

Abstract
Efforts to improve the clinical outcome of highly aggressive triple-negative breast cancer (TNBC) have been hindered by the lack of effective targeted therapies. Thus, it is important to identify the specific gene targets/pathways driving the invasive phenotype to develop more effective therapeutics. Here we show that ubiquitin-associated and SH3 domain-containing B (UBASH3B), a protein tyrosine phosphatase, is overexpressed in TNBC, where it supports malignant growth, invasion, and metastasis largely through modulating epidermal growth factor receptor (EGFR). We also show that UBASH3B is a functional target of anti-invasive microRNA200a (miR200a) that is down-regulated in TNBC. Importantly, the oncogenic potential of UBASH3B is dependent on its tyrosine phosphatase activity, which targets CBL ubiquitin ligase for dephosphorylation and inactivation, leading to EGFR up-regulation. Thus, UBASH3B may function as a crucial node in bridging multiple invasion-promoting pathways, thereby providing a potential therapeutic target for TNBC.
Triple-negative breast cancer (TNBC)—so named for its absent expression of estrogen receptor (ER), progesterone receptor, and human epidermal growth factor receptor 2 (HER2)—is among the most aggressive of breast cancer subtypes, with a high propensity for metastasis and poor prognosis (1). Current treatment modalities for TNBC are limited to surgery, radiation, and systemic chemotherapy, given the absence of more specific therapeutic targets. However, patients often experience early relapse from distant tumor metastasis, although they may initially respond well to the treatments (2).
Over the past few decades, tremendous effort has been expended in the search for molecular targeted therapy for TNBC, with only limited success. Epidermal growth factor receptor (EGFR)-targeted therapy is an example of these investigational therapies. Although TNBC often harbors EGFR overexpression (3), anti-EGFR treatments have shown only limited favorable response in clinical trials, however (4). Another example is poly-(ADP ribose) polymerase inhibitor, which can induce synthetic lethality in BRCA1-deficient TNBC tumors, although its clinical efficacy in a Phase II trial remains to be determined (5). Although the reasons for the ineffectiveness of these molecular targeted therapies are unclear, one hypothesis is that breast tumors, particularly TNBCs, are highly heterogeneous and use multiple mechanisms, including invasion and metastasis, to enable the aggressive phenotypes. These pathways then may become integrated as specific signaling steps that collectively contribute to the disease progression. Pathways and mechanisms involved in invasion and metastasis often involve transcriptional reprogramming induced by deregulated transcriptional and epigenetic regulators, as well as miRNAs such as miR200 (6–11). Although the breast cancer transcriptome has been investigated extensively, how these pathways are interconnected in disease progression is unclear, and the regulatory elements crucial for network integrity remain to be identified. The availability of improved genomic technologies allows us to readdress these issues.
The goal of the present study was to identify “druggable” targets in TNBC that may inform new treatment options. Using the latest gene array technology, we have identified the overexpression of Ubiquitin-Associated and SH3 Domain-Containing B (UBASH3B) tyrosine phosphatase as an oncogenic driver in TNBC invasion and metastasis. We show that UBASH3B acts as a crucial downstream target of invasive regulator miR200a, and that its oncogenic function requires phosphatase activity. Our work thus identifies an oncogenic protein phosphatase as a potential therapeutic target for TNBC.
Results
Functional Genomics Identifies Overexpression of UBASH3B in TNBC.
To identify specific targets associated with TNBC, we used the Illumina HumanHT-12 V4.0 expression Beadchip to profile the gene expression of a panel of TNBC and non-TNBC human breast cancer cell lines and primary tumor tissues. Toward this end, we identified a set of 103 genes as overexpressed in both TNBC cell lines and patient tissues compared with respective non-TNBC counterparts (Fig. 1A, Fig. S1A, and Table S1). Among these genes, 19 were annotated as enzymes and thus considered potential “druggable” targets (Fig. 1A). Moreover, seven of these genes showed a significant association with disease outcome in at least one examined breast cancer cohort (Fig. S1B).
Fig. 1.
