human colon cancer cell line DLD1 was transfected with a lentiviral vector for expressing cytoplasmic ovalbumin (cOVA) and GFP (DLD1-cOVA-GFP) (Fig. S1). DLD1-cOVA-GFP cancer cells express CD47 and can be recognized by both CD47 mAbs, clones B6H12 and 2D3 (Fig. S1). Anti-CD47 B6H12 (blocking) mAb blocks the interaction between CD47 and SIRP-α, whereas anti-CD47 2D3 (non-blocking) antibody binds CD47 but does not block its interaction with SIRP-α. Macrophages phagocytose DLD1-cOVA-GFP cancer cells in the presence of anti-CD47 B6H12, but not anti-CD47 2D3 mAbs, demonstrating that phagocytosis is dependent on the blockade of CD47/SIRPα interactions and not entirely due to antibody opsonization effects (Fig. 1 and Fig. S2). Anti-CD47 mediated phagocytosis of DLD1-cOVA-GFP cancer cells by macrophages leads to cross-presentation of ovalbumin peptide onto MHC-I, as assessed by staining for the SIINFEKL-H2kb complex on the cell surface (Fig. S3). Costimulatory molecule CD86 is up-regulated, but not coinhibitory molecule B7-H1 (Fig. S4). Anti-CD47 B6H12–mediated phagocytosis of cancer cells leads to macrophage release of proinflammatory cytokines. For example, IL-12p40, TNF-α, regulated upon activation normal T cell expressed and secreted (RANTES), and monocyte chemotactic protein-3 (MCP-3) cytokine levels increase after anti-CD47 B6H12-mediated phagocytosis (Fig. S5). Next, the ability of the APCs, macrophages and dendritic cells, were tested for phagocytic activity in response to anti-CD47 mAbs. Compared to dendritic cells, macrophages effectively phagocytose DLD1-cOVA-GFP cancer cells in the presence of anti-CD47 B6H12 mAb (Fig. 1). Consistent with this result, SIRP-α, the ligand for CD47, is expressed at high levels on macrophages but at lower levels on dendritic cells (Fig. S6).
Fig. 1.

Macrophages effectively phagocytose cancer cells in the presence of anti-CD47 B6H12 antibody. (A) RFP+ macrophages (Mac) were cocultured with DLD1-cOVA-GFP cancer cells in the presence of IgG or anti-CD47 B6H12 (blocking) or 2D3 (nonblocking) mAbs. Percentage of phagocytosis was determined by the percentage of GFP+ cells within RFP+ macrophage cell gate. (B) RFP+ macrophages versus dendritic cells (DCs) were cocultured with DLD1-cOVA-GFP cancer cells in the presence of IgG, anti-CD47 B6H12, or anti-CD47 2D3 mAbs. The experiment was performed three times with similar results.
Macrophages Prime OT-I (CD8+) T Cells After Phagocytosis of Cancer Cells by Anti-CD47 Blocking Antibody.
To assess priming of CD8+ T cells after anti-CD47–mediated phagocytosis by macrophages, a carboxyfluorescein succinimidyl ester (CFSE)-dilution assay was used to measure the proliferative response of OVA-specific CD8+ T cells (OT-I). Red fluorescent protein-positive (RFP+) macrophages were cocultured with DLD1-cOVA-GFP cancer cells in the presence of IgG, anti-CD47 B6H12 (blocking), or anti-CD47 2D3 (non-blocking) mAbs. Lymph nodes were harvested from OT-I (CD8+) transgenic mice and were labeled with CFSE (0.5 μM) and CD8+ cells enriched by magnetic separation. On day 3, the percentage of proliferating OT-I T cells was quantified based on the percentage of cells that had diluted the CFSE dye (CFSE-low). The percentage of proliferating OT-I T cells increased following anti-CD47 B6H12-mediated phagocytosis of cancer by macrophages (Fig. 2A). To verify that the proliferative response of OT-I T cells was an antigen-specific response, macrophages were allowed to phagocytose DLD1-cOVA-GFP versus DLD1-GFP cancer cells (the latter not expressing OVA) in the presence of anti-CD47 B6H12 mAb before the addition of CFSE-labeled OT-I T cells. Increased OT-I T-cell proliferation was observed only after anti-CD47–mediated phagocytosis of DLD1-cOVA-GFP cancer cells but not DLD1-GFP cancer cells, indicating an antigen-specific effect (Fig. 2B).
Fig. 2.

