

Abstract
A fluorescent peptide substrate was used to measure dephosphorylation by protein tyrosine phosphatases (PTP) in cell lysates, and single cells and to investigate the effect of environmental toxins on PTP activity in these systems. Dephosphorylation of the substrate by PTPN1 and PTPN2 obeyed Michaelis-Menten kinetics, with KM values of 770 ± 250 nM and 290 ± 54 nM, respectively. Dose-response curves and IC50 values were determined for the inhibition of these two enzymes by the environmental toxins Zn2+ and 1,2-naphthoquinone, as well as pervanadate. In A431 cell lysates, the reporter was a poor substrate for peptidases (degradation rate of 100 ± 8.2 fmol min−1 mg−1) but an excellent substrate for phosphatases (dephosphorylation rate of 1.4 ± 0.3 nmol min−1 mg−1). Zn2+, 1,2-naphthoquinone and pervanadate inhibited dephosphorylation of the reporter in cell lysates with IC50 values of 470 nM, 35 μM, and 100 nM, respectively. Dephosphorylation of the reporter following loading into living single cells occurred at rates of at least 2 pmol min−1 mg−1. When single cells were exposed to 1,2-naphthoquinone (50 μM), Zn2+ (100 μM), and pervandate (1 mM), dephosphorylation was inhibited with median values and first and third quartile values of 41 (Q1 = 0%, Q3 = 96%), 50 (Q1 = 46%, Q3 = 74%), and 53% (Q1 = 36%, Q3 = 77%), respectively, demonstrating both the impact of these toxic exposures on cell signaling and the heterogeneity of response between cells. This approach will provide a valuable tool for the study of PTP dynamics, particularly in small, heterogeneous populations such as human biopsy specimens.
INTRODUCTION
The human genome encodes 107 protein tyrosine phosphatases (PTPs),1 enzymes containing a catalytic cysteine residue capable of removing phosphoryl moieties from tyrosine residues on proteins. This enzyme family plays a critical role in maintaining proper cell signaling through its opposition of protein tyrosine kinases and dysregulation of these key enzymes is associated with many diseases, including type I diabetes mellitus, cancer, and rheumatoid arthritis.2 PTP inhibition is also linked to the toxicity associated with inhalation of diesel exhaust particles (DEP), an important component of air pollution.3 Combustion of diesel fuel by vehicles and industrial equipment produces fine particulate matter (diameter < 2.5 μm) containing a mixture that includes elemental carbon, polyaromatic hydrocarbons, and metals.4 The deposition of these particles in small airways and alveoli is associated with numerous human health hazards including heart attacks, arrhythmias, chronic obstructive pulmonary disease (COPD), and asthma.5 In vitro studies have linked DEP components to reactive oxygen species production6 as well as direct inhibition of PTPs.7
Although studies on purified PTPs, cell lysates, and fixed tissue samples have provided valuable insight into the role of diesel exhaust particles in human disease, analysis of ex vivo samples acquired from human subjects remains challenging. Bronchial brushing (BB) is a well-established method of obtaining airway cells for analysis by dislodging them with a brush in conjunction with fiberoptic bronchoscopy.8 Although BB samples are a valuable source of primary airway cells for research, the sample size is typically very limited (0.8–3.6 × 104 cells).9 The small sample size is further complicated by the specimen heterogeneity which in addition to the intact airway epithelia contains mucus, stromal tissue, and the remnants of cells killed during dislodging. To date, direct analysis of these specimens has focused on genetic techniques including fluorescence in situ hybridization (FISH),10 RNA microarrays,11 and polymerase chain reaction (PCR).12 Although these approaches provide valuable information, new analytical tools for these BB specimens are required to provide additional information about the biochemistry of human airway cells.