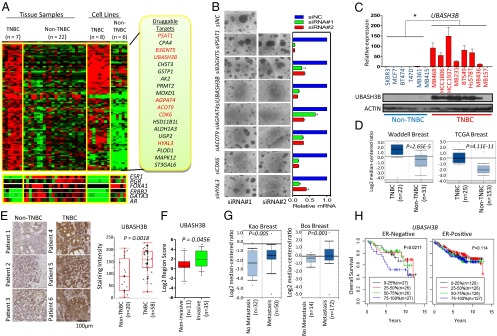
Functional genomics identifies the overexpression of UBASH3B in TNBC. (A) Heat map representation of 103 genes expressed more than twofold higher in TNBC compared with non-TNBC breast cancer tissues and cell lines. Expressions of ER (ESR1) and its related genes FOXA1 and GATA3, as well as ERBB2, AR, and PGR, are also shown. A total of 19 druggable genes were identified (Right); the genes in red were associated with poor patient survival. (B) Representative images of MB231 cells cultured in 3D Matrigel. (Left) Cells were depleted of indicated genes using two independent siRNA sequences. (Right) Knockdown efficiency was assessed by qPCR. (C) qPCR and Western blot analysis of UBASH3B expression in a panel of breast cancer cell lines. *P < 0.05, unpaired two-tailed t test. (D) Boxplots showing the mRNA expression of UBASH3B in two Oncomine datasets, Waddell and TCGA breast. (E) Representative images of IHC staining of UBASH3B on paraffin-embedded breast cancer tissues from the John Wayne Cancer Institute patient cohort. Staining was graded based on the intensity, and P values were calculated by the Wilcoxon test. (F) IHC staining of UBASH3B on a tissue microarray consisting of invasive and noninvasive breast cancer tissues. The proportion of positive-stained cytoplasm was scored and plotted. P values were calculated by the unpaired two-tailed t test. (G) Boxplots showing the mRNA expression of UBASH3B in two Oncomine datasets, Kao (metastatic event at 3 y) and Bos (metastatic event at 5 y). (H) Kaplan–Meier analyses of overall survival of breast cancer patients in the Oxford/Stockholm/Uppsala datasets. Patients were stratified based on quartile mRNA expression of UBASH3B. P values were calculated by the Wald test. Error bars represent mean ± SEM.
To examine whether the foregoing genes could have roles in TNBC, we knocked down their expression individually in the highly aggressive breast cancer cell line MDA-MB-231 (MB231 hereinafter) by transfecting two independent siRNA oligos. We monitored the efficiency of gene knockdown by quantitative PCR (qPCR) and assessed the phenotypic changes on 3D Matrigel growth as an indicator of malignant growth. Our results showed that five of the seven genes (UBASH3B, AGPAT4, ACOT9, CDK6, and HYAL3) may be required for TNBC progression, given that their knockdown strongly reduced the Matrigel growth of MB231 cells (Fig. 1B). Among these genes, UBASH3B, also called Suppressor of T-Cell Receptor Signaling 1 (STS-1), is of particular interest for further study, as it was top-ranked in causing phenotypic change upon knockdown and known to regulate EGFR endocytosis and degradation (12). Moreover, UBASH3B was recently shown to have tyrosine protein phosphatase activity (13). Nevertheless, a role of UBASH3B in promoting tumorigenesis has yet to be described.
Further validation analysis using qPCR and Western blot analysis revealed high levels of both UBASH3B mRNA and protein in highly invasive TNBC cell lines compared with the less-invasive luminal lines (Fig. 1C). In contrast, Ubiquitin-Associated and SH3 Domain-Containing A (UBASH3A), a close family member of UBASH3B, showed no such difference (Fig. S1C). Interestingly, UBASH3B was overexpressed in invasive prostate cancer cells as well (Fig. S1D). Furthermore, gene expression analysis based on Oncomine datasets also showed the overexpression of UBASH3B in TNBC and metastatic prostate cancer specimens (Fig. 1D and Fig. S1E). The overexpression of UBASH3B in TNBC was further verified by immunohistochemistry (IHC) analysis of clinical tumor specimens (P = 0.0018) (Fig. 1E). Furthermore, IHC analysis of a cohort of primary and invasive breast tumors on a tissue microarray revealed higher UBASH3B expression in invasive breast cancer compared with noninvasive counterparts (P = 0.0456) (Fig. 1F). More strikingly, higher expression of UBASH3B was consistently observed in patients undergoing early metastasis events (Fig. 1G). These results indicate an association between UBASH3B and breast cancer invasion and metastasis in patients with breast cancer.
Finally, we performed a meta-analysis by combining three independent patient cohorts to assess the overall breast cancer patient survival in relation to the expression level of UBASH3B. The results indicated that high expression of UBASH3B confers poor overall survival (particularly 5-y survival) in patients with ER-negative breast cancer (Fig. 1H), but no significant survival disadvantage in patients with ER-positive breast cancer. This observation suggests that high UBASH3B expression may predict early relapse in ER-negative patients. Taken together, these findings suggest a potential role of UBASH3B in TNBC invasion and metastasis.
UBASH3B Is a Functional Target of miR200a.
To identify the molecular events leading to UBASH3B up-regulation associated with invasion, we examined the regulatory regions of UBASH3B. We found that several miR200a target sequences were present in the 3′ UTR of UBASH3B (Fig. 2A). miR200s are known to be anti-invasive, and their expression is down-regulated in aggressive breast cancer and prostate cancer (14, 15).
Fig. 2.