Macrophages prime CD8+ T cells to proliferate after phagocytosis of cancer cells by anti-CD47 B6H12 mAb. (A) RFP+ macrophages were cocultured with DLD1-cOVA-GFP colon cancer cells in the presence of IgG, anti-CD47 B6H12 (blocking), or anti-CD47 2D3 (nonblocking) mAbs. The next day, CD8+ T cells from OT-I transgenic mice were magnetically enriched and labeled with CFSE (0.5 μM). Analysis was performed on day 3, and the percentage of proliferating cells was determined. Macrophages were pulsed with OT-I peptide (OVA257-264, SIINFEKL) as a positive control. The experiment was performed three times with similar results. (B) RFP+ macrophages were cocultured with DLD1-cOVA-GFP cancer cells or DLD1-GFP cancer cells not expressing cOVA. (Left) Phagocytosis was determined by the percentage of GFP+ cells within the RFP+ macrophage cell gate. (Right) CFSE-labeled CD8+ T cells from OT-I mice were added to cultures, and the percentage of proliferating cells was determined.
Macrophages Do Not Prime OT-II (CD4+) T cells After Phagocytosis of Cancer Cells by Anti-CD47 B6H12 Blocking Antibody.
To assess priming of CD4+ T cells after anti-CD47–mediated phagocytosis by macrophages, a CFSE-dilution assay was used to measure the proliferative response of OVA-specific CD4+ T cells (OT-II). Macrophages were cocultured with DLD1-cOVA-GFP cancer cells in the presence of IgG, anti-CD47 B6H12 (blocking), or anti-CD47 2D3 (non-blocking) mAbs, and CFSE-labeled OT-II (CD4+) T cells were added to cultures. Interestingly, the percentage of proliferating OT-II T cells was reduced following anti-CD47-mediated phagocytosis compared with baseline levels (Fig. 3A). Because of the possibility that cell-surface MHC II (I-Ab+) on macrophages might be limiting OT-II (CD4+) T cell proliferation after anti-CD47-mediated phagocytosis, we measured the percentage of macrophages expressing MHC II on the cell surface. Interestingly, the percentage of I-Ab+ macrophages increased after anti-CD47 B6H12–mediated phagocytosis of cancer cells, despite the decrease in CD4+ T-cell priming (Fig. S7). To determine whether IFN-γ stimulation of macrophages could overcome the decrease in CD4+ T cell proliferation, IFN-γ was used to up-regulate surface MHC II levels on macrophages for evaluation in phagocytosis and T cell proliferation assays. IFN-γ–stimulated macrophages efficiently phagocytosed cancer in the presence of anti-CD47 B6H12 mAb, but the OT-II CD4+ T-cell response was also diminished compared with baseline (Fig. 3B).
Fig. 3.

After phagocytosis of cancer cells by anti-CD47, macrophages do not prime CD4+ T cells to proliferate. (A) RFP+ macrophages were cocultured with DLD1-cOVA-GFP cancer cells in the presence of IgG, anti-CD47 B6H12 (blocking), or anti-CD47 2D3 (nonblocking) mAbs. The next day, CD4+ T cells were isolated from OT-II transgenic mice and were labeled with CFSE (0.5 μM). Analysis was performed on day 4, and the percentage of proliferating cells was determined. Macrophages were pulsed with OVA peptide 323–339 as a positive control. (B) RFP+ macrophages were stimulated with IFN-γ to up-regulate MHC II levels. Phagocytosis and priming of OT-II CD4+ cells were determined in the presence of anti-CD47 mAbs.
Reduction in Forkhead Box P3-Positive Regulatory T Cells After Anti-CD47–Mediated Phagocytosis of Cancer by Macrophages.
To assess the functional effects of anti-CD47 B6H12–mediated phagocytosis on CD4+ regulatory T cells, we crossed OT-II transgenic mice with forkhead box 3P (Foxp3)-GFP reporter mice to generate double-transgenic mice (Fig. S8). These mice express 25% Foxp3-GFP+ cells within the CD4+ CD25+ population and exhibit T cell receptor V alpha 2 (Vα2) restriction (Fig. S8A). In addition, CD4+ Foxp3-GFP+ T cells are responsive to OVA peptide 323–339 and can be induced to differentiate in the context of TGF-β and all-trans-retinoic acid (Fig. S8B). RFP+ macrophages were cocultured with DLD1-cOVA-GFP cancer cells in the presence of IgG, anti-CD47 B6H12 (blocking), or anti-CD47 2D3 (non-blocking) mAbs. The next day, CD4+ T cells from OT-II/Foxp3-GFP+ double-transgenic mice were magnetically enriched and were added to the cultures. After 4 d, the percentage of regulatory T-cells was quantified by the percentage of CD4+ Foxp3-GFP+ cells (Fig. 4). A reduction in Foxp3+ regulatory T cells was observed after anti-CD47-mediated phagocytosis of cancer cells.
Fig. 4.