The direct measurement of cell processes by chemical cytometry has the potential to provide unique advantages in the study of BB specimens. Although a wide range of chemical cytometry approaches exist,13 the measurement of PTP activity in airway specimens must be compatible with very small numbers of heterogeneous primary cells, and single-cell CE-LIF is a promising approach to address this challenge. Sample heterogeneity is readily addressed by the selection and analysis of individual cells, while the high separation efficiency and detection limits approaching 10−21 mol14 have made it possible to study many aspects of single cells, including protein and lipid kinase activity, proteolysis, nitric oxide release, and response to oxidative stress.14–15 Because PTPs are part of a complicated signaling network, the ability to multiplex measurement of several enzymes is very attractive, and CE has been used for monochromatic detection of up to 20 analytes simultaneously16 and multicolor systems have also been described.17
Herein we demonstrate the utility of single-cell capillary electrophoresis for the measurement of PTP activity in single intact cells, a technology well-suited for analysis of small heterogeneous samples such as that from a BB. We developed and validated a fluorescent peptide reporter of PTP activity and measured the kinetics of dephosphorylation by two common model phosphatases. Additionally, we observed inhibition of these phosphatases by three toxins known to be present in diesel exhaust and then generated dose-response curves for the environmental toxins. A separation method for the peptide and all possible fluorescent cleavage products was developed so that peptide cleavage in cell lysates and intact cells could be quantified. The lifetime of the peptide, dephosphorylation rate and influence of environmental toxins was assessed in A431 cell lysates. Finally, the peptide lifetime and the dephosphorylation rate in single A431 cells were quantified in the presence and absence of environmental toxins.
EXPERIMENTAL SECTION
Peptide Synthesis
The full-length substrate peptide, termed “pTS13” when phosphorylated and “TS13” when nonphosphorylated, was synthesized using standard solid-phase peptide synthesis techniques on TGR resin, while peptide acid fragments were synthesized on 2-chlorotritylchloride resin, both using an automated peptide synthesizer (PS3, Protein Technologies, Tuscon, AZ).
Capillary Electrophoresis
Separation of the peptide reporter based on phosphorylation status was performed using 140 mM borate, 150 mM SDS, 50 mM NaCl in a bare fused-silica capillary [30 μm inner diameter, 350 μm outer diameter, (Polymicro Technologies, Phoenix, AZ)] . Separations were performed on either a ProteomeLab PA800 (Beckman Coulter, Fullerton, CA) with a 30 cm long capillary, or on a customized system with a 38 cm long capillary.
Recombinant Phosphatase Activity Assay
Dephosphorylation of peptide by recombinant human PTPN1 and PTPN2 was observed with and without the PTP inhibitors pervanadate, 1,2-naphthoquinone, and zinc. The PTPN1 used was a human, N-terminal GST-tagged construct (residues 1-321) while the PTPN2 was a human construct (residues 1-341), both expressed in E. coli. For some reactions, PTP was incubated with the environmental toxins pervanadate, 1,2-naphthoquinone, or zinc pyrithione for 10 min at 21 °C prior to addition of pTS13.
Cell Culture
A431 cells, a human epidermoid carcinoma cell line expressing high levels of EGFR,18 were grown in a humidified atmosphere at 37 °C and 5% CO2. Cells were cultured in 75 cm2 tissue culture flasks in DMEM (10 (v/v) % FBS, 1 (v/v) % P/S) and were passaged at 60–80% confluency using 0.25% trypsin.
Cell lysate experiments
A431 cells from two 75 cm2 culture flasks were trypsinized for 5 min, diluted in DMEM, centrifuged 2 min at 800 × g, and then washed twice with PBS and pelleted. After removal of the supernatant, the pellet was resuspended in lysis buffer (20 mM HEPES, 1% Triton X-100, 10% Glycerol, 200mM NaCl, 5 mM β-glycerophosphate) with 1 (v/v) % Sigma Protease Inhibitor Cocktail. Lysate was mixed end-over-end for 20 min, followed by centrifugation at 15000× g for 40 min. The supernatant was transferred to a fresh 1.5 mL tube and protein concentration was measured as previously reported19. Lysates were then diluted to 10 mg/mL total protein and stored at −20 °C for up to 1 month.