UBASH3B is a target of miR200a. (A) Schematic diagram of UBASH3B gene structure. TAA, stop codon; MRE, miRNA response element. (B) qPCR analysis of miR200a expression in a panel of breast cancer cell lines. *P < 0.05, unpaired two-tailed t test. (C) Schematic diagram showing the four MREs in the 3′ UTR of the UBASH3B gene base-paired to an miR200a mature sequence. Two regions (P1 and P2) were cloned into a luciferase reporter, pMIR-REPORT. (Left) P1 mutation (mut) and P2 mut were constructed by mutating the MRE to the mutant sequence as indicated. (Right) Luciferase reporter assay in cells cotransfected with miR200a or miR200c mimics and pMIR-REPORT containing WT or mutant P1 or P2. *P < 0.05, paired two-tailed t test. (D) qPCR and Western blot analysis of UBASH3B expression in MB231- and BT549-overexpressing miR200a or miR200c mimics. Protein expression of ZEB1 and ZEB2 was assessed as well. *P < 0.05, paired two-tailed t test. (E) qPCR and Western blot analysis of UBASH3B expression in indicated cells transfected with miR200a antagomir. *P < 0.05, paired two-tailed t test. (F) qPCR analysis of miR200a and UBASH3B expression in TNBC and non-TNBC tumor samples. (G) (Upper) Transwell Matrigel invasion assay conducted on MB231 transfected with miR200a or miR200c mimics in the presence of EGF as a chemoattractant. (Lower) UBASH3B expression assessed by Western blot analysis. *P < 0.05, paired two-tailed t test. Error bars indicate mean ± SEM.
We next investigated the possibility of UBASH3B serving as a target of miR200s. qPCR analysis revealed the down-regulation of miR200a/b/c in mesenchymal TNBC cell lines (Fig. 2B and Fig. S2A). Consistent with the foregoing analysis indicating that the UBASH3B 3′ UTR contains sites for miR200a, but not for miR200b/c, we were able to demonstrate direct and specific targeting and inhibition of UBASH3B by miR200a, but not by miR200c, using UBASH3B 3′ UTR reporters containing WT or a variant in which the miR200a-binding sequences were mutated (Fig. 2C). As expected, enforced expression of miR200a, but not of miR200c, was able to reduce the UBASH3B mRNA and protein expression in invasive breast and prostate cancer cells (Fig. 2D and Fig. S2C), although both miR200a and miR200c could repress the expression of ZEB1 and ZEB2 (Fig. 2D), two well-known common targets of miR200a/b/c (7, 16). Conversely, miR200a antagomir treatment of MCF7 and T47D cells that express high levels of miR200a resulted in up-regulation of UBASH3B mRNA and protein levels (Fig. 2E). We also confirmed the down-regulation of miR200a (P = 0.032), together with miR200b/c, in clinical TNBC samples compared with non-TNBC samples (Fig. 2F and Fig. S2B), in concordance with the up-regulation of UBASH3B in TNBC samples (P = 0.0007) (Fig. 2F).
We performed a rescue invasion assay to investigate the functional association between UBASH3B and miR200a., and found that treatment with either miR200a or miR200c could reduce the invasive capacity of MB231 cells (Fig. 2G). Ectopic expression of UBASH3B, which could not be targeted by miR200a, effectively restored the reduced invasion by miR200a, but not that by miR200c. Thus, UBASH3B is a functional target of miR200a, supporting a miR200a–UBASH3B functional axis in promoting cell invasion in TNBC.
UBASH3B Knockdown Reduces Malignancy by Modulating EGFR.
We next assessed the relevance of UBASH3B overexpression in tumorigenesis. Because UBASH3B is implicated in antagonizing EGFR degradation during endocytosis on EGF treatment (12), we asked whether UBASH3B has a role in EGF-induced invasion, a process of particular relevance in TNBC. UBASH3B knockdown by two independent siRNAs resulted in marked inhibition of EGF-induced Transwell invasion in invasive breast and prostate cancer cells (Fig. 3A, Upper and Fig. S3A). Consistently, UBASH3B knockdown facilitated EGF-induced EGFR protein degradation and abolished EGF-induced EGFR phosphorylation (Fig. 3A, Lower), These findings are consistent with the known role of EGFR as a well-established invasive effector in TNBC and other invasive tumors (17–19).
Fig. 3.

UBASH3B knockdown reduces malignant phenotypes and facilitates EGFR degradation (A) (Upper) Transwell Matrigel invasion assays were performed on MB231 and BT549 depleted of UBASH3B using two independent siRNAs in the presence or absence of EGF as a chemoattractant. (Lower) EGFR and UBASH3B expression was assessed by Western blot analysis. *P < 0.05, paired two-tailed t test. (B) Contingency analysis of UBASH3B and EGFR IHC staining on 73 breast cancer tissues from the John Wayne Cancer Institute patient cohort. P values were calculated with Fisher’s exact test. (C) (Left) UBASH3B expression as assessed by Western blot analysis. (Right) Representative images of indicated cells grown in 3D Matrigel. (D) Western blot analysis of EGFR and UBASH3B expression (Left) and Transwell invasion assay (Right) using MB231-LN cells with UBASH3B depletion and EGFR overexpression. *P < 0.05, paired two-tailed t test.