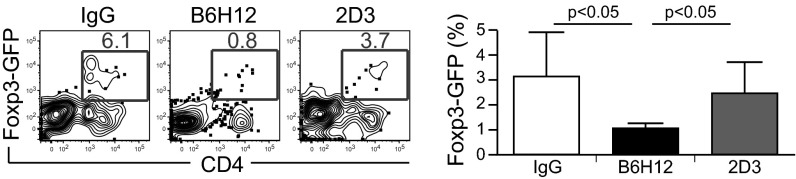
A reduction in Foxp3+ regulatory T cells occurs after anti-CD47 B6H12–mediated phagocytosis of cancer cells by macrophages. RFP+ macrophages were cocultured with DLD1-cOVA-GFP cancer cells in the presence of IgG or anti-CD47 B6H12 (blocking) or 2D3 (nonblocking) mAbs. The next day, CD4+ T cells from OT-II/Foxp3-GFP+ transgenic mice were magnetically enriched and were added to cultures. On day 4, the percentage of CD4+ Foxp3-GFP+ cells was quantified.
After Anti-CD47–Mediated Phagocytosis of Cancer Cells, Macrophages Prime OT-I (CD8+) T Cells to Proliferate in Vivo.
To evaluate the effects of anti-CD47 B6H12–mediated phagocytosis on CD8+ T-cell priming in vivo, OT-I (CD8+) T cells (CD45.2) were CFSE-labeled and adoptively transferred to CD45.1 recipient mice (Fig. 5A). The next day, RFP+ macrophages were cocultured with DLD1-cOVA-GFP cancer cells in the presence of IgG or anti-CD47 B6H12 mAb. Macrophages were isolated by magnetic enrichment, and phagocytosis was verified by FACS analysis before subcutaneous (subQ) transfer into the footpad. After 4 d, the popliteal lymph node was analyzed for the percentage of proliferating cells (CFSE-low) within the CD45.2+ gate. There was an increase in proliferating OT-I T cells in mice receiving macrophages that phagocytose cancer cells via anti-CD47 B6H12 mAb (Fig. 5B).
Fig. 5.

After anti-CD47–mediated phagocytosis of cancer cells, macrophages prime CD8+ T cells in vivo. (A) Experimental setup. BMDM, bone marrow-derived macrophages. (B) Adoptively transferred CFSE+ OT-I T cells were analyzed in the draining lymph node by gating on CD45.2+ cells. The percentage of proliferating cells was determined by gating on the CFSE-low population. n = 5 mice per group.
Macrophages Prime an Antitumor CD8+ T-Cell Response in Vivo After Anti-CD47–Mediated Phagocytosis of Cancer Cells.
We next evaluated the functional effects of OT-I (CD8+) T-cell priming after anti-CD47–mediated phagocytosis of cancer cells by macrophages. To assess the efficacy of CD8+ T-cell killing of OVA peptide-displaying targets, CD8+ T cells were isolated from OT-I transgenic mice and were i.v. transferred to recipient mice (Fig. 6A). RFP+ macrophages were cocultured with DLD1-cOVA-GFP cancer cells in vitro in the presence of IgG or anti-CD47 B6H12 mAb. After 2-h incubation, macrophages were isolated and injected into the footpad. After 4 d, mice were challenged with target cells (CD45.1 splenocytes) to assess cytotoxic activity. CFSE-high splenocytes were pulsed with 1 μM OVA class I-restricted peptide (OVA257-264, SIINFEKL) to make them targets for OT-I cytotoxic T cells and then were mixed in a 1:1 ratio with non–peptide-pulsed CFSE-low cells before i.v. transfer. Analysis of draining lymph nodes 16 h later showed increased cell killing of peptide-pulsed CFSE-high lymphocytes in mice receiving macrophages that had phagocytosed cancer cells by anti-CD47 B6H12 mAb (Fig. 6A).
Fig. 6.

After anti-CD47–mediated phagocytosis of cancer cells, macrophages prime an antitumor CD8+ T-cell response in vivo. (A) After anti-CD47–mediated phagocytosis of cancer cells, macrophages prime effector cytotoxic T cells. CD8+ T cells were isolated from OT-I transgenic mice and were transferred i.v. to recipient mice. Macrophages (Mac) were cocultured with DLD1-cOVA-GFP cancer cells in vitro in the presence of IgG or anti-CD47 B6H12 (blocking) mAb. Macrophages were isolated by magnetic separation and were transferred subcutaneously (subQ) on the next day. After 4 d, target cells (CD45.1 splenocytes) were labeled as CFSE-high (10 μM) or -low (1 μM). CFSE-high cells were pulsed with 1 μM OVA class I-restricted peptide (SIINFEKL) to make them targets for OT-I cytotoxic T-cell function. CFSE-high (peptide-pulsed) and -low (unpulsed) cells were mixed in a 1:1 ratio and transferred i.v. Draining lymph nodes were analyzed 16 h later to determine the percentage of CFSE-high versus CFSE-low cells. The percentage of cell killing was determined as described in Materials and Methods. n = 10 mice. (B) After anti-CD47–mediated phagocytosis of cancer cells, macrophages prime an antitumor CD8+ T-cell response. OT-I CD8+ T cells were transferred i.v. to recipient mice. Macrophages were cocultured with DLD1-cOVA-GFP cancer cells in vitro in the presence of IgG or anti-CD47 B6H12 mAb, and then macrophages were transferred on days 1 and 10. Animals were challenged with EG.7 (EL4 mouse lymphoma cells expressing ovalbumin) cancer cells on day 14, and tumor growth was monitored over time. n = 5 mice per group. *P < 0.05; **P < 0.01.