For phosphatase assays, cell lysates were diluted to 111 ng/μL total protein in 90 μL of reaction buffer (described above) with or without environmental toxins. After 10 min of pre-incubation, pTS13 in water was added to a final concentration of 280 nM and the reaction was allowed to proceed for 90 s. Reactions were stopped by addition of an equal volume of 200 μM HCl. Samples were analyzed by CE to establish percent dephosphorylation and to determine if fluorescent peaks in addition to the intact phosphorylated and nonphosphorylated peptide appeared on the electropherograms. Dephosphorylation rates were reported as nmol peptide per minute per mg total cell protein (nmol min−1 mg−1). For inhibition experiments, dose response curves were generated over a range of inhibitor concentrations and IC50 values were interpolated by regression of the linear portion of the semi-log plot of percent inhibition versus log10 of inhibitor concentration.
Single Cell Phosphatase Measurement
A431 cells were grown in DMEM on cell chambers comprising a round 25 mm diameter No. 1 glass coverslip bonded to a silicone O-ring (18 mm internal diameter) with PDMS. Pretreatment of cells, if any, was performed by removing media from a cell chamber, rinsing twice with 37 °C serum-free DMEM and then adding the appropriate inhibitor dissolved in serum-free DMEM and incubating at 37 °C for 10–20 min. After pretreatment, a cell chamber was placed on the microscope stage of the customized single-cell CE system,20 a cell was visualized and microinjected with a mixture of peptide reporter and internal standard using a Transjector 5246 microinjection system (Eppendorf AG, Hamburg, Germany). The cell chamber temperature was maintained at 37 °C using a constant flow of warmed extracellular buffer (ECB; 10 mM HEPES, 135 mM NaCl, 5 mM KCl, 1 mM CaCl2, 1 mM MgCl2, pH 7.4) during microinjection, reporter incubation, and cell lysis. At 60 s post-microinjection, the cell was lysed with a 532 nm ND:YAG pulsed laser21 and electrokinetically injected into the capillary by applying a negative potential to the capillary outlet reservoir (5 s at 79 V/cm). The capillary inlet was then repositioned into a reservoir of electrophoretic buffer (140 mM borate, 150 mM SDS, 50 mM NaCl, pH 7.0), electrophoresis was performed (263 V/cm), and data was analyzed using Matlab and Origin. Data from untreated and inhibitor treated cells was compared using bootstrapping22 (details in Supplemental Information) and p-values were reported.
RESULTS AND DISCUSSION
Peptide Selection and Separation
In order to measure PTP activity in single cells, a phosphotyrosine-containing peptide reporter sequence (Glu-Glu-Leu-Glu-Asp-Asp-pTyr-Glu-Asp-Asp-Nle-Glu-Glu-amide, where Nle is norleucine) was chosen. The sequence was adapted from a peptide substrate of epidermal growth factor receptor (EGFR),23 a receptor tyrosine kinase opposed by phosphatases including PTPN1, PTPN2, SHP-1, and Cdc25A.24–25 6-FAM was conjugated to the N-terminus to permit fluorescence detection. Since incubation of peptides in cells or cell lysates can result in their metabolism by peptidases with cleavage at any peptide bond, the ability to electrophoretically separate pTS13, TS13 and their fluorescent fragments which are similar in charge/mass was critical for use of pTS13 as a reporter in cell lysates and single cells. Moreover it was critical that the sample matrix used for the separation be that of a physiologic buffer since the ultimate goal of this work was measurement of single-cell, reporter dephosphorylation during toxin exposure.