We further confirmed the specificity of knockdown effects on invasion by a rescue experiment using ectopic expression of UBASH3B and the third siRNA targeting the 3′ UTR of UBASH3B (Fig. S3B). Conversely, ectopic UBASH3B overexpression was able to increase EGFR protein abundance in a noncancerous human mammary epithelial MCF10A cells, although it could not induce transformation or enhance invasive growth (Fig. S3C). This indicates that UBASH3B expression is required but is insufficient to induce malignancy. The positive correlation between UBASH3B and EGFR in primary breast tumors was further validated by IHC analysis of 73 patient tissues revealing that patients with higher EGFR expression were significantly enriched with higher UBASH3B expression (P = 0.034) (Fig. 3B).
In addition to affecting cell invasion, UBASH3B knockdown also reduced the ability of breast and prostate cancer cells to form tumorspheres (Fig. S3 D and E), a growth feature associated with cancer stem cells. Notably, UBASH3B knockdown did not affect cell proliferation when grown on monolayers (Fig. S3F). This indicates that UBASH3B is not required for cell proliferation in general, and is more involved in regulating cellular phenotypes associated with malignancy. Consistent with these observations, we found that UBASH3B expression is further up-regulated in a more aggressive subline of MB231 derived from lymph node metastasis (MB231-LN) (20), which displayed a more disorganized morphology compared with the parental cells under 3D Matrigel growth conditions (Fig. 3C). As expected, UBASH3B knockdown in MB231-LN cells attenuated the aggressive morphology (Fig. 3C).
To examine whether the phenotypic changes after UBASH3B knockdown are related to the reduction of EGFR protein, we performed a rescue assay by stably ectopic expression of EGFR in MB231 cells depleted of UBASH3B. We found that the ectopic EGFR overexpression largely restored the invasive capacity of UBASH3B-depleted cells (Fig. 3D and Fig. S3 G and H), consistent with our hypothesis that UBASH3B, at least in large part, drives invasion through EGFR.
UBASH3B Knockdown Reduces Breast Cancer Metastasis.
To determine whether UBASH3B is required for tumor metastasis, we used a lung metastatic MB231 subline (LM2) (21) that expresses luciferase reporter, and generated short hairpin (sh) RNA knockdown (KD) shNC, shUBASH3B1 (KD1), and shUBASH3B2 (KD2) stable cell lines (Fig. 4A). Tail vein injection of these cells resulted in lung metastasis in nude mice. Bioluminescent imaging (BLI) measurement revealed significant reduction in lung metastasis compared with controls, with a 50% and 77% reduction of metastasis burden at week 4 after injection of KD1 and KD2, respectively (Fig. 4 B–D), closely corresponding to the respective knockdown efficiency in these cells. The reduced metastasis provided a significant survival advantage in these mice. Knockdown of UBASH3B substantially reduced the risk of mortality in mice, by 3.51-fold (95% CI, 1.10–11.18) with KD1 and by 6.99-fold (95% CI, 1.46–33.50) with KD2, which significantly prolonged survival in these mice (P = 0.02 for KD1; P = 0.004 for KD2) (Fig. 4E).
Fig. 4.

UBASH3B knockdown reduces breast cancer metastasis. (A) UBASH3B knockdown by shRNAs in LM2 cells as detected by Western blot analysis. (B) BLI curves of lung metastasis development in female athymic nude mice injected via the lateral tail vein with control and UBASH3B KD cells. Data are mean ± SEM; n = 9. *P < 0.05; **P < 0.01, Mann–Whitney U test. (C) Representative BLI images of mice in B at 4 wk postinjection. (D) Whole lung images of mice from a repeat experiment as in B, at 4 wk postinjection. (E) Kaplan–Meier curves of mice from B. n = 9. *P < 0.05; Cox proportional HR, 3.51 (95% CI, 1.10–11.18). **P < 0.005; Cox proportional HR, 6.99 (95% CI, 1.46–33.50). (F) UBASH3B knockdown by shRNA in 4T1 cells detected by Western blot analysis. (G) Boxplot showing the number of lung metastasis nodules from experimental metastases generated by control or UBASH3B KD of 4T1 cells at 2 wk after tail vein injection. n = 7–8. P values were determined by the Student t test. (H) Representative gross images of lung metastasis from mice as in G. (I) (Upper) Size of primary tumors generated by control or UBASH3B KD 4T1 cells at 24 d after mammary fat pad injection. (Lower) Boxplot showing the number of lung metastasis nodules from spontaneous metastases generated by control or UBASH3B KD 4T1 cells after mammary fat pad injection. n = 8. **P < 0.01, Student t test. (J) Representative gross images of spontaneous lung metastasis from mice as in I. (K) Representative images (Left) and BLI signals (Right) of lung metastasis of MB231-LN cells of control (NC) and UBASH3B KD2, with ectopic expression of vector or EGFR.