Next, the ability of CD8+ effector T cells to prime an antitumor immune response was evaluated. CD8+ T cells from OT-I mice were transferred into recipient animals (Fig. 6B). Macrophages were cocultured with DLD1-cOVA-GFP cancer cells in vitro in the presence of IgG or anti-CD47 B6H12 mAb; then the macrophages were transferred into the footpad on days 1 and 10. Animals were challenged with EG.7 (OVA-expressing EL4) cancer cells on day 14, and tumor growth was monitored over time. CD8+ T cells primed by macrophages following anti-CD47-mediated phagocytosis protected mice from tumor challenge (Fig. 6B).
Discussion
Recent studies from the I.L.W. laboratory have shown that malignant cancers universally up-regulate the “don’t eat me” signal CD47, presumably in their progression to allow escape from endogenous “eat me” signals that were induced as part of programmed cell death and programmed cell removal (5, 6, 13, 14, 16–18). These results indicate a role for human CD47-blocking antibodies in cancer therapy via induced phagocytosis. Our previous experiments involved xenotransplantation of primary human cancers into immunodeficient mice. We now have examined the possible role of anti-CD47–enabled phagocytosis in antigen presentation of tumor peptides to T cells of the adaptive immune system. Here we have demonstrated that CD47 serves as an “invisibility cloak” for both innate and adaptive immunity. Treatment with anti-CD47 blocking mAbs led to adaptive T-cell immune responses, thereby providing an additional mechanism of action for anti-CD47 antibodies. OVA-specific OT-I (CD8+) and OT-II (CD4+) T-cell clones were used to follow the outcomes of antigen presentation by macrophages after anti-CD47–mediated phagocytosis of cancer cells engineered to express cytoplasmic OVA. Using in vitro and in vivo assays, we show that antigens are presented to CD8+ T cells effectively, but the proliferative response of OT-II CD4+ T cells to loaded macrophages was diminished compared with baseline levels. The baseline level of OT-I CD8+ proliferation and OT-II CD4+ proliferation was 20% (Figs. 2 and 3), likely because of OVA released from cancer cells that become endocytosed or pinocytosed by macrophages and then processed for presentation to both MHC I and MHC II pathways. Together, our results suggest that anti-CD47–mediated phagocytosis of cancer cells results in the presentation of negative signals to CD4+ T cells and positive signals to CD8+ T cells. In addition, the CD4+ T-cell response was characterized by reduced regulatory T cells. This reduction might be attributed either to decreased proliferation of regulatory T cells in response to peptide or to less efficient regulatory T-cell differentiation. The in vivo priming of an antitumor T-cell response by macrophages after anti-CD47–mediated phagocytosis of cancer protects mice from tumor challenge. Anti-CD47 mAbs may represent a therapeutic strategy for overcoming the regulatory T-cell contribution to immune evasion by cancer and initiating an effective antitumor cytotoxic T-cell response.
In this system macrophages are the primary APCs that phagocytose cancer in response to anti-CD47 antibody and present antigen to CD8+ T cells. Perhaps this observation is due to higher levels of SIRP-α expressed on macrophages than on dendritic cells or because macrophages phagocytose whole cells more effectively. Because subsets of dendritic cells have been reported to vary in their levels of SIRP-α expression (19), it is possible that other dendritic cell subsets may more effectively phagocytose cancer in response to anti-CD47 antibody in vivo. While dendritic cells are well known for their function in antigen presentation, in this study we show that macrophages also have the capacity to stimulate a CD8+ T-cell immune response in the context of blocking the CD47-SIRPalpha axis. Prior to this work, the ability to present exogenous antigens to CD8+ T cells has been largely attributed to the CD8+ subtype of DCs. Understanding the roles of tissue-specific macrophages and dendritic cells in response to anti-CD47 mAbs warrants further investigation both in mouse and human systems (20–22).