Initial methods development focused on the separation of pTS13 and TS13. A survey of separation buffers previously reported for peptides and highly negative analytes comprised borate with and without sodium dodecyl sulfate (SDS) or cetyltrimethlammonium bromide (CTAB), sodium citrate, sodium phosphate with SDS, tris(hydroxymethyl)aminomethane (Tris), and 1,1-bis(hydroxymethyl)ethyl)glycine (Tricine).26 The best resolution observed during this screening was 1.3 ± 0.1 for 140 mM borate, 70 mM SDS, pH 7.5. This concentration of SDS is well above its critical micelle concentration (7.9–8.3 mM)27 and thus the mode of separation was expected to be micellar electrokinetic capillary chromatography, modified by borate complexation. For both lysate and single-cell analyses, the sample matrix was not the electrophoretic buffer but a high-salt biocompatible buffer. When pTS13 and TS13 were loaded into the capillary using a high-salt, neutral-pH buffer (ECB) as the sample matrix, the peaks were no longer resolved (Figure 1a). The loss in separation capacity was most likely due to destacking of the analytes in the high ionic strength sample matrix. To minimize destacking when using high salt samples, 50 mM sodium chloride was added to the electrophoretic buffer to more closely match the ionic strength of the sample matrix i.e. physiologic buffer, and SDS concentration was increased to 150 mM to increase micellar sweeping effects. Although the added salt minimized destacking, it resulted in a greatly increased current flow through the capillary necessitating electrophoretic separations at very low field strengths (≤ 160 V/cm) and consequent long separation times (1680 ± 54 s for TS13, 1760 ± 63 s for pTS13). For this reason, all subsequent separations were performed in a 30 μM ID capillary to maintain acceptable field strengths (260 V/cm) at low currents and shorter separation times (1170 ± 22 s for TS13, 1230 ± 26 s for pTS13). These conditions yielded separation of pTS13 and TS13 with a resolution of 3.4 ± 0.1, and efficiencies of 7.3 ± 0.6 × 104 (TS13) and 6.7 ± 0.5 × 104 (pTS13) theoretical plates (Figure 1b). Importantly, the ratio of the peak areas of pTS13 and TS13 to that of the total peptide was highly reproducible with an RSD of 0.032 and 0.023, respectively. Although these efficiencies are lower than for many peptide separations by CE, the highly negative analytes and high-salt sample matrix make this a challenging separation. Most important, the resolution was sufficient for the intended biological application. The limit of detection for pTS13, defined as the smallest amount of analyte detectable with a signal-to-noise ratio greater than 3, was determined to be 4.4 × 10−20 mol.
Figure 1.
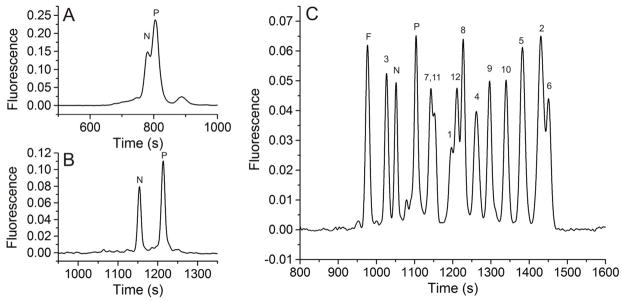
Separation of pTS13 (P) and TS13 (N) in 140 mM borate, 70 mM SDS, pH 7.4 in a 50–μm diameter capillary (A) or with 140 mM borate, 150 mM SDS, 50 mM NaCl, pH 7.4 in a 30-μm diameter capillary (B). Reporter species were also separated from all fluorescent fragments (C), indicated by length in residues (e.g. fragment 1 is 6-FAM-Glu-COOH), as well as from the 6-FAM internal standard (F).
The phospho- and nonphospho-peptides migrated 60 s apart; however, this spacing was not sufficient to consistently identify single peaks given the variability in the migration time especially in the presence of potentially biofouling constituents such as protein or cell debris. For this reason, 6-FAM was used as an internal standard for all biologic samples. This dye is detectable by LIF and migrates near TS13 and pTS13. The 12 possible fluorescent fragments generated by removal of C-terminal residues from TS13 were synthesized. Separation of all analytes simultaneously (Figure 1c) demonstrated that no fragments co-migrated with 6-FAM, pTS13, or TS13, and thus any degradation of the reporter within the biologic sample will produce additional CE peaks.