To further test the generality of the prometastatic function of UBASH3B, we also knocked down UBASH3B in the 4T1 mouse mammary tumor cell line and tested the effects in both experimental and orthotopic spontaneous metastasis models by tail vein and mammary gland injection, respectively. In both models, UBASH3B knockdown strongly inhibited lung metastasis (Fig. 4 F–J). Nevertheless, depletion of UBASH3B had little effect on primary tumor growth, as shown in the model of 4T1 cell line mammary fat pad injection (Fig. 4I). Interestingly, we found that ectopic expression of EGFR partially rescued inhibited metastasis in UBASH3B-depleted cells, although this rescue was seen only in the later weeks of metastasis progression (Fig. 4K). These data from both in vitro and in vivo studies demonstrate an important role of UBASH3B, at least in part through EGFR, in the progression of TNBC invasion and metastasis.
Oncogenic Role of UBASH3B Requires Phosphatase Activity Targeting CBL Phosphorylation and EGFR Degradation.
Considering that UBASH3B recently has been identified as a tyrosine phosphatase (13), we explored whether phosphatase activity is required for its invasion-promoting capability. To do this, we created a phosphatase-dead mutant UBASH3B (H391A) (13) and compared it with the WT UBASH3B in terms of the ability to promote cell invasion. Although the overexpression of WT UBASH3B in BT549 and MB231 cells robustly promoted both basal and EGF-induced invasion, overexpression of UBASH3B (H391A) did not (Fig. 5A). Consistently, EGF-induced EGFR degradation over time was dampened by ectopic overexpression of WT UBASH3B, but not of UBASH3B (H391A) (Fig. 5B). We further validated the functionality of this observation using a lung metastasis model. Consistent with in the vitro data, WT UBASH3B, but not the mutant UBASH3B lacking phosphatase activity, enhanced lung metastasis and concurrently reduced mouse survival (Fig. 5C). These in vitro and in vivo results demonstrate that the phosphatase activity of UBASH3B is required for its oncogenic activity to promote EGFR protein abundance, invasion, and metastasis in TNBC.
Fig. 5.
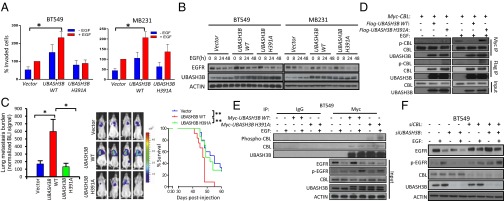
Oncogenic role of UBASH3B requires phosphatase activity targeting CBL phosphorylation and EGFR degradation (A) Transwell invasion of cells expressing WT or mutant UBASH3B (H391A) in the presence or absence of EGF as a chemoattractant. *P < 0.05, paired two-tailed t test. (B) EGFR and UBASH3B expression assessed by Western blot analysis. (C) (Left) BLI values of lung metastasis development in female NOD/SCID mice at 6 wk after injection with control and WT and mutant UBASH3B cells via the lateral tail vein. Data are mean ± SEM; n = 12 per category. *P < 0.05, unpaired two-sided Student t test. (Center) Representative BLI images of mice at 6 wk postinjection. (Right) Kaplan–Meier analysis. **P < 0.01, log-rank test. (D) Coimmunoprecipitation assays conducted on HEK293T with transient expression of CBL and WT or mutant UBASH3B in the presence or absence of EGF. (E) Coimmunoprecipitation assays conducted on BT549 stably expressing WT or mutant UBASH3B in the presence or absence of EGF. (F) Western blot analysis of EGFR, UBASH3B, and CBL protein expression in BT549 depleted with UBASH3B and/or CBL. Error bars represent mean ± SEM.
It has been reported previously that UBASH3B promotes EGFR protein stability by binding to and inhibiting CBL ubiquitin ligase-mediated EGFR degradation (12); however, whether this UBASH3B inhibition of CBL is mediated by its tyrosine phosphatase activity through dephosphorylating CBL is unknown. We found that overexpression of the WT UBASH3B, but not of the mutant UBASH3B (H391A), resulted in a loss of EGF-induced tyrosine phosphorylation of ectopic CBL in HEK293T cells, as assessed by probing the immunoprecipitated CBL with a pan-phospho-Tyr antibody, although interaction was observed between CBL and both WT and mutant UBASH3B (Fig. 5D). Moreover, in BT549 cells, EGF failed to induce CBL phosphorylation on overexpression of WT UBASH3B, but did induce robust CBL phosphorylation in the presence of UBASH3B (H391A) mutant (Fig. 5E). We further showed that UBASH3B knockdown-induced EGFR degradation and abolished EGFR phosphorylation could be restored by the concomitant CBL knockdown (Fig. 5F), indicating an antagonistic role of CBL in promoting UBASH3B-induced EGFR stabilization.