Antibody-mediated uptake of antigens via Fcγ receptor-mediated endocytosis by dendritic cells can prime CD4+ and CD8+ T-cell responses in some circumstances (23, 24). In contrast, anti-CD47–mediated phagocytosis by macrophages predominantly primes CD8+ T cells. This predominant CD8+ T cell response is not explained entirely by opsonization of cancer cells, because both the anti-CD47 clones B6H12 and 2D3 bind DLD1-cOVA-GFP cancer cells, but only B6H12 is capable of inducing phagocytosis through its ability to block the interaction between CD47 on cancer cells and SIRP-α on macrophages. The preferential activation of CD8+ T cells may rely on routing phagocytosed cancer antigens toward the MHC I pathway or might be explained by SIRP-α’s role as a negative regulator of macrophage function. Engagement of SIRP-α by CD47 leads to phosphorylation of SIRP-α’s cytoplasmic ITIM motifs by SHP-1 and SHP-2. This pathway is well described to inhibit macrophage phagocytosis, but more recently, SIRP-α also has been shown to attenuate macrophage activation by LPS by sequestering SHP-2, which is needed for activation of the MAPK and NF-κB pathways (25). In addition, inhibition of SHP-1 in dendritic cells has been reported to enhance CD8+ and CD4+ T-cell responses and reduce regulatory T cells, a response that protected mice against tumor challenge (26). SIRP-α also may negatively regulate cross-presentation by a mechanism not yet understood, perhaps involving recruitment of cross-presentation machinery to phagosomes. Alternatively, prophagocytic signals such as calreticulin have been described to elicit antitumor immunity (27–29). It may be that the unmasking of such prophagocytic signals may route antigens preferentially toward the cross-presentation pathway.
Other groups examining the effect of SIRP-α on dendritic cells interacting with CD47 on T cells have reported T-cell activation or inhibition, depending on the context (30–33). In contrast, our experiments were designed to study the impact of antigen presentation after anti-CD47 mAb–mediated phagocytosis of cancer cells by macrophages. In our system, anti-CD47 B6H12 mAb did not have a direct effect on T-cell activation or inhibition (Figs. 2 and 3).
The involvement of both the innate and adaptive immune systems in the mechanism of action of anti-CD47 antibody has several clinical implications. First, our findings suggest a novel role of anti-CD47 blocking mAbs as a vaccination strategy to enhance CD8+ effector T cells recognizing antigens on phagocytosed target cells. Second, in designing clinical trial protocols for testing anti-CD47 therapy in patients, immune monitoring of T cells may be important for understanding clinical response to treatment and clinical outcomes. Third, anti-CD47 mAbs might be used clinically in combination with adoptive T-cell therapy or T-cell–activating antibodies to enhance the adaptive immune response against tumor antigens and minimize toxicity. We conclude that anti-CD47–mediated phagocytosis of cancer not only functions in directly clearing cancer cells but also can initiate an antitumor T-cell response to eliminate cancers. Patients receiving anti-CD47 therapy may benefit from both the innate and adaptive immune responses against cancer.
Materials and Methods
Mice.
Mice, including C57BL/Ka (CD45.2), C57BL/Ka (CD45.1), and C57BL/Ka Rosa26-mRFP1 mice, were bred and maintained at the Stanford University Research Animal Facility in accordance with the Administrative Panel on Laboratory Animal Care. All the animals were housed in sterile microinsulators and were given water and rodent chow ad libitum. OT-I TCR transgenic mice, OT-II TCR transgenic mice, and Foxp3-GFP mice were purchased from the Jackson Laboratory. OT-I mice have transgenic TCRs specific for OVA257-264 in the context of H2-kb. OT-II mice have TCRs specific for OVA323-339 in the context of IAb. Both OT-I and OT-II mice have transgenic Vα2 Vβ5 TCRs.
Molecular Biology.
cOVA was cloned from pCI-neo-cOVA (plasmid 25097; AddGene) and was shuttled into the lentiviral pCDH-EF1-MCS-T2A-copGFP vector (System Biosciences) using EcoRI and BamHI restriction sites. Lentiviral production and concentration were accomplished using standard protocols.
Generation of Macrophages and Dendritic Cells.
Whole bone marrow cells were isolated from C57BL/Ka (CD45.2) or C57BL/Ka Rosa26-mRFP1 mice. Macrophages were generated by incubating whole bone marrow in macrophage colony-stimulating factor (10 ng/mL) for 7 d and harvesting the adherent fraction. Dendritic cells were generated in granulocyte-macrophage colony stimulating factor (GM-CSF) (1,000 U/mL); on days 2 and 4 cells were washed and medium was replaced with fresh cytokine-containing medium. Nonadherent cells were replated on day 6 and harvested on day 7.
In Vitro Phagocytosis Assay.