Kinetics of in vitro Dephosphorylation
The human enzymes PTPN1 (PTP-1B) and PTPN2 (TC-PTP) favor substrates with acidic side-chains proximal to the phosphotyrosine residue, and are implicated in airway inflammation.28–30 To determine if phosphatase activity could be measured using pTS13, the reporter at varying concentrations was incubated with recombinant PTPN1 and PTPN2. Product formation was quantified by CE and the percent dephosphorylated peptide was calculated as the ratio of the integrated area of the TS13 peak to the combined area of the pTS13 and TS13 peaks. The Michaelis constant (KM), maximal reaction velocity (Vmax), and turnover number (kcat) were determined for pTS13 dephosphorylation by PTPN1 and PTPN2 (Table 1) by fitting the initial rates of product formation to the Michaelis-Menten equation. PTPN1 and PTPN2 showed first order kinetics with respect to pTS13 dephosphorylation. Previous studies of peptide substrates for PTPN1 have reported KM values of 2.6–23 μM, with kcat values of 55.7–78.9 s−1,31–32 comparable to the values observed for pTS13. The kinetics of PTPN2 are less well documented, but a KM for phosphotyrosine was measured to be 300 μM,33 which is approximately 1000-fold higher than that seen for pTS13. This is consistent with previous studies comparing PTP dephosphorylation for peptide substrates and phosphotyrosine alone.32 In summary, Michaelis-Menten kinetics measured with recombinant phosphatases and pTS13 are consistent with the existing literature, establishing the utility of this method for studying PTP activity in vitro.
Table 1.
Kinetic constants KM and Vmax for pTS13 dephosphorylation by recombinant PTPN1 and PTPN2 were determined by fitting the Michaelis-Menten equation to initial reaction rates obtained over a range of substrate concentrations. Kcat was calculated as Vmax/[E] where [E] is enzyme concentration.
| Enzyme | KM (nM) | Vmax (nM s−1 ng−1) | kcat (s−1) |
|---|---|---|---|
| PTPN1 | 770 ± 250 | 2.2 ± 0.25 | 21 ± 2.4 |
| PTPN2 | 290 ± 54 | 3.1 ± 0.18 | 30 ± 1.7 |
Inhibition of Recombinant Enzymes by Toxins From Diesel Exhaust
Inhibitors of PTP activity were chosen to represent three major mechanisms of environmentally relevant PTP inhibition: active site cysteine oxidation, covalent modification, and non-oxidative inhibition by transition metals.34 Vanadium is a heavy metal found in fossil fuels and is released during combustion.35 Several oxidation states of vanadium are established PTP inhibitors, including pervanadate, which acts through direct oxidation of the catalytic cysteine.36 The polycyclic aromatic hydrocarbon 1,2-naphthoquinone is a naphthalene derivative generated by diesel combustion and petroleum processing which covalently and irreversibly arylates cysteine and histidine residues in the PTP active site.7 Finally, zinc is a major transition metal component of diesel exhaust particles that is theorized to inhibit PTP action by a non-redox mechanism involving direct interaction with vital active site residues.37
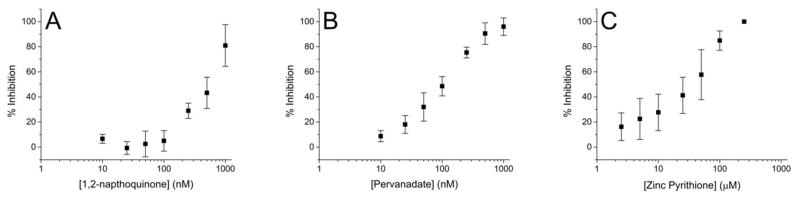
To establish whether PTP inhibition could be measured using our phosphatase activity reporter, pTS13 was incubated with recombinant PTPN1 and PTPN2 in the presence of the aforementioned inhibitors across a range of concentrations. The enzyme was pretreated with sodium pervanadate, 1,2-naphthoquinone, or Zn2+, then pTS13 dephosphorylation was assessed by CE. Dose-response curves were generated (Figure 2) to establish this system as a useful method for measurement of PTP inhibition in vitro. For PTPN1 inhibition, IC50 values for 1,2-naphthoquinone, pervanadate and Zn2+ were determined as 520 nM, 59 nM, and 28 μM respectively. IC50 values for these compounds with PTPN2 were found to be 53 nM, 39 nM, and 87 μM. These data are similar to previous reports of PTPN1 inhibition with 1,2-naphthoquinone (IC50: 1.6–5 μM),7, 38 pervanadate (IC50: 400 nM),36 and PTPN2 inhibition with Zn2+ (85% inhibition at 100 μM).39 The difference in inhibition between PTPN1 and PTPN2 for 1,2-napthoquinone is likely due to the complex mechanism of inhibition associated with this agent.
