

Abstract
Local alignment-free sequence comparison arises in the context of identifying similar segments of sequences that may not be alignable in the traditional sense. We propose a randomized approximation algorithm that is both accurate and efficient. We show that under D2 and its important variant 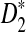 as the similarity measure, local alignment-free comparison between a pair of sequences can be formulated as the problem of finding the maximum bichromatic dot product between two sets of points in high dimensions. We introduce a geometric framework that reduces this problem to that of finding the bichromatic closest pair (BCP), allowing the properties of the underlying metric to be leveraged. Local alignment-free sequence comparison can be solved by making a quadratic number of alignment-free substring comparisons. We show both theoretically and through empirical results on simulated data that our approximation algorithm requires a subquadratic number of such comparisons and trades only a small amount of accuracy to achieve this efficiency. Therefore, our algorithm can extend the current usage of alignment-free–based methods and can also be regarded as a substitute for local alignment algorithms in many biological studies.
as the similarity measure, local alignment-free comparison between a pair of sequences can be formulated as the problem of finding the maximum bichromatic dot product between two sets of points in high dimensions. We introduce a geometric framework that reduces this problem to that of finding the bichromatic closest pair (BCP), allowing the properties of the underlying metric to be leveraged. Local alignment-free sequence comparison can be solved by making a quadratic number of alignment-free substring comparisons. We show both theoretically and through empirical results on simulated data that our approximation algorithm requires a subquadratic number of such comparisons and trades only a small amount of accuracy to achieve this efficiency. Therefore, our algorithm can extend the current usage of alignment-free–based methods and can also be regarded as a substitute for local alignment algorithms in many biological studies.
Key words: algorithms, alignment, dynamic programming, metagenomics
1. Introduction
Sequence alignment is perhaps the most well-known and intensively studied computational problem in modern molecular biology. The need to quickly and reliably identify relations between sequences has been a driving force in the field of computational biology, both on the statistical (Dayhoff et al., 1978; Karlin and Altschul, 1990; Waterman and Vingron, 1994) and algorithmic fronts (Johnson et al., 2008; Pearson and Lipman, 1988; Smith and Waterman, 1981; Zhang et al., 1998). Sequence alignment arises in a wide variety of data analysis contexts, ranging from theoretical studies of evolution to the practical use of sequencing instrumentation.
Alignment-based sequence similarity makes a correspondence between letters (bases or residues) in sequences but also requires this correspondence to preserve the order of letters. Alignment-free sequence comparison ignores positional information in strings and compares sequences without regard for the order (or orientation) of elements within the sequences. The use of such methods is motivated partially by technical issues of more rapidly screening candidates in very large-scale database searching (Haubold et al., 2011; Mahmood et al., 2012) and also by biological issues that suggest more functional information can be leveraged when order and orientation of sequence elements are not constrained (Sims and Kim, 2011).
To date every optimal sequence alignment algorithm requires a core step, based on dynamic programming, that requires quadratic time in the length of the sequences. Although heuristics have been extensively applied to improve the computational efficiency (Altschul et al., 1990, 1997), the inherent quadratic behavior remains unchanged when the sequence similarity is weak and there is no prior knowledge available about sequences. On the other hand, for global sequence comparison, alignment-free methods can run in time that is a linear function of the sequence lengths. Even in cases when alignment-free similarity is not as biologically meaningful as alignment-based similarity, the additional speed makes alignment-free approaches attractive for large-scale database filtering to reduce the number of pairwise sequence alignments that must be computed (Altschul et al., 1997; Lippert et al., 2002).
There are also certain biological phenomena overlooked by alignment and hence motivate the use of alignment-free methods as a substitute. Among these problems is the study of horizontal gene transfer (HGT), the transfer of genetic material between two organisms to acquire new traits. HGT is believed to be a vital step in bacterial adaptation and virulent niches (Alm et al., 2006). A recent study suggests HGT frequency in oceanic bacteria is up to a hundred million times greater than previously estimated, implying that the diversity gained by HGT might be dramatically underestimated (McDaniel et al., 2010). On the other hand, horizontal gene transfer as a localized recombination phenomenon scatters DNA fragments and makes the homologous sequences difficult to align (Domazet-Lošo and Haubold, 2011). Recent results have shown alignment-free methods are able to accurately reconstruct evolutionary relationships between metagenomic populations (Song et al., 2012).
Just as local alignment is used to identify locally similar parts of sequences, the concept of alignment-free comparison may be applied in a local sense. The essence of such methods emerges when the conserved functional elements are harbored in certain regions of the divergent sequences hindering the global methods to discover them. One clear example with this complication is the identification of transcription factor–binding sites in gene regulatory regions with low level of sequence conservation (Berman et al., 2002). Gene expression is often controlled by regulatory modules typically seen as short stretches of DNA sequence with highly variable distances from the target gene. Each of these modules, called a cis-regulatory module (CRM), contains one or a combined set of binding sites that is recognized by transcription factors (Kazemian et al., 2011). Identification of CRMs has been addressed as a challenging computational problem, especially for organisms in which high binding-site turnover takes place (Meader et al., 2010; Sinha and Siggia, 2005; Venkataram and Fay, 2010). This decreases the performance of alignment-based methods significantly because the order of functional elements is not necessarily preserved between similarly functioning sequences (Taher et al., 2011).
Much of the literature is on alignment-free sequence comparison addressing the statistical features of word frequencies in sequences. Sequences are often represented by word-count vectors, and inferences are made using similarity scores defined for those vectors. In this context, it is natural to count the number of k-letter words (k-mers) that a pair of sequences have in common for small values of k. This results in a well-studied and often applied statistic called D2 (Torney et al., 1990). Several investigations have analyzed the properties of this statistic and its variants (Forêt et al., 2009; Kantorovitz et al., 2007; Liua et al., 2011). One important concern is to estimate the asymptotic distribution of this statistic under the null hypothesis that two sequences are generated by a particular model (e.g., Markovian dependence or i.i.d. sequences). Once the distribution of D2 (or its variants) is approximated for such sequences, meaningful thresholds can be established to measure the deviation from the null hypothesis signaling biological relatedness (Lippert et al., 2002; Reinert et al., 2009; Wan et al., 2010). Due to the simplicity and time efficiency for the calculation of this statistic, D2 and its transformations have been previously used for EST sequence searches (Lippert et al., 2002). A formal treatment of D2 and two of its particular variants 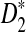 and
and 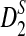 are given in the next section.
are given in the next section.
We present an algorithm for local alignment-free sequence comparison (Liua et al., 2011) under a class of similarity measures that we describe as dot product measures. The well-known D2 measure falls within this class, as does the specific standardized variant called 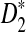 . We construct a framework to transform this string problem to a classical problem from computational geometry: to find the bichromatic closest pair (BCP) of points in a high-dimensional metric space (Agarwal et al., 1991; Indyk, 2001; Khuller and Matias, 1995). Our framework provides a general means of transforming dot product similarity into metric distance in the context of many similarity optimization problems, and we expect this framework will find applications outside of alignment-free sequence comparison.
. We construct a framework to transform this string problem to a classical problem from computational geometry: to find the bichromatic closest pair (BCP) of points in a high-dimensional metric space (Agarwal et al., 1991; Indyk, 2001; Khuller and Matias, 1995). Our framework provides a general means of transforming dot product similarity into metric distance in the context of many similarity optimization problems, and we expect this framework will find applications outside of alignment-free sequence comparison.