For the in vitro phagocytosis assay, 2 × 104 macrophages or dendritic cells per well were plated in a 96-well ultra-low-adherent plate (Corning), along with 2 × 104 cancer cells (DLD1-cOVA-GFP) in serum-free RPMI medium.
The indicated antibodies (10 μg/mL) were added and incubated for 4 h at 37°. Macrophages were washed twice and analyzed using a BD LSR Fortessa Analyzer. The percentage of phagocytosis was calculated as the percentage of GFP+ cells within RFP+ macrophages or F4/80+ macrophages. For in vivo transfer assays, 5 × 105 macrophages and cancer cells were cocultured in the presence of control IgG1 or anti-CD47 B6H12 mAb (10 μg/mL) and were incubated for 2 h. Macrophages then were separated from the cultures during anti–Mac-1 magnetic beads (Miltenyi Biotec).
T Cell Priming Assay.
For in vitro T cell priming assays, 104 macrophages were cocultured overnight with equal numbers of DLD1-cOVA-GFP cancer cells in serum-free RPMI medium. The next day, equal volume of RPMI+ 20% (vol/vol) FCS was added to the cultures. Peripheral lymph nodes were harvested from OT-I or OT-II TCR transgenic mice and labeled with 0.5 mM CFSE (Molecular Probes). T cells were isolated using biotinylated anti-CD8 or anti-CD4 antibodies, followed by enrichment with anti-biotin magnetic beads (Miltenyi Biotec). Then 5 × 104 T cells were added to the cultures and analyzed on day 3 (for OT-I T cells) or day 4 (for OT-II T cells). For in vivo T cell priming assays, 2 × 106 CFSE-labeled OT-I T cells (CD45.2) were adoptively transferred i.v. into recipient mice (CD45.1). Macrophages were isolated from coculture with cancer cells as previously described and were injected into the footpads of mice. Popliteal lymph nodes were analyzed on day 4 for CFSE dilution within CD45.2+ cells.
Antibodies and Flow Cytometry Analysis.
Mouse anti-human anti-CD47 mAb B6H12 (IgG1) was obtained from Bio-XCell. Mouse anti-human anti-CD47 mAb 2D3 (IgG1) and mouse IgG1 mAb were obtained from eBioscience. For verification of binding of anti-CD47 B6H12 and 2D3 to DLD1-cOVA-GFP cancer cells, the cells were labeled with a saturating concentration of anti-CD47 antibody, followed by phycoerythrin-conjugated donkey-anti-mouse IgG (H&L) (eBioscience). Data were acquired using a BD LSR Fortessa Analyzer and analyzed using FlowJo software.
In Vivo Cell-Killing Assay.
In brief, splenocytes from C57BL/Ka (CD45.1) mice were labeled with 10 μM CFSE (CFSE-high) or 1 μM CFSE (CFSE-low). CFSE-high splenocytes then were pulsed in a six-well plate with 1 μM SIINFEKL peptide for 1 h. Cells then were mixed in a 1:1 ratio with non–peptide-pulsed CFSE-low cells before i.v. transfer. To account for variation in the CFSE-high/-low ratio in the absence of peptide-specific lysis, control mice received CFSE-high and -low splenocytes not pulsed with SIINFEKL peptide and mixed in a 1:1 ratio. Draining lymph nodes were analyzed 16 h later. The percentage of cytotoxicity was calculated as (1 − percentage of CFSE-high/percentage of CFSE-low) normalized to the ratio in control mice.
Tumor Challenge.
CD8-enriched OT-I T cells (1 × 106) were adoptively transferred i.v. into recipient C57BL/Ka mice. Macrophages from syngeneic C57BL/Ka mice were cocultured with DLD1-cOVA-GFP cancer cells as previously described and then were isolated by magnetic enrichment and injected into the footpads of mice. The tumor cell line E.G7 (EL.4 cells expressing the chicken OVA cDNA) was used for tumor challenge of mice (Amercan Type Culture Collection). E.G7 cells (1 × 105) were injected s.c. into the right hindlimb of the mice in a 1:1 ratio with regular Matrigel. Tumor size was measured every day using fine calipers, and tumor volume was calculated based on length × width × height × π/6.
Cytokine Assay.
Macrophages were cocultured overnight with equal numbers of DLD1-cOVA-GFP cancer cells in serum-free RPMI medium. The next day, supernatants were harvested and submitted to the Stanford Human Immune Monitoring Core for cytokine analysis by mouse 26-plex Luminex assay (Affymetrix).