We further streamlined the functional connection of UBASH3B, CBL, and EGFR by measuring 3D Matrigel invasive growth. We found that the WT UBASH3B, but not the UBASH3B (H391A) mutant, promoted invasive growth, which was rescued by concomitant EGFR knockdown (Fig. S4). In addition, UBASH3B-promoted invasive growth was also rescued by cotransfection of ectopic CBL Y371E mutant, which is known to have a constitutive E3 ligase activity toward EGFR (22) (Fig. S4). Taken together, our biochemical and phenotypical analysis data support the hypothesis that UBASH3B promotes invasion and aggressive growth by promoting EGFR stability through dephosphorylation and inhibition of CBL function. Thus, the oncogenic phosphatase activity of UBASH3B makes this gene an excellent therapeutic target for cancer invasion and metastasis.
Discussion
Unlike for ER- or HER2-positive breast tumors, there currently are no targeted therapies for TNBC. Given the high genetic heterogeneity of TNBC and the complex gene network driving malignancy, along with the limited benefits of current targeted therapies, continued efforts to identify specific enzymes that represent crucial nodal points of a control network for pharmacologic intervention are of particular relevance. Given our finding that UBASH3B overexpression is associated with early relapse of TNBC, along with its fundamental role as a tyrosine phosphatase in invasive progression and disease outcome, we contend that UBASH3B overexpression is an important oncogenic event in TNBC. Importantly, UBASH3B acts to bridge several key invasive pathways, raising the possibility that therapeutic targeting UBASH3B may provide additional benefits for patients with TNBC.
UBASH3B up-regulation in TNBC can be the result of deregulation of miR200a, an important member of the miR200 family whose loss is often associated with cancer invasion through promotion of epithelial-mesenchymal transition (EMT). We have shown that UBASH3B is a direct and functional target of miR200a, and thus up-regulation of UBASH3B could be a result of miR200a down-regulation, as seen in TNBC. Although miR200s can suppress EMT by repressing the expression of ZEB1/2, thereby facilitating the spread of cancer cells (7), UBASH3B does not seem to affect EMT; its knockdown does not change cell morphology or the expression of EMT markers such as E-cadherin and vimentin. Thus, in early stages of disease progression, UBASH3B may coordinate with other miR200 targets to promote invasive capacity by enabling extravasation and subsequent migration to distal sites. We have previously shown that when circulating cancer cells colonize a distal site (eg, lung), miR200 needs to be reexpressed to induce mesenchymal-to-epithelial transition (MET), a rate-limiting step that allows for attachment and colonization of the new niche (23). Thus, UBASH3B expression might not be required during the late stages of metastasis progression, and its role in metastasis colonization may require further investigation.
In the present study, we have shown that UBASH3B functions as an oncogenic protein tyrosine phosphatase. At first glance, this finding may appear counterintuitive, considering that TNBC is known to harbor an overall induction of protein tyrosine phosphorylation activity owing to activation of a number of receptor tyrosine kinases, such as EGFR and MET, as well as nonreceptor tyrosine kinase Src (24). Moreover, other protein phosphatases, such as PTEN and PTPN12, often function as tumor suppressors that are inactivated in TNBC (25, 26). Thus, it is conceivable that UBASH3B as a tyrosine phosphatase likely exerts its oncogenic function by selectively targeting a specific tumor-suppressive tyrosine phosphorylation event. An example of this is tyrosine phosphatase PTP1B, which has been shown exert oncogenic effects by inhibiting growth-inhibitory Src Y530 phosphophorylaton in prostate and colon cancers (27). Of note, a recent study implicated another oncogenic tyrosine phosphatase, SHP2, in breast cancer progression (28), although SHP2’s association with TNBC, as well as the direct targets for dephosphorylation, remain to be elucidated.
A possible mechanism by which UBASH3B promotes invasive growth and metastasis in TNBC is at least partially through CBL-mediated EGFR regulation. We have shown that UBASH3B directly targets CBL phosphorylation to promote EGFR stability. CBL is long known to have dual roles in oncogenesis. In its tumor-suppressive role, CBL acts as an ubiquitin ligase to degrade multiple oncogenic tyrosine kinases, including EGFR (29). CBL’s oncogenic role has been linked to its capability to be recruited to activate receptors and to serve as adaptor for the docking of downstream kinases cascade to relay oncogenic signaling (30). Tumor-suppressive ubiquitin ligase activity is associated with phosphorylation at tyrosine residue 371, although other phosphorylation sites also have been linked to its oncogenic activity (31). We have shown that UBASH3B is able to bind to and dephosphorylate CBL using a pan-tyrosine phosphorylation antibody, leading to EGFR stabilization. It will be interesting to investigate whether UBASH3B specifically targets Y371 or other phosphorylation sites. Although UBASH3B may have effects on both tumor-suppressive and oncogenic phosphorylation sites, it may preferentially modulate CBL toward oncogenic activity when it is overexpressed in aggressive cancers like TNBC.