In the next section, we introduce technical concepts and provide formal definitions used throughout subsequent sections. Section 3 elucidates the general framework for transforming the problem of finding maximum local similarity to BCP. In Section 4, we explain a randomized algorithm to solve BCP in subquadratic time with bounded error. We present empirical results related to the practical performance of our algorithm in Section 5.
2. Background
We assume all strings are over a fixed alphabet 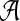 . For any string S, let S[i.j] denote the substring of S beginning at position i and having length j − i + 1. A k-mer is a string of length k, and we let
. For any string S, let S[i.j] denote the substring of S beginning at position i and having length j − i + 1. A k-mer is a string of length k, and we let 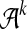 denote the set of possible k-mers. Define the count of k-mer z in string Si as
denote the set of possible k-mers. Define the count of k-mer z in string Si as
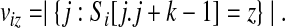 |
The k-mer count vector associated with Si is
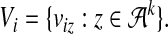 |
The similarity measure D2 between strings S1 and S2 is defined as
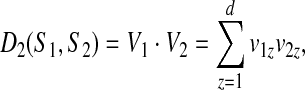 |
(1) |
that is the dot product between k-mer count vectors for S1 and S2. Here 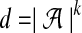 is the dimension of each vector, and we treat count vectors as points in
is the dimension of each vector, and we treat count vectors as points in 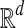 . Dot products are computed as usual, the norm of point p is
. Dot products are computed as usual, the norm of point p is 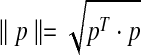 , and the angle between points p and q is
, and the angle between points p and q is 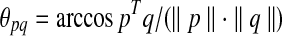 .
.
The D2 similarity measure was first applied to molecular sequences by Torney et al. (1990). Neglecting to account for statistical properties of k-words inherent in various kinds of molecular sequences has proven problematic (Lippert et al., 2002), and augmented measures have been designed to improve upon D2. If we view Si as a random sequence, letting pia denote the probability of drawing letter 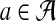 when generating Si, then for any
when generating Si, then for any  ,
,
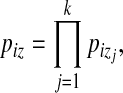 |
so 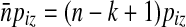 , with n = |Si|, approximates the expected number of occurrences of z in Si (ignoring autocorrelations). The associated standard deviation is denoted σz. The
, with n = |Si|, approximates the expected number of occurrences of z in Si (ignoring autocorrelations). The associated standard deviation is denoted σz. The 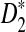 measure introduced by Reinert et al. (2009) is defined
measure introduced by Reinert et al. (2009) is defined
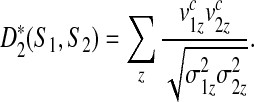 |
(2) |
where 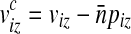 . Since frequencies of k-mers in sequences can be approximated using a Poisson approximation,
. Since frequencies of k-mers in sequences can be approximated using a Poisson approximation,
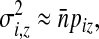 |
and we can simplify Equation (2). Let
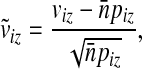 |
and 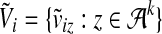 . Then we can also express
. Then we can also express 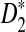 as a dot product of vectors:
as a dot product of vectors:
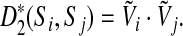 |
(3) |
We will also consider 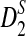 a variant of D2 introduced by Reinert et al. (2009):
a variant of D2 introduced by Reinert et al. (2009):
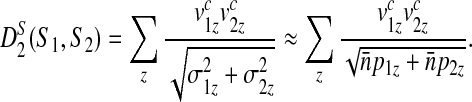 |
(4) |
Local similarity: We are interested in local similarity between two sequences, analogous to local alignment. While local alignments seek to optimize both the locations and the lengths of the aligned substrings, we restrict ourselves to fixed-sized windows. So in pairwise sequence comparison, we seek to identify windows of fixed width, one in each of two strings, such that the similarity between the pair of windows is maximal over all possible pairs of substrings.
Local alignment-free pairwise sequence similarity
Input: Two strings S1 and S2, both of length n, a similarity measurement score s and positive integers w ≤ n and k ≤ w.
Question: What is the maximum value of s(S1[i.i+w−1], S2[j.j+w−1]) over all 1≤i, j≤n−w+1?
Here w is the window size, which is fixed, and for all practical applications k = o(w) and w = o(n). The similarity measure s is assumed to be based on the k-mer counts vectors. In particular, we study dot-product-based similarity measures, for example D2 and 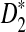 .
.
For each of the similarity measures in this study, the naive or brute-force algorithm for solving the local pairwise similarity problem iterates over all possible pairs of windows in each of the two given sequences and explicitly computes dot products for each. For sequences of length n, this requires Θ(λn2) time, where λ is the time required to compute each dot product. This time depends on how we represent count vectors (including the standardized vectors), but will generally be Θ(d).
When we think of each S[i.i+w−1] window as a point in k-mer counts space, the sequence of such points has an interesting geometric interpretation. Each point in the sequence differs from the previous point by at most one unit (i.e., a single count) in at most two dimensions. One new k-mer is included, and the corresponding count increases. Another, possibly the same, k-mer is excluded, and the corresponding count is decreased. We refer to this dynamic of our points as the “sliding property” and use it later to improve the running time of our algorithm. The naive algorithm can also take advantage of this property. For each update it identifies two “in” and “out” k-mers separately in logarithmic time and modifies the corresponding counts. Since there are Θ(n2) updates required to find the largest local dot-product between two sequences, the naive algorithm takes 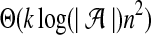 using sliding property. By encoding the sequences as suffix trees and making clever use of suffix links, the factor of k can be eliminated.
using sliding property. By encoding the sequences as suffix trees and making clever use of suffix links, the factor of k can be eliminated.
3. A Geometric Framework for Maximizing Similarity
In this section we place the local alignment-free sequence comparison problem in a geometric context that can transform a large class of similarity measures to distances satisfying the triangle inequality. Our framework is sufficiently general that it can be used for many global alignment-free similarity optimization problems. In this section we assume that the similarity measure is the basic D2. We remark that our framework can be applied to other variants of this statistic with only slight modification. First we recast the local alignment-free similarity problem as the maximial dot product problem.
Maximal dot product (MDP)
Input: Two sets 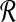 and
and 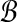 of vectors in
of vectors in 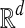 .
.
Question: What is the maximum value of r · b over any pair 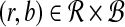 ?
?
The transformation from local alignment-free pairwise similarity under D2 is clear: the vectors in 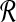 are the count vectors for the length w windows in S1 and the vectors in
are the count vectors for the length w windows in S1 and the vectors in 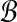 are the count vectors for the length w windows in S2. When referring to these sets of points, unless otherwise stated we assume
are the count vectors for the length w windows in S2. When referring to these sets of points, unless otherwise stated we assume 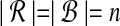 , which is equivalent to assuming |S1| = |S2| in the original problem instance.
, which is equivalent to assuming |S1| = |S2| in the original problem instance.