Figure 2.

(A–C) Recombinant PTPN1 and (D–F) PTPN2 were pretreated with a range of inhibitor concentrations for 10 min and then incubated with pTS13 for 10 min. Percent inhibition was calculated relative to the dephosphorylation observed in the absence of inhibitor.
Lifetime of pTS13/TS13 in Cell Lysates
To assess reporter susceptibility to intracellular degradation, TS13 was incubated with A431 cell lysates and samples were analyzed by CE. One additional peak appeared over 3 hours. This breakdown product was identified as the 8 residue fragment (6FAM-EELEDDYE-COOH) based on co-migration with the synthetic fragment. The average rate of formation, and thus reporter breakdown, was 100 ± 8.2 fmol min−1 mg−1. This rate is approximately 104 times slower than the dephosphorylation rate measured in A431 lysates (discussed below), suggesting that TS13 is sufficiently robust to degradation for its intended applications. Several peptides have been shown to undergo rapid degradation in cell lysates at rates ranging from 1.4–13 nmol min−1 mg−1,40–41 approximately 104–105 fold faster than observed for TS13. The peptidase resistance of TS13 is consistent with the finding that both glutamic and aspartic acid, the two most prevalent residues in this reporter, are moieties not preferred by peptidases.41
PTP activity and Inhibition in A431 Cell Lysates
Unlike an isolated recombinant enzyme, cellular PTP activity is opposed by tyrosine kinases. To determine if PTP activity could be measured in the context of cellular contents, pTS13 was incubated with whole cell lysates generated from A431 epidermoid carcinoma cells with and without the previously described environmental toxins (Figure 3). In the absence of inhibitors, pTS13 dephosphorylation occurred at a rate of 1.4 ± 0.3 nmol min−1 mg−1. Toxin inhibition of total PTP activity in A431 lysates was similar to that seen in recombinant PTPs, with IC50 values of 470 nM, 100 nM, and 35 μM for 1,2-naphthoquinone, pervanadate, and Zn2+ respectively. These values are comparable to those seen for recombinant PTPs, and agree with previously reported C6 cell lysate inhibition by Zn2+ (IC50: 31 μM).37
Figure 3.
(A–C) A431 cell lysates (10 μg total protein) were pretreated with a range of inhibitor concentrations for 10 min and then incubated with pTS13 for 90 s. Percent inhibition was calculated relative to the dephosphorylation observed in the absence of inhibitor.