Materials and Methods
Primary breast tumor specimens were obtained from John Wayne Cancer Institute (Santa Monica, CA). All animal procedures were approved by the Institutional Animal Care and Use Committees at the University of Princeton and National University of Singapore.
Statistical Analysis.
All statistical analyses were performed using GraphPad Prism. Significance was evaluated by Student's t test or one-way ANOVA.
Supplementary Material
Acknowledgments
We thank the Histopathology Department, Institute of Molecular and Cell Biology, Agency for Science, Technology, and Research for their assistance with immunohistochemistry staining and analysis. This work was supported by the Agency for Science, Technology, and Research and Fashion Footwear Association of New York, Gonda Foundation (D.H.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission. C.L.A. is a guest editor invited by the Editorial Board.
Data deposition: The data reported in this paper have been deposited in the Gene Expression Omnibus (GEO) database, www.ncbi.nlm.nih.gov/geo (accession no. GSE36693).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1300873110/-/DCSupplemental.
References
- 1.Foulkes WD, Smith IE, Reis-Filho JS. Triple-negative breast cancer. N Engl J Med. 2010;363(20):1938–1948. doi: 10.1056/NEJMra1001389. [DOI] [PubMed] [Google Scholar]
- 2.Berrada N, Delaloge S, Andre F. Treatment of triple-negative metastatic breast cancer: Toward individualized targeted treatments or chemosensitization? Ann Oncol. 2010;21(Suppl 7):vii30–vii35. doi: 10.1093/annonc/mdq279. [DOI] [PubMed] [Google Scholar]
- 3.Corkery B, Crown J, Clynes M, O'Donovan N. Epidermal growth factor receptor as a potential therapeutic target in triple-negative breast cancer. Ann Oncol. 2009;20(5):862–867. doi: 10.1093/annonc/mdn710. [DOI] [PubMed] [Google Scholar]
- 4.Peddi PF, Ellis MJ, Ma C. Molecular basis of triple negative breast cancer and implications for therapy. Int J Breast Cancer. 2012;2012:217185. doi: 10.1155/2012/217185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Veldhuis JD, Anderson SM, Iranmanesh A, Bowers CY. Testosterone blunts feedback inhibition of growth hormone secretion by experimentally elevated insulin-like growth factor-I concentrations. J Clin Endocrinol Metab. 2005;90(3):1613–1617. doi: 10.1210/jc.2004-1303. [DOI] [PubMed] [Google Scholar]
- 6.Chase A, Cross NC. Aberrations of EZH2 in cancer. Clin Cancer Res. 2011;17(9):2613–2618. doi: 10.1158/1078-0432.CCR-10-2156. [DOI] [PubMed] [Google Scholar]
- 7.Gregory PA, et al. The miR200 family and miR-205 regulate epithelial-to-mesenchymal transition by targeting ZEB1 and SIP1. Nat Cell Biol. 2008;10(5):593–601. doi: 10.1038/ncb1722. [DOI] [PubMed] [Google Scholar]
- 8.Hahne JC, et al. Ets-1 expression promotes epithelial cell transformation by inducing migration, invasion and anchorage-independent growth. Oncogene. 2005;24(34):5384–5388. doi: 10.1038/sj.onc.1208761. [DOI] [PubMed] [Google Scholar]
- 9.Javelaud D, et al. TGF-β/SMAD/GLI2 signaling axis in cancer progression and metastasis. Cancer Res. 2011;71(17):5606–5610. doi: 10.1158/0008-5472.CAN-11-1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Naugler WE, Karin M. NF-kappaB and cancer-identifying targets and mechanisms. Curr Opin Genet Dev. 2008;18(1):19–26. doi: 10.1016/j.gde.2008.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sethi N, Kang Y. Notch signalling in cancer progression and bone metastasis. Br J Cancer. 2011;105(12):1805–1810. doi: 10.1038/bjc.2011.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kowanetz K, et al. Suppressors of T-cell receptor signaling Sts-1 and Sts-2 bind to Cbl and inhibit endocytosis of receptor tyrosine kinases. J Biol Chem. 2004;279(31):32786–32795. doi: 10.1074/jbc.M403759200. [DOI] [PubMed] [Google Scholar]