We can say a few things immediately about MDP problem instances obtained from instances of pairwise local alignment-free similarity. First, the dimensions correspond to the k-mers over the sequence alphabet, 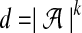 . This should immediately raise concerns among those familiar with high-dimensional optimization problems, which frequently require time that is exponential in the dimension of the space. Second, when the similarity measure is D2, the ℓ1 norm of each of these vectors is precisely determined by the values of w and k:
. This should immediately raise concerns among those familiar with high-dimensional optimization problems, which frequently require time that is exponential in the dimension of the space. Second, when the similarity measure is D2, the ℓ1 norm of each of these vectors is precisely determined by the values of w and k:
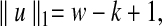 |
for all 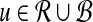 , since each possible k-mer in a window contributes one count in the vectors. More generally, for other similarity measures in the original problem, the ℓ1 norms can usually be bounded. We can also bound the ℓ2 norms of these vectors. In the case of D2, the maximum possible norm is achieved when all counts are for the same k-mer. This can only happen for
, since each possible k-mer in a window contributes one count in the vectors. More generally, for other similarity measures in the original problem, the ℓ1 norms can usually be bounded. We can also bound the ℓ2 norms of these vectors. In the case of D2, the maximum possible norm is achieved when all counts are for the same k-mer. This can only happen for 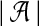 distinct substrings, corresponding to runs of the same letter. We have the following bound under D2, which will be useful later:
distinct substrings, corresponding to runs of the same letter. We have the following bound under D2, which will be useful later:
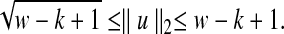 |
(5) |
The naive solution for MDP is the quadratic time evaluation of all pairs of elements from two sets. Nothing is trivially gained by transforming local alignment-free similarity into MDP, and our definition of MDP does not reflect the potentially useful structural property relating count vectors for consecutive windows. The dot product between vectors is related to the angle between those vectors, and optimization involving angles between vectors has received attention in the context of comparing documents based on the cosine similarity metric (Tan et al., 2006). Identifying the pair of points with maximum cosine similarity is equivalent to finding the pair with minimum angle but ignores the magnitude of those points. Instead of directly solving the MDP problem, we transform it into the well-studied bichromatic closest pair.
Bichromatic closest point (BCP)
Input: Two sets 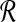 and
and 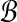 of vectors in
of vectors in 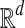 .
.
Question: What is the minimum value of ∥r − b∥ over any pair 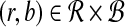 ?
?
The BCP problem was first addressed by Yao (1982) under the name nearest foreign neighbor in the context of geometric spanning trees. We will show how to efficiently approximate MDP if an oracle for BCP is available, and in later sections we will explain how to rapidly solve BCP for our problem.
We begin with the following straight-forward observation, which provides motivation for our approach. Suppose ∥r∥ = xr for all 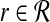 and ∥b∥ = xb for all
and ∥b∥ = xb for all 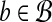 . Then, by the cosine law,
. Then, by the cosine law,
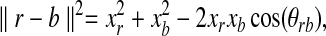 |
where θrb is the angle between r and b relative to the origin. So if the ℓ2 norms for all points in 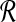 and
and 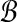 are fixed, any bichromatic pair of points with maximal dot product also has minimal Euclidean distance:
are fixed, any bichromatic pair of points with maximal dot product also has minimal Euclidean distance:
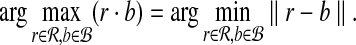 |
As explained above, the ℓ1 norm is fixed for all points when the original similarity measure is D2, but the ℓ2 norm can vary (Equation (5)). We introduce the sequential layering framework (SLF) to transform the original vectors in a way that constrains their ℓ2 norms.
We now explain how the SLF is used to partition the set 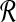 . The procedure is identical for
. The procedure is identical for 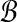 but the two sets must be partitioned separately. Define the set
but the two sets must be partitioned separately. Define the set 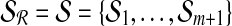 of hyperspheres centered at the origin. Note that we have dropped the subscript from
of hyperspheres centered at the origin. Note that we have dropped the subscript from 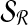 for convenience, and
for convenience, and 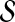 with no subscript is assumed to be defined relative to
with no subscript is assumed to be defined relative to 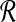 . We will explain below how we compute the value of m. Radii of the hyperspheres are defined recursively as follows:
. We will explain below how we compute the value of m. Radii of the hyperspheres are defined recursively as follows:
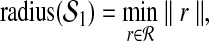 |
and for 2 ≤ i ≤ m + 1,
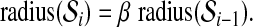 |
We will explain later how the constant β>1 is determined. For each 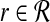 , define the orthogonal projection
, define the orthogonal projection
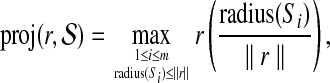 |
which effectively “shrinks” r by the smallest amount so that it resides on a hypersphere in 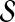 . Then define, for 1 ≤ i ≤ m,
. Then define, for 1 ≤ i ≤ m,
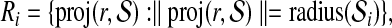 |
and let 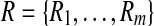 . We define
. We define 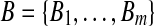 similarly based on
similarly based on 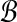 and
and 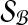 .
.
Our procedure is as follows. As mentioned previously, we assume an oracle for BCP. The m-partitions of 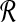 and
and 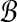 by the SLF are used to create m2 subproblems. We solve each of these subproblems by identifying pairs of points from each Ri × Bj that solve BCP(Ri, Bj). Dot products are only actually computed for the m2 point pairs returned as solutions for BCP from each of the m2 subproblems. We retain the maximum dot product from among these m2 as the solution to MDP. Pseudocode for this procedure is presented in Algorithm 1. In the pseudocode we use the notation proj−1 to denote the inverse of projections used above to define R and B from
by the SLF are used to create m2 subproblems. We solve each of these subproblems by identifying pairs of points from each Ri × Bj that solve BCP(Ri, Bj). Dot products are only actually computed for the m2 point pairs returned as solutions for BCP from each of the m2 subproblems. We retain the maximum dot product from among these m2 as the solution to MDP. Pseudocode for this procedure is presented in Algorithm 1. In the pseudocode we use the notation proj−1 to denote the inverse of projections used above to define R and B from 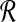 and
and 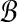 . What remains is to determine an appropriate value for m and to explain why the bichromatic point pair produced by this algorithm is guaranteed to have dot product within a certain factor of the optimal.
. What remains is to determine an appropriate value for m and to explain why the bichromatic point pair produced by this algorithm is guaranteed to have dot product within a certain factor of the optimal.
| Algorithm 1: Approximation for MDP via SLF with an oracle for BCP |
|---|
Input: Sets 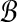 and and 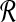 of points and constant β > 1. of points and constant β > 1. |
Output: A pair 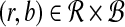 with approximately maximal dot product with approximately maximal dot product |
1: Construct 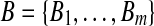 and and 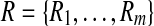 from from 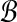 and and 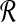
|
2: 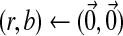
|
| 3: fori = 1 tomdo |
| 4: forj = 1 tomdo |
| 5: (p, q) ← BCP(Bi, Rj) |
6: 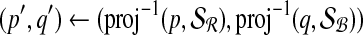
|
| 7: if p′ · q′ > r · bthen |
| 8: (r, b) ← (p′, q′) |
| 9: return (r, b) |
Proposition 1. If we select
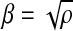 for some ρ > 1 in Algorithm 1, then the optimal dot product value is at most ρ times the value returned by the algorithm.
for some ρ > 1 in Algorithm 1, then the optimal dot product value is at most ρ times the value returned by the algorithm.