Supplementary Material
Acknowledgments
We thank Theresa Storm and Libuse Jerabek for excellent laboratory management, Tejaswitha Naik for antibody conjugation, Aaron McCarty for mouse breeding and management, Patty Lovelace and Jennifer Ho for technical assistance with flow cytometry, the Stanford Human Immune Monitoring Core and Yael Rosenberg-Hasson for assistance with the Luminex assay, and Suparna Dutt for intellectual discussions. Funding support for this work was provided by the Student Training and Research in Tumor Immunology (STaRT) Program of the Cancer Research Institute (D.T.), the Virginia and D. K. Ludwig Fund for Cancer Research (I.L.W., D.T., M.M.), the Joseph and Laurie Lacob Gynecologic/Ovarian Cancer Fund (J.V.P.), and by Grants R01 CA86017 (I.L.W.), P01 CA139490 (I.L.W., S.W., H.C.-T., K.W.), P30 CA124435 (I.L.W.), and F30 CA168059 (K.W.) from the National Institutes of Health. D.T. is the recipient of a Howard Hughes Medical Institute medical student fellowship. We also thank the Stanford Program in Cancer Biology and Stanford Medical Scientist Training Program for their support (D.T. and K.W.).
Footnotes
Conflict of interest statement: I.L.W. owns Amgen Inc. stock and is a Director of Stem Cells, Inc.
See Commentary on page 10886.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1305569110/-/DCSupplemental.
References
- 1.Unanue ER. The regulatory role of macrophages in antigenic stimulation. Adv Immunol. 1972;15:95–165. doi: 10.1016/s0065-2776(08)60684-7. [DOI] [PubMed] [Google Scholar]
- 2.Burgdorf S, Kautz A, Böhnert V, Knolle PA, Kurts C. Distinct pathways of antigen uptake and intracellular routing in CD4 and CD8 T cell activation. Science. 2007;316(5824):612–616. doi: 10.1126/science.1137971. [DOI] [PubMed] [Google Scholar]
- 3.Bonifaz LC, et al. In vivo targeting of antigens to maturing dendritic cells via the DEC-205 receptor improves T cell vaccination. J Exp Med. 2004;199(6):815–824. doi: 10.1084/jem.20032220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Callahan MK, Wolchok JD, Allison JP. Anti-CTLA-4 antibody therapy: Immune monitoring during clinical development of a novel immunotherapy. Semin Oncol. 2010;37(5):473–484. doi: 10.1053/j.seminoncol.2010.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Majeti R, et al. CD47 is an adverse prognostic factor and therapeutic antibody target on human acute myeloid leukemia stem cells. Cell. 2009;138(2):286–299. doi: 10.1016/j.cell.2009.05.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Willingham SB, et al. The CD47-signal regulatory protein alpha (SIRPa) interaction is a therapeutic target for human solid tumors. Proc Natl Acad Sci USA. 2012;109(17):6662–6667. doi: 10.1073/pnas.1121623109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brown EJ, Frazier WA. Integrin-associated protein (CD47) and its ligands. Trends Cell Biol. 2001;11(3):130–135. doi: 10.1016/s0962-8924(00)01906-1. [DOI] [PubMed] [Google Scholar]
- 8.Oldenborg PA, et al. Role of CD47 as a marker of self on red blood cells. Science. 2000;288(5473):2051–2054. doi: 10.1126/science.288.5473.2051. [DOI] [PubMed] [Google Scholar]
- 9.Kharitonenkov A, et al. A family of proteins that inhibit signalling through tyrosine kinase receptors. Nature. 1997;386(6621):181–186. doi: 10.1038/386181a0. [DOI] [PubMed] [Google Scholar]
- 10.Fujioka Y, et al. A novel membrane glycoprotein, SHPS-1, that binds the SH2-domain-containing protein tyrosine phosphatase SHP-2 in response to mitogens and cell adhesion. Mol Cell Biol. 1996;16(12):6887–6899. doi: 10.1128/mcb.16.12.6887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Timms JF, et al. Identification of major binding proteins and substrates for the SH2-containing protein tyrosine phosphatase SHP-1 in macrophages. Mol Cell Biol. 1998;18(7):3838–3850. doi: 10.1128/mcb.18.7.3838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tsai RK, Discher DE. Inhibition of “self” engulfment through deactivation of myosin-II at the phagocytic synapse between human cells. J Cell Biol. 2008;180(5):989–1003. doi: 10.1083/jcb.200708043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chao MP, et al. Therapeutic antibody targeting of CD47 eliminates human acute lymphoblastic leukemia. Cancer Res. 2011;71(4):1374–1384. doi: 10.1158/0008-5472.CAN-10-2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chao MP, et al. Extranodal dissemination of non-Hodgkin lymphoma requires CD47 and is inhibited by anti-CD47 antibody therapy. Blood. 2011;118(18):4890–4901. doi: 10.1182/blood-2011-02-338020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim D, et al. Anti-CD47 antibodies promote phagocytosis and inhibit the growth of human myeloma cells. Leukemia. 2012;26(12):2538–2545. doi: 10.1038/leu.2012.141. [DOI] [PubMed] [Google Scholar]