- 13.Mikhailik A, et al. A phosphatase activity of Sts-1 contributes to the suppression of TCR signaling. Mol Cell. 2007;27(3):486–497. doi: 10.1016/j.molcel.2007.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cao Q, et al. Coordinated regulation of polycomb group complexes through microRNAs in cancer. Cancer Cell. 2011;20(2):187–199. doi: 10.1016/j.ccr.2011.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Iliopoulos D, et al. Loss of miR200 inhibition of Suz12 leads to polycomb-mediated repression required for the formation and maintenance of cancer stem cells. Mol Cell. 2010;39(5):761–772. doi: 10.1016/j.molcel.2010.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Korpal M, Lee ES, Hu G, Kang Y. The miR200 family inhibits epithelial-mesenchymal transition and cancer cell migration by direct targeting of E-cadherin transcriptional repressors ZEB1 and ZEB2. J Biol Chem. 2008;283(22):14910–14914. doi: 10.1074/jbc.C800074200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Foley J, et al. EGFR signaling in breast cancer: Bad to the bone. Semin Cell Dev Biol. 2010;21(9):951–960. doi: 10.1016/j.semcdb.2010.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mitsudomi T, Yatabe Y. Epidermal growth factor receptor in relation to tumor development: EGFR gene and cancer. FEBS J. 2010;277(2):301–308. doi: 10.1111/j.1742-4658.2009.07448.x. [DOI] [PubMed] [Google Scholar]
- 19.Morishige M, et al. GEP100 links epidermal growth factor receptor signalling to Arf6 activation to induce breast cancer invasion. Nat Cell Biol. 2008;10(1):85–92. doi: 10.1038/ncb1672. [DOI] [PubMed] [Google Scholar]
- 20.Jenkins DE, Hornig YS, Oei Y, Dusich J, Purchio T. Bioluminescent human breast cancer cell lines that permit rapid and sensitive in vivo detection of mammary tumors and multiple metastases in immune deficient mice. Breast Cancer Res. 2005;7(4):R444–R454. doi: 10.1186/bcr1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Minn AJ, et al. Genes that mediate breast cancer metastasis to lung. Nature. 2005;436(7050):518–524. doi: 10.1038/nature03799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kassenbrock CK, Anderson SM. Regulation of ubiquitin protein ligase activity in c-Cbl by phosphorylation-induced conformational change and constitutive activation by tyrosine to glutamate point mutations. J Biol Chem. 2004;279(27):28017–28027. doi: 10.1074/jbc.M404114200. [DOI] [PubMed] [Google Scholar]
- 23.Korpal M, et al. Direct targeting of Sec23a by miR200s influences cancer cell secretome and promotes metastatic colonization. Nat Med. 2011;17(9):1101–1108. doi: 10.1038/nm.2401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hochgräfe F, et al. Tyrosine phosphorylation profiling reveals the signaling network characteristics of Basal breast cancer cells. Cancer Res. 2010;70(22):9391–9401. doi: 10.1158/0008-5472.CAN-10-0911. [DOI] [PubMed] [Google Scholar]
- 25.Marty B, et al. Frequent PTEN genomic alterations and activated phosphatidylinositol 3-kinase pathway in basal-like breast cancer cells. Breast Cancer Res. 2008;10(6):R101. doi: 10.1186/bcr2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sun T, et al. Activation of multiple proto-oncogenic tyrosine kinases in breast cancer via loss of the PTPN12 phosphatase. Cell. 2011;144(5):703–718. doi: 10.1016/j.cell.2011.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bjorge JD, Pang A, Fujita DJ. Identification of protein-tyrosine phosphatase 1B as the major tyrosine phosphatase activity capable of dephosphorylating and activating c-Src in several human breast cancer cell lines. J Biol Chem. 2000;275(52):41439–41446. doi: 10.1074/jbc.M004852200. [DOI] [PubMed] [Google Scholar]
- 28.Aceto N, et al. Tyrosine phosphatase SHP2 promotes breast cancer progression and maintains tumor-initiating cells via activation of key transcription factors and a positive feedback signaling loop. Nat Med. 2012;18(4):529–537. doi: 10.1038/nm.2645. [DOI] [PubMed] [Google Scholar]
- 29.Joazeiro CA, et al. The tyrosine kinase negative regulator c-Cbl as a RING-type, E2-dependent ubiquitin-protein ligase. Science. 1999;286(5438):309–312. doi: 10.1126/science.286.5438.309. [DOI] [PubMed] [Google Scholar]
- 30.Thien CB, Langdon WY. Cbl: many adaptations to regulate protein tyrosine kinases. Nat Rev Mol Cell Biol. 2001;2(4):294–307. doi: 10.1038/35067100. [DOI] [PubMed] [Google Scholar]
- 31.Levkowitz G, et al. Ubiquitin ligase activity and tyrosine phosphorylation underlie suppression of growth factor signaling by c-Cbl/Sli-1. Mol Cell. 1999;4(6):1029–1040. doi: 10.1016/s1097-2765(00)80231-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.