Proof. Denote the optimal pair (ropt, bopt) and suppose 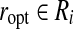 and
and 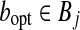 . Let (r, b) be the output of BCP for the same subproblem. For convenience we assume
. Let (r, b) be the output of BCP for the same subproblem. For convenience we assume 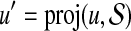 for every point u. For any point u we have
for every point u. For any point u we have 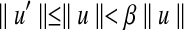 . Since the angle between any pair of points is preserved when both are projected toward the origin,
. Since the angle between any pair of points is preserved when both are projected toward the origin,
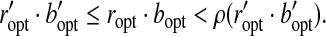 |
On the other hand, BCP guarantees 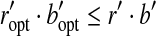 . Therefore,
. Therefore,
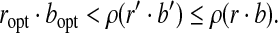 |
If (r, b) is replaced in a subsequent iteration, the resulting dot product is increased and the above inequality holds for the replacing pair of points. ▪
We now explain how the size m of partitions of 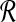 and
and 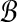 is determined based on our desired performance ratio.
is determined based on our desired performance ratio.
Proposition 2. There exists a set of Θ(logρ w) hyperspheres partitioning
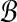 such that for all
such that for all
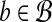 ,
,
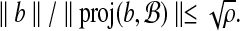 |
Proof. We show m = Θ(logρ
w) is sufficient for orthogonal projection of all points in 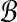 with the performance ratio ρ > 1. Denote bmin (bmax) the point with the minimum (maximum) norm in
with the performance ratio ρ > 1. Denote bmin (bmax) the point with the minimum (maximum) norm in 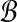 . As we previously described (Eq. 5),
. As we previously described (Eq. 5), 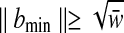 and
and 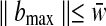 where
where 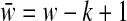 . Since bmin and bmax are projected to
. Since bmin and bmax are projected to 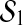 and
and 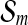 respectively, ∥bmax∥
respectively, ∥bmax∥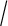 ∥bmin∥ can not be greater than radius(
∥bmin∥ can not be greater than radius(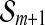 )/radius(
)/radius(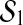 ) and thereby
) and thereby
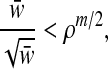 |
and the desired bound for m follows. ▪
In fact, 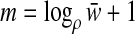 hyperspheres suffice for partitioning
hyperspheres suffice for partitioning 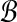 with the performance ratio ρ. Note that since k = o(w), we can remove the dependency of m on k and write m = Θ(logρw). In the statement of the next result we explicitly bound the constant c between 1 and 2. While this may seem artificial, it simplifies the proof compared with a more general statement and also corresponds to actual bounds: we can never do better than linear time, and the naive algorithm takes quadratic time.
with the performance ratio ρ. Note that since k = o(w), we can remove the dependency of m on k and write m = Θ(logρw). In the statement of the next result we explicitly bound the constant c between 1 and 2. While this may seem artificial, it simplifies the proof compared with a more general statement and also corresponds to actual bounds: we can never do better than linear time, and the naive algorithm takes quadratic time.
Theorem 1. Given an oracle that solves BCP(Ri, Bj) in O((|Ri| + |Bj|)c) time, for some constant 1 ≤ c ≤ 2, MDP can be approximated with performance ratio ρ in time O(logρ (w)nc).
Proof. Let TBCP be the time complexity for BCP and similarly define TSLF(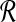 ,
, 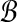 ) for the runtime for solving MDP via SLF. Partitioning
) for the runtime for solving MDP via SLF. Partitioning 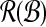 is linear. Therefore,
is linear. Therefore,
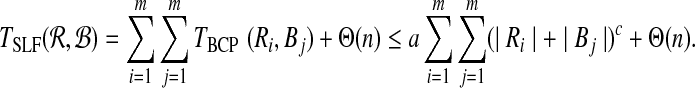 |
for some constant a. Let 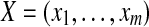 and
and 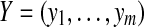 and define
and define
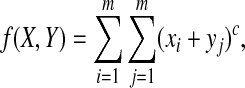 |
subject to xi ≥ 0, yi ≥ 0 and 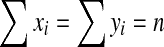 . Using the method of Lagrange multipliers, we obtain
. Using the method of Lagrange multipliers, we obtain
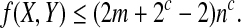 |
Since the sizes of members of 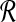 and
and 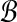 are under the same constraints as X and Y, we can substitute the bound on f(
are under the same constraints as X and Y, we can substitute the bound on f(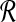 ,
, 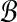 ) and conclude
) and conclude
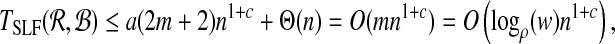 |
since logρ(w) is the number of hyperspheres required to ensure the performance ratio. ▪
The sequential layering framework transforms MDP into the well-studied BCP problem with a performance ratio of ρ and an additional factor of logρw time. BCP can be considered well-solved when the number of dimensions is low. Elegant and efficient o(n2) time algorithms have been designed specifically for 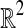 and
and 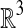 points (Preparata and Shamos, 1985). However, few options exist for solving the general BCP efficiently in high dimensions. Recall that the dimensions of our problem instances are already exponential in a natural parameter of our original problem: d = 4k. The time complexity of existing algorithms are either exponential in the number of dimensions or rapidly approach quadratic as d grows. The randomized algorithm of Khuller and Matias (1995) is an example of the former, requiring Ω(3d) time for a filtering step. As an example of the latter, Agarwal et al. (1991) proposed a randomized algorithm to solve BCP in expected
points (Preparata and Shamos, 1985). However, few options exist for solving the general BCP efficiently in high dimensions. Recall that the dimensions of our problem instances are already exponential in a natural parameter of our original problem: d = 4k. The time complexity of existing algorithms are either exponential in the number of dimensions or rapidly approach quadratic as d grows. The randomized algorithm of Khuller and Matias (1995) is an example of the former, requiring Ω(3d) time for a filtering step. As an example of the latter, Agarwal et al. (1991) proposed a randomized algorithm to solve BCP in expected 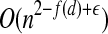 time, for any positive ε, where f (d) = 2/( ⌈d/2⌉ + 1).
time, for any positive ε, where f (d) = 2/( ⌈d/2⌉ + 1).
In the following section, we describe an algorithm based on hashing for BCP and show that it provides the subquadratic oracle in our algorithm. This is, however, just one approach to solving BCP and SLF can be regarded as a general framework to find the maximum local alignment-free score under dot-product-based similarity measures.
4. Solving BCP in High Dimensions
In this section, we present an algorithm for solving BCP in high dimensions based on random hashing. We show that under certain assumptions about the statistical properties of the inputs, the algorithm has a subquadratic time complexity. Later we investigate the behavior of the algorithm when relaxing some constraints on the input.
We conduct our analysis under the assumption of i.i.d. sequences—an assumption commonly used in sequence analysis. We assume all of the sequences are generated under the null hypothesis that each alphabet letter is identically and independently distributed and all sequences are independent. We name points extracted from such sequences i.i.d.-induced points.
In our context, even if the sequences are i.i.d., we have two sources of dependency: within a count vector the overlapping k-mers in the underlying string are not independent, and between count vectors from the same original string there is an overlap of the window size w making consecutive points highly dependent. We refer to the former as the dependency associated with parameter k and the latter as dependency associated with parameter w. We first conduct our analysis ignoring these two sources of dependence, and then explicitly address the dependency associated with w. For the dependency associated with k, we present simulation results to indicate that in practice the behavior of our algorithm is not affected by the latter form of dependency for a broad range of values for k.