- 16.Edris B, et al. Antibody therapy targeting the CD47 protein is effective in a model of aggressive metastatic leiomyosarcoma. Proc Natl Acad Sci USA. 2012;109(17):6656–6661. doi: 10.1073/pnas.1121629109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chao MP, et al. Anti-CD47 antibody synergizes with rituximab to promote phagocytosis and eradicate non-Hodgkin lymphoma. Cell. 2010;142(5):699–713. doi: 10.1016/j.cell.2010.07.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chao MP, et al. Calreticulin is the dominant pro-phagocytic signal on multiple human cancers and is counterbalanced by CD47. Sci Transl Med. 2010;2(63):63ra94. doi: 10.1126/scitranslmed.3001375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Saito Y, et al. Regulation by SIRPα of dendritic cell homeostasis in lymphoid tissues. Blood. 2010;116(18):3517–3525. doi: 10.1182/blood-2010-03-277244. [DOI] [PubMed] [Google Scholar]
- 20.Asano K, et al. CD169-positive macrophages dominate antitumor immunity by crosspresenting dead cell-associated antigens. Immunity. 2011;34(1):85–95. doi: 10.1016/j.immuni.2010.12.011. [DOI] [PubMed] [Google Scholar]
- 21.Harshyne LA, Watkins SC, Gambotto A, Barratt-Boyes SM. Dendritic cells acquire antigens from live cells for cross-presentation to CTL. J Immunol. 2001;166(6):3717–3723. doi: 10.4049/jimmunol.166.6.3717. [DOI] [PubMed] [Google Scholar]
- 22.Allan RS, et al. Migratory dendritic cells transfer antigen to a lymph node-resident dendritic cell population for efficient CTL priming. Immunity. 2006;25(1):153–162. doi: 10.1016/j.immuni.2006.04.017. [DOI] [PubMed] [Google Scholar]
- 23.Harbers SO, et al. Antibody-enhanced cross-presentation of self antigen breaks T cell tolerance. J Clin Invest. 2007;117(5):1361–1369. doi: 10.1172/JCI29470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rafiq K, Bergtold A, Clynes R. Immune complex-mediated antigen presentation induces tumor immunity. J Clin Invest. 2002;110(1):71–79. doi: 10.1172/JCI15640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kong XN, et al. LPS-induced down-regulation of signal regulatory protein alpha contributes to innate immune activation in macrophages. J Exp Med. 2007;204(11):2719–2731. doi: 10.1084/jem.20062611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ramachandran IR, et al. The phosphatase SRC homology region 2 domain-containing phosphatase-1 is an intrinsic central regulator of dendritic cell function. J Immunol. 2011;186:3934–3945. doi: 10.4049/jimmunol.1001675. [DOI] [PubMed] [Google Scholar]
- 27.Basu S, Srivastava PK. Calreticulin, a peptide-binding chaperone of the endoplasmic reticulum, elicits tumor- and peptide-specific immunity. J Exp Med. 1999;189(5):797–802. doi: 10.1084/jem.189.5.797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nair S, et al. Calreticulin displays in vivo peptide-binding activity and can elicit CTL responses against bound peptides. J Immunol. 1999;162(11):6426–6432. [PubMed] [Google Scholar]
- 29.Obeid M, et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat Med. 2007;13(1):54–61. doi: 10.1038/nm1523. [DOI] [PubMed] [Google Scholar]
- 30.Ticchioni M, et al. Integrin-associated protein (CD47/IAP) contributes to T cell arrest on inflammatory vascular endothelium under flow. FASEB J. 2001;15(2):341–350. doi: 10.1096/fj.99-0833com. [DOI] [PubMed] [Google Scholar]
- 31.Reinhold MI, Lindberg FP, Kersh GJ, Allen PM, Brown EJ. Costimulation of T cell activation by integrin-associated protein (CD47) is an adhesion-dependent, CD28-independent signaling pathway. J Exp Med. 1997;185(1):1–11. doi: 10.1084/jem.185.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Waclavicek M, et al. T cell stimulation via CD47: Agonistic and antagonistic effects of CD47 monoclonal antibody 1/1A4. J Immunol. 1997;159(11):5345–5354. [PubMed] [Google Scholar]
- 33.Latour S, et al. Bidirectional negative regulation of human T and dendritic cells by CD47 and its cognate receptor signal-regulator protein-alpha: Down-regulation of IL-12 responsiveness and inhibition of dendritic cell activation. J Immunol. 2001;167(5):2547–2554. doi: 10.4049/jimmunol.167.5.2547. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.