Lifetime of pTS13/TS13 in Single Cells
The degradation resistance of the PTP reporter in living cells was tested by microinjection of single A431 cells with TS13 and single-cell analysis by CE. A single degradation product was observed and corresponded to the 8 residue fragment based on migration relative to TS13. This suggests that degradation patterns for TS13 in intact A431 cells and lysates are similar. The degradation rate was linearly correlated (R2 = 0.8) with amount of TS13 injected, suggesting first-order kinetics. The amount of peptide delivered ranged from 0.2–35 amol and the median degradation rate was 39 fmol min−1 mg−1 with first and third quartiles (Q1 and Q3) of 22 and 440 fmol min−1 mg−1, similar to the rate observed in A431 cell lysates. The median half-life for TS13 was 35 min (Q1 = 23 min, Q3 = 39 min), approximately 250-fold longer than a previous report for a native peptide in living cells.42
Single A431 Cell PTP Activity
Cell lysates lack the organization and compartmentalization of the intact cell and represent the pooled average of all processes occurring within a population, obscuring any intercellular heterogeneity. To demonstrate the utility of pTS13 as a reporter of enzyme activity in single cells, individual A431 cells were analyzed with and without pretreatment with environmental toxins (Figure 4). In untreated cells, a median of 0% (Q1 = 0%, Q3 = 11%) remaining phosphorylation was observed after 60 s. For the majority of these cells, 100% dephosphorylation had occurred within this time frame, so rates could not be calculated. A lower limit to the rate was estimated by assuming a total protein concentration of 1 g/mL and a 500 fL cell volume43 for the average mammalian cell. The median rate was 2.1 pmol min−1 mg−1 (Q1 and Q3 of 1.1 and 8.0 pmol min−1 mg−1), significantly lower than seen in lysates, however this difference is overestimated by the inability to calculate exact rates for single cells, i.e. if 100% dephosphorylation is achieved in less than 60 s, actual rates will be higher than those calculated. Compartmentalization of PTPs within the intact architecture of living cells could also contribute to lower dephosphorylation rates in single cells. Plots of percent dephosphorylation vs. amount of peptide delivered showed that across the mass range observed (0.5–33 amol), PTP activity was not saturated. Treatment of A431 cells with sodium pervanadate (1 mM) prior to microinjection resulted in a median of 53% (Q1 = 36%, Q3 = 77%) phosphorylation remaining in 60 s. Results of 1,2-naphthoquinone (50 μM) and zinc pyrithione (100 μM) pretreatment were similar, yielding 41% (Q1 = 0%, Q3 = 96%) and 50% (Q1 = 46%, Q3 = 74%) median phosphorylation remaining after 60 s, respectively. Comparison of dephosphorylation for each treatment group versus untreated controls found statistically significant inhibition with p-values of 0.035, 0.0072, and 0.002 for 1,2-napthoquinone, zinc pyrithione, and pervanadate. The measurement of the relative peak area of pTS13 to that of total peptide peak area in the presence of a lysed single cell was highly reproducible (RSD = 0.018). Prior work has shown that the termination of intracellular reactions by laser lysis44 and the introduction of cell contents into the capillary by electrokinetic injection21 are highly reproducible. Thus the most likely source of the measured variability in the PTP activity is the cellular heterogeneity in signaling.
Figure 4.
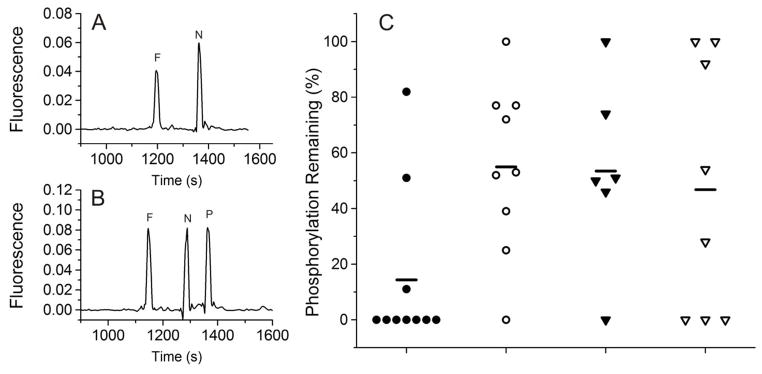
Single A431 cells were incubated in serum-free medium with and without inhibitors, microinjected with pTS13, and analyzed by CE. Representative electropherograms of (A) full dephosphorylation in an untreated cell and (B) partial dephosphorylation in a pervanadate treated cell. (C) Summary of PTP inhibition seen after treatment with (●) no inhibitor, (○) 1 mM sodium pervanadate, (▼)100 μM zinc pyrithione, or (▽) 50 μM 1,2-naphthoquinone. The bars represent the median for each measurement.