A previous study showed that under 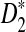 , the count of each k-mer has an asymptotic standard normal distribution for i.i.d. sequences (Reinert et al., 2009). It is well-known that if the counts of each coordinate of a vector have independent standard normal distributions, then the normalized vector is uniformly distributed on the surface of the hypersphere (Muller, 1959). Therefore, ignoring dependencies associated with k and w reduces our problem to solving BCP when the input is two independent sets of i.i.d.-induced points, each having a uniform distribution on the surface of the unit hypersphere.
, the count of each k-mer has an asymptotic standard normal distribution for i.i.d. sequences (Reinert et al., 2009). It is well-known that if the counts of each coordinate of a vector have independent standard normal distributions, then the normalized vector is uniformly distributed on the surface of the hypersphere (Muller, 1959). Therefore, ignoring dependencies associated with k and w reduces our problem to solving BCP when the input is two independent sets of i.i.d.-induced points, each having a uniform distribution on the surface of the unit hypersphere.
Our algorithm relies on the concept of locality sensitive hashing, which has been successfully applied to solve a closely related problem of finding the nearest neighbor in high dimensions in various contexts (Buhler, 2001; Dutta et al., 2006; Haveliwala et al., 2000). The basic idea is to hash points using a function that ensures nearby points are more likely to hash into the same bucket. Here we use a family of such hash functions that uses the angle between points as the measure of proximity.
Suppose u is a point sampled uniformly at random on the d-dimensional unit hypersphere centered at the origin. Assuming a source of random bits, simple algorithms are known for generating such points (Muller, 1959). For any 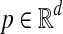 define
define
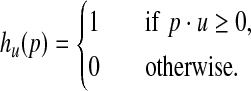 |
(6) |
The function hu has the property that for any two points p and q,
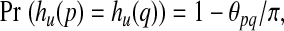 |
(7) |
where 0 ≤ θpq ≤ π. Let 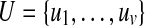 be a set of random points in
be a set of random points in  with unit norm and define the hash function
with unit norm and define the hash function
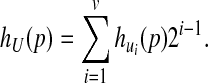 |
(8) |
In words, the closer the two points are on the unit hypersphere, the more likely they are to have a common image under the hash function hU. Hash functions with this behavior are called “locality sensitive” (Indyk and Motwani, 1998). This particular function, which can be considered locality sensitive when distances are measured as angles, originated in the elegant analysis due to Goemans and Williamson (Goemans and Williamson, 1995) and was first applied in the context of locality sensitive hashing by Charikar (2002). Subsequently, this function has been applied in different contexts including document similarity search (Ture et al., 2011) and natural language processing (Ravichandran et al., 2005).
Recall from Algorithm 1 that we need to solve BCP(Ri, Bj), where Ri and Bj are obtained by transforming subsets of 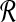 and
and 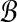 . The solution to BCP(Ri, Bj) is achieved by performing L hashing iterations as follows. In one iteration, a set U of random vectors is generated, with |U| = v. Each p in Ri ∪ Bj is hashed using the function hU(p) of Equation (8). Then for every bucket that contains points of both colors, we solve the BCP problem restricted to the (unprojected) pre-images of points that were hashed into that bucket. The maximum dot product is retained over all buckets and over each of the L iterations.
. The solution to BCP(Ri, Bj) is achieved by performing L hashing iterations as follows. In one iteration, a set U of random vectors is generated, with |U| = v. Each p in Ri ∪ Bj is hashed using the function hU(p) of Equation (8). Then for every bucket that contains points of both colors, we solve the BCP problem restricted to the (unprojected) pre-images of points that were hashed into that bucket. The maximum dot product is retained over all buckets and over each of the L iterations.
This is a randomized algorithm, and the parameters L and v determine the relationship between the time complexity of the algorithm and the probability of missing the closest bichromatic pair. In what follows, we analyze this algorithm and show how L and v can be chosen to achieve a favorable balance between running time and accuracy of the algorithm.
A random string S consists of |S|/w independent substrings of length w. Under our geometric interpretation, as k-mer count vectors these are |S|/w points without any dependency associated with w. First we consider these points and analyze our algorithm for BCP(Ri, Bj) assuming all members of Ri and Bj are independent random points.
Without loss of generality, let |Ri| = |Bj| = n and assume each point has unit norm—thanks to the SLF in the previous section we need only be concerned with angles between points. According to Equation (8), the hashing value of a point p is determined by the relative position of p to v random vectors in U. For any 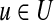 , the geometric loci of all points p such that p · u > 0 is obtained by (1) partitioning the hypersphere into two equal parts by the hyperplane orthogonal to u and (2) selecting the half that contains u. We may consider this random partitioning as a Bernoulli trial, and therefore two points p and q are hashed to the same bucket if they fall on the same side of a random hyperplane v times or equivalently when the outcome of v independent Bernoulli trials for p and q are identical.
, the geometric loci of all points p such that p · u > 0 is obtained by (1) partitioning the hypersphere into two equal parts by the hyperplane orthogonal to u and (2) selecting the half that contains u. We may consider this random partitioning as a Bernoulli trial, and therefore two points p and q are hashed to the same bucket if they fall on the same side of a random hyperplane v times or equivalently when the outcome of v independent Bernoulli trials for p and q are identical.
Observation 1. Let v = log n and hash point set X using v random vectors and the hash family h. Then occupancy of any bucket follows a binomial distribution with parameters n and p = 1/n.
This observation, which follows directly from the uniformity assumption on the distribution of points, casts our analysis as a classic occupancy problem. The following upper bound on the maximum occupancy of the buckets can then be established (similar to Theorem 3.1 in Motwani and Raghavan, 1995).
Proposition 3. There exists an absolute constant c < 2.91 such that if n independent i.i.d.-induced points are hashed to n buckets, the probability that any bucket occupancy exceeds c log n is at most 1/n2.
Consider all of L iterations of this hashing-based procedure. The probability that no bucket occupancy ever exceeds c log n is at least
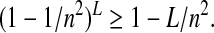 |
Assume sublinear number of iterations is sufficient to find the bichromatic minimum angle (i.e., L = o(n)), then the above formula establishes an important fact about our algorithm: The number of naive dot-product computations in any bucket does not exceed O(log2n) (with high probability), and thereby BCP algorithm mostly performs O(nL log2n) dot-product computations to obtain and retain the closest bichromatic pair.
It remains to bound the probability that the algorithm fails to identify the closest pair when L = o(n). Suppose points 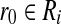 and
and 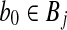 achieve the minimum angle θmin in a given instance of BCP. According to Equation (7), the probability that r0 and b0 hash to different buckets in all L iterations is
achieve the minimum angle θmin in a given instance of BCP. According to Equation (7), the probability that r0 and b0 hash to different buckets in all L iterations is
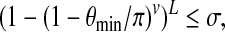 |
for some constant error probability threshold σ > 0. For v = log n this inequality establishes a trade-off between θmin and L. In particular, it is straightforward to show that L = O(nγ) where γ < 1 is sufficient to satisfy this inequality if θmin < π/2.
Proposition 4. Suppose set R of n independent points is uniformly distributed on the unit d-hypersphere. For any point b on the unit d-hypersphere and any θ satisfying
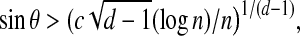 |
for some constant c, with probability at least 1 − 1/n there exists an
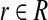 such that θrb < θ.
such that θrb < θ.
A detailed proof is given in the Supplementary Material (available online at www.liebertonline.com/cmb). This proposition asserts that even for small subproblems with a few points, there exists a bichromatic pair 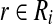 and
and 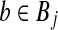 such that sin θrb < 1 and thus the minimum bichromatic angle should be bounded away from π/2. We extend these findings considering dependency between our points associated with w and conclude a subquadratic algorithm for solving BCP.
such that sin θrb < 1 and thus the minimum bichromatic angle should be bounded away from π/2. We extend these findings considering dependency between our points associated with w and conclude a subquadratic algorithm for solving BCP.
Theorem 2. For i.i.d.-induced points, BCP problem with two point sets each having n points can be solved using O(n1+γ log n) hashing and
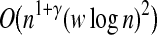 dot-product computations for some γ < 1.
dot-product computations for some γ < 1.
Proof. For each point, the algorithm requires Lv hash function evaluations overall. To find the number of dot-product computations, we notice that the maximum bucket occupancy is O(w log n). This is because of the fact that during the hashing process the number of the independent i.i.d.-induced points in any bucket is O(log n) with high probability. On the other hand, for each point there are at most w − 1 points with some shared k-mers and hence proposing some dependency associated with w. Assuming v = log n, we have n buckets in each iteration and therefore the number of dot-product computations does not exceed O(Ln(w log n)2). Substituting L = O(nγ) completes the proof.
Up to this point, we ignored the existing dependency associated with the parameter k. In the Supplementary Material, we present the empirical evaluation of the behavior of our algorithm considering the dependency associated with k. More specifically, (1) we show the maximum bucket occupancy remains O(log n) for i.i.d.-induced points if w/d is sufficiently large and (2) we argue that with high probability, the minimum bichromatic angle in any BCP subproblem is strictly less than π/2 if both sequences are generated from the same model.
5. Results
We present empirical results to demonstrate both the accuracy and efficiency of our method. We show that under a reasonable random data model (i.i.d. sequences) our algorithm is typically much more accurate than the theoretical guarantees established above. Moreover, our simulations show that under a planted motif model, our algorithm performs almost as accurately as the naive algorithm. Therefore, to the degree that 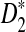 describes important sequence similarities, our algorithm is an effective means of identifying regions of local similarity.
describes important sequence similarities, our algorithm is an effective means of identifying regions of local similarity.
Simulation setup: Our simulation experiments require specifying a triple (n, w, k) of parameters. For simulations under the null hypothesis, when the sequences share no interesting local similarity, a pair S1 and S2 of length n sequences is randomly generated by sampling letters i.i.d. from the DNA alphabet. We use a planted motif model for the alternate hypothesis, which first generates sequences S1 and S2 as for the null hypothesis, but then modifies the sequences. For each of the two sequences, a random location is chosen and the corresponding window of length w is replaced by a sequence that has additional planted occurrences of a specific k-mer (called the motif). This k-mer is randomly sampled from all of the d = 4k possible k-mers for each experiment. Nonoverlapping motif occurrences are inserted randomly, and we use the parameter α to indicate the density of these occurrences; the selected length w window will contain αw/k occurrences of the motif.
When evaluating an algorithm under the alternate hypothesis, we ask whether the algorithm has identified the pair of windows in which the motif has been planted. The local alignment-free comparison involves identifying a pair of maximally similar windows within the sequences being compared. We compute the amount of overlap between the windows identified by the algorithm (WO1 and WO2, in sequences S1 and S2, respectively) and the windows generated during the simulation (WM1 and WM2 in S1 and S2, respectively). Assume x is the starting position where motifs have been inserted in S1 and y is the analogous position in S2. We define sD as follows:
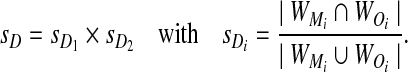 |
Here, WM1 starts at x-th position in the first sequence and ends at position x + w − 1. It is important to note that there is some chance in the simulation that sD will be less than 1 even for the naive algorithm, if the optimal windows are not exactly those that have been targeted in the simulation process.
Study of the naive algorithm: We conducted a set of experiments repeating each of them 100 times while we increment the length of the sequence from 2,000 to 20,000 base pairs. Using the naive algorithm, we compare the performance of D2 and two of its variants 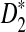 and
and 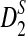 for different values of α. Figure 1A shows the results for α = 0.05. We also tested the performance of these three statistics using the same process for different values of w, k, and α (see Supplementary Materials). Based on the results, local
for different values of α. Figure 1A shows the results for α = 0.05. We also tested the performance of these three statistics using the same process for different values of w, k, and α (see Supplementary Materials). Based on the results, local 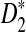 achieves the best performance (the largest sD) in discrimination of the regions containing planted motifs from the background sequence. Furthermore,
achieves the best performance (the largest sD) in discrimination of the regions containing planted motifs from the background sequence. Furthermore, 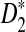 has the minimum variance among these three statistics, meaning that it is the most robust and stable one in local identification of the sequence relatedness.
has the minimum variance among these three statistics, meaning that it is the most robust and stable one in local identification of the sequence relatedness.
FIG. 1.
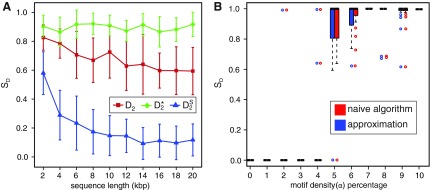
(A) A comparison of D2 and two of its important variants 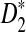 and
and 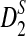 with w = 1000 and k = 5 for a range of sequence lengths. (B) The power of the approximation algorithm compared with the naïve algorithm under
with w = 1000 and k = 5 for a range of sequence lengths. (B) The power of the approximation algorithm compared with the naïve algorithm under 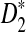 . Data was simulated as described in the text for a set of α values; (n, w, k) = (100k, 1k, 5) and for each fixed α the experiment was repeated 20 times.
. Data was simulated as described in the text for a set of α values; (n, w, k) = (100k, 1k, 5) and for each fixed α the experiment was repeated 20 times.
Accuracy of the algorithm on simulated data: We evaluated sD of the approximation algorithm while α is adjusted from 0 to 0.1 at increments of 0.01. Each experiment was repeated 20 times and the results were compared to the naive algorithm. Interestingly, in almost all of the experiments the performance of the approximation algorithm is identical with the naïve, demonstrating the accuracy of our algorithm. The results shown in Figure 1B are all based on parameter combinations of (n, w, k) = (100k, 1k, 5).
Evaluating the speed of the algorithm: We expect to observe quadratic behavior for the naive algorithm and a subquadratic running time for the approximation algorithm. Figure 2A shows results for both algorithms when no motif is planted and both sequences are i.i.d. In the broad range of input sequence lengths from 100 kbp to 500 kbp, these results match our expectations based on the analysis presented in Section 4.
FIG. 2.
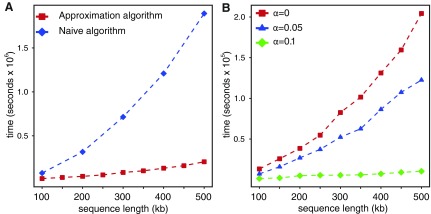
(A) The sequences are i.i.d., and their length increases from 100 kbp to 500 kbp while (w, k) = (1000, 5), demonstrating different asymptotic behavior for the naïve and approximation algorithms. (B) The relation between planted motif density and the run time of the algorithm for a range of sequence lengths. Note that for α = 0, the curve is defined to be identical to that of panel (A) where there is no planted motif.