The concentrations of 1,2-naphthoquinone and pervanadate necessary to ellicit 50% inhibition of PTP activity in single cells are 3 orders of magnitude higher than for lysates, while there is little difference seen for zinc pyrithione. Although the ionophore pyrithione aids entry of Zn2+ into cells, pervanadate has no such ionophore or any established membrane transporter, potentially limiting its access to the cytoplasm. 1,2-naphthoquinone is highly lipophilic and may partition extensively in the cell membrane. Additionally, unlike for Zn2+, inhibition of PTPs by pervanadate requires oxidation, and the antioxidative machinery of intact cells has not been diluted or disorganized as is the case in a lysate. Exposure of intact cells to these toxins clearly inhibits PTP activity, and the variability between cells observed in these studies is consistent with previous reports of phenotypic heterogeneity in clonal populations,45 underlining the value of single-cell measurements as compared to bulk analysis of many cells.
CONCLUSION
A novel chemical cytometry approach to measure PTP activity was demonstrated for analysis of recombinant enzymes in vitro, cell lysates, and single cells. In each system, total PTP activity was assessed by measuring dephosphorylation of a fluorescent substrate peptide. Inhibition of PTP activity by environmental toxins was observed for recombinant enzymes and lysates at levels similar to those previously reported. In single A431 cells, pretreatment with all toxins reduced PTP activity relative to untreated cells, with substantial variation between cells. This heterogeneity seen in cultured cells illustrates the value of analyzing individual cells rather than population averages.
This approach provides several advantages over existing techniques. The simplicity of sample preparation and assay protocols makes this technique an attractive option for analysis of recombinant enzymes and cell lysates, and the high sensitivity and flexibility is well suited to analysis of intact single cells. This system does not require genetic manipulation of cells, making it useful for analysis of primary cells. The ability to pre-select individual cells using microscopy permits analysis of desired subpopulations of viable cells in low viability or highly complex samples as well as very small samples. These characteristics in particular suggest this technique will be valuable in the analysis of specimens such as airway brush biopsies and tumor samples. Additionally, enzyme activity is measured directly without the need for antibodies which, combined with the analytical separation and tunability of a peptide substrate reporter, allows for simultaneous multiplexed measurements of several signaling events. The ability to analyze the effects of extracellular insults on signaling in small, heterogeneous samples of primary cells promises to be a valuable new research tool in environmental health research in particular as well as the larger field of cell signaling.
Supplementary Material
Acknowledgments
The authors wish to thank James Samet, PhD for material support and discussion. RP wishes to thank Angela Proctor, PhD, Pavak K. Shah, and Michelle L. Kovarik, PhD for valuable discussion and technical assistance. This work was funded by the NIH (R01CA139599, T32GM008719 and 5F30ES019829).
Footnotes
SUPPORTING INFORMATION AVAILABLE
This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Vang T, Miletic AV, Arimura Y, Tautz L, Rickert RC, Mustelin T. Annu Rev Immunol. 2008;26:29–55. doi: 10.1146/annurev.immunol.26.021607.090418. [DOI] [PubMed] [Google Scholar]
- 2.Mustelin T. Adv Exp Med Biol. 2006;584:53–72. doi: 10.1007/0-387-34132-3_5. [DOI] [PubMed] [Google Scholar]
- 3.Tal TL, Bromberg PA, Kim Y, Samet JM. Toxicol Appl Pharmacol. 2008;233:382–388. doi: 10.1016/j.taap.2008.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Miller MR, Shaw CA, Langrish JP. Future Cardiol. 2012;8:577–602. doi: 10.2217/fca.12.43. [DOI] [PubMed] [Google Scholar]
- 5.U.S. EPA. Integrated Science Assessment for Particulate Matter (Final Report) 2009. [Google Scholar]
- 6.Gurgueira SA, Lawrence J, Coull B, Murthy GG, Gonzalez-Flecha B. Environ Health Perspect. 2002;110:749–755. doi: 10.1289/ehp.02110749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Iwamoto N, Sumi D, Ishii T, Uchida K, Cho