According to the theory, if the number of shared k-mers is increased then the minimum bichromatic angle, and consequently the number of required iterations for BCP, will decrease. We compared the running time of our algorithm in three cases of α ε {0, 0.05, 0.1}. The results, presented in Figure 2B, demonstrate that these heuristics lead to substantial improvement in running time as the local similarity increases. Interestingly the relative error for all experiments carried out here remained less than three percent while the performance ratio of SLF was set to ρ = 1.05. Overall, the running time and the accuracy of the approximation algorithm to identify the regions with the maximum 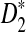 makes it applicable for solving many real biological problems efficiently. For example, our algorithm enables comparisons of large orthologous intergenic regions to identify locally similar intervals of several hundred to a few thousand bases, which are candidate enhancer regions bearing similar sets of transcription factor binding sites.
makes it applicable for solving many real biological problems efficiently. For example, our algorithm enables comparisons of large orthologous intergenic regions to identify locally similar intervals of several hundred to a few thousand bases, which are candidate enhancer regions bearing similar sets of transcription factor binding sites.
6. Discussion
Alignment-free sequence comparison is becoming increasingly important because it can accelerate similarity searching (e.g., either alone or as a filtering step prior to alignment). It can also detect biological signals that evade alignment-based methods, for example analogously functioning regulatory regions whose similarity is based on convergent evolution with individual sites whose order, orientation and multiplicity allow flexibility. Statistical measures to describe similarity without alignment have received attention, but there has been little attention to the development of algorithms for rapid alignment-free comparison. Here, focusing on a local variant of the alignment-free sequence comparison problem, we introduce an algorithmic framework that substantially accelerates the sequence comparisons while providing a means of achieving a balance between accuracy and speed. We designed this framework in the context of the D2 and 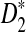 statistics, but we remark that our framework is equally applicable to other measures based on dot-product similarity (Göke et al., 2012).
statistics, but we remark that our framework is equally applicable to other measures based on dot-product similarity (Göke et al., 2012).
The essence of our framework is a transformation that maps the original string problem to a geometric problem and then decomposes dot-product similarity measures by separating the influence of vector angles and norms. This framework, which we call SLF, converts a nonmetric similarity measure to the Euclidean distance, which is accompanied by the triangle inequality. This transformation increases time complexity by a logarithmic factor. Following this transformation, the triangle inequality allows inherent locality in the data to be leveraged. Besides allowing us to draw from a large body of existing algorithmic results for searching in metric spaces, the SLF has another major advantage for local alignment-free sequence comparison: it permits heuristics in the flavor of branch-and-bound to be implemented at several stages. Such heuristics for pruning the search space are not apparent as extensions of the naive algorithm, which performs all pairs of comparisons between windows in the two sequences. Our empirical results show that these heuristics lead to substantial speed-ups even when the similarity in the sequences is weak.
In order to solve local alignment-free sequence comparison, the SLF must be coupled with a procedure to solve BCP problem instances that result from the transformation. We described a simple method based on random hashing and showed favorable performance both in theory and in practice. However, any approach for solving BCP could be used, and in some situations other BCP algorithms may be more appropriate. Under the null hypothesis, nonoverlapping windows of sequences are transformed into a set of points that may satisfy certain sparsity constraints (e.g., Preparata and Shamos, 1985), and we believe such sparsity could form the basis for alternative efficient algorithms to find the minimum bichromatic angle.
Our approach has certain limitations, and in our analyses we made specific assumptions that are not always met in practice. First, we assumed generally that our sequence background is i.i.d. with equiprobable letters. While neither of these simplifying assumptions hold for biological sequences, both have been used effectively in the past (Lippert et al., 2002; Reinert et al., 2009) and are reasonable approximations. Second, we ignored the overlap between k-mers in the analysis of our algorithm. Even for i.i.d. sequences, especially for sparse k-mer count vectors, the dependency between overlapping k-mers means the spatial distribution of hashed points will not be uniform in the domain and therefore bucket occupancy may not be balanced. Our upper bound on the maximum bucket occupancy after hashing assumed independence between different coordinates of the vectors. Previous studies (Reinert et al., 2009) have shown that the covariance matrix describing the effects of these k-mer overlap dependencies does not disappear even for the asymptotic distribution of 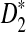 . Fortunately, our empirical results indicate that this specific issue has little impact on the efficiency of our method for values of k we tested (Supplementary Fig. S1). Finally, we note that without too much difficulty, our analysis for
. Fortunately, our empirical results indicate that this specific issue has little impact on the efficiency of our method for values of k we tested (Supplementary Fig. S1). Finally, we note that without too much difficulty, our analysis for 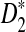 can be extended to sequences generated by a homogeneous Markov chain as the asymptotic distribution of
can be extended to sequences generated by a homogeneous Markov chain as the asymptotic distribution of 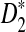 has been already generalized for this case (Reinert et al., 2009).
has been already generalized for this case (Reinert et al., 2009).
Supplementary Material
Acknowledgments
We thank Fengzhu Sun, Reza Ardekani, Phil Uren, Tim Daley, and Jenny Qu for many helpful discussions.
Author Disclosure Statement
No competing financial interests exist.
References
- Agarwal P. Edelsbrunner H. Schwartzkopf O., et al. Euclidean MST and bichromic closest pairs. Disc. Comput. Geom. 1991;6:407–422. [Google Scholar]
- Alm E. Huang K. Arkin A. The evolution of two-component systems in bacteria reveals different strategies for niche adaptation. PLoS Comput. Biol. 2006;2:e143. doi: 10.1371/journal.pcbi.0020143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul S. Gish W. Miller W., et al. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Altschul S. Madden T. Schaffer A., et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman B. Nibu Y. Pfeiffer B., et al. Exploiting transcription factor binding site clustering to identify cis-regulatory modules involved in pattern formation in the Drosophila genome. Proc. Natl. Acad. Sci. 2002;99:757–762. doi: 10.1073/pnas.231608898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buhler J. Efficient large-scale sequence comparison by locality-sensitive hashing. Bioinformatics. 2001;17:419–428. doi: 10.1093/bioinformatics/17.5.419. [DOI] [PubMed] [Google Scholar]
- Charikar M. Similarity estimation techniques from rounding algorithms. Proceedings of the thiry-fourth annual ACM symposium on theory of computing; 2002. pp. 380–388. [Google Scholar]
- Dayhoff M. Schwartz R. Orcutt B. A model of evolutionary change in proteins. Atlas of Protein Sequence and Structure. 1978;5:345–352. [Google Scholar]
- Domazet-Lošo M. Haubold B. Alignment-free detection of local similarity among viral and bacterial genomes. Bioinformatics. 2011;27:1466–1472. doi: 10.1093/bioinformatics/btr176. [DOI] [PubMed] [Google Scholar]
- Dutta D. Guha R. Jurs P., et al. Scalable partitioning and exploration of chemical spaces using geometric hashing. J. Chem. Inf. Model. 2006;46:321–333. doi: 10.1021/ci050403o. [DOI] [PubMed] [Google Scholar]
- Forêt S. Wilson S. Burden C. Empirical distribution of k-word matches in biological sequences. Pattern Recognition. 2009;42:539–548. [Google Scholar]
- Goemans M. Williamson D. Improved approximation algorithms for maximum cut and satisfiability problems using semidefinite programming. Journal of the ACM. 1995;42:1115–1145. [Google Scholar]
- Göke J. Schulz M. Lasserre J., et al. Estimation of pairwise sequence similarity of mammalian enhancers with word neighbourhood counts. Bioinformatics. 2012;28:656–663. doi: 10.1093/bioinformatics/bts028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haubold B. Reed F. Pfaffelhuber P. Alignment-free estimation of nucleotide diversity. Bioinformatics. 2011;27:449–455. doi: 10.1093/bioinformatics/btq689. [DOI] [PubMed] [Google Scholar]
- Haveliwala T. Gionis A. Indyk P. Scalable techniques for clustering the web. Proceeding of the 3rd intl. workshop on web and databases; 2000. pp. 129–134. [Google Scholar]
- Indyk P. High-dimensional computational geometry. [Ph.D. thesis] Department of Computer Science, Stanford University; Stanford, CA: 2001. [Google Scholar]
- Indyk P. Motwani R. Approximate nearest neighbors: Towards removing the curse of dimensionality. Proceedings of the 30th Annual ACM Symposium on Theory of Computing; 1998. pp. 604–613. [Google Scholar]
- Johnson M. Zaretskaya I. Raytselis Y., et al. NCBI BLAST: a better web interface. Nucleic Acids Res. 2008;36:W5–W9. doi: 10.1093/nar/gkn201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kantorovitz M. Robinson G. Sinha S. A statistical method for alignment-free comparison of regulatory sequences. Bioinformatics. 2007;23:1249–1255. doi: 10.1093/bioinformatics/btm211. [DOI] [PubMed] [Google Scholar]
- Karlin S. Altschul S. Methods for assessing the statistical significance of molecular sequence features by using general scoring schemes. Proc. Natl. Acad. Sci. 1990;87:2264–2268. doi: 10.1073/pnas.87.6.2264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazemian M. Zhu Q. Halfon M., et al. Improved accuracy of supervised CRM discovery with interpolated markov models and cross-species comparison. Nucleic Acids Res. 2011;39:9463–9472. doi: 10.1093/nar/gkr621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khuller S. Matias Y. A simple randomized sieve algorithm for the closest-pair problem. Information and Computation. 1995;118:34–37. [Google Scholar]
- Lippert R. Huang H.Y. Waterman M. Distributional regimes for the number of k-word matches between two random sequences. Proc. Natl. Acad. Sci. 2002;100:13980–13989. doi: 10.1073/pnas.202468099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liua X. Wan L. Li J., et al. New powerful statistics for alignment-free sequence comparison under a pattern transfer model. J. Theor. Biol. 2011;284:106–116. doi: 10.1016/j.jtbi.2011.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahmood K. Webb G. Song J., et al. Efficient large-scale protein sequence comparison and gene matching to identify orthologs and co-orthologs. Nucleic Acid Res. 2012;40:e44. doi: 10.1093/nar/gkr1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDaniel L. Young E. Delaney J., et al. High frequency of horizontal gene transfer in the oceans. Science. 2010;330:50. doi: 10.1126/science.1192243. [DOI] [PubMed] [Google Scholar]
- Meader S. Ponting C. Lunter G. Massive turnover of functional sequence in human and other mammalian genomes. Genome Res. 2010;20:1335–1343. doi: 10.1101/gr.108795.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motwani R. Raghavan P. Randomized algorithms. Cambridge University Press; Cambridge, United Kingdom: 1995. [Google Scholar]
- Muller M. A note on a method for generating points uniformly on n-dimensional spheres. Communications of the Association for Computing Machinery. 1959;2:19–20. [Google Scholar]
- Pearson W. Lipman D. Improved tools for biological sequence comparison. Proc. Natl. Acad. Sci. 1988;85:2444–2448. doi: 10.1073/pnas.85.8.2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preparata F. Shamos M. Computational geometry: an introduction. Springer-Verlag; New York: 1985. [Google Scholar]
- Ravichandran D. Pantel P. Hovy E. Randomized algorithms and NLP: using locality sensitive hash function for high speed noun clustering. Proceedings of the 43rd Annual Meeting on Association for Computational Linguistics; 2005. pp. 622–629. [Google Scholar]
- Reinert G. Chew D. Sun F., et al. Alignment-free sequence comparison (I): statistics and power. J. Comp. Biol. 2009;16:1615–1634. doi: 10.1089/cmb.2009.0198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims G. Kim S. Whole-genome phylogeny of Escherichia coli/Shigella group by feature frequency profiles (FFPs) Proc. Natl. Acad. Sci. 2011;108:8329–8334. doi: 10.1073/pnas.1105168108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha S. Siggia E. Sequence turnover and tandem repeats in cis-regulatory modules in Drosophila. Mol. Biol. Evol. 2005;22:874–885. doi: 10.1093/molbev/msi090. [DOI] [PubMed] [Google Scholar]
- Smith T. Waterman M. Identification of common molecular subsequences. J. Mol. Biol. 1981;147:195–197. doi: 10.1016/0022-2836(81)90087-5. [DOI] [PubMed] [Google Scholar]
- Song K. Ren J. Zhai Z., et al. Alignment-free sequence comparison based on next generation sequencing reads: extended abstract. RECOMB. 2012:272–285. doi: 10.1089/cmb.2012.0228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taher L. McGaughey D. Maragh S., et al. Genome-wide identification of conserved regulatory function in diverged sequences. Genome Res. 2011;21:1139–1149. doi: 10.1101/gr.119016.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan P. Steinbach M. Kumar V. Introduction to data mining (Chapter 8) Addison-Wesley; Boston, MA: 2006. [Google Scholar]
- Torney D. Burks C. Davison D., et al. Computation of D2: a measure of sequence dissimilarity. Computers and DNA. 1990:109–125. [Google Scholar]
- Ture F. Elsayed T. Lin J. No free lunch: brute force vs locality-sensitive hashing for cross-lingual pairwise similarity. Proceedings of ACM Special Interest Group on Information Retrieval; 2011. pp. 943–952. [Google Scholar]
- Venkataram S. Fay J. Is transcription factor binding site turnover a sufficient explanation for cis-regulatory sequence divergence? Genome Biol Evol. 2010;2:851–858. doi: 10.1093/gbe/evq066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan L. Reinert G. Sun F., et al. Alignment-free sequence comparison (II): theoretical power of comparison statistics. J. Comput. Biol. 2010;17:1467–1490. doi: 10.1089/cmb.2010.0056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterman M. Vingron M. Rapid and accurate estimates of statistical significance for sequence data base searches. Proc. Natl. Acad. Sci. 1994;91:4625–4628. doi: 10.1073/pnas.91.11.4625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao A. On constructing minimum spanning trees in k-dimensional spaces and related problems. SIAM J. Comput. 1982;11:721–736. [Google Scholar]
- Zhang Z. Schaffer A. Miller W., et al. Protein sequence similarity searches using patterns as seeds. Nucleic Acids Res. 1998;26:3986–3990. doi: 10.1093/nar/26.17.3986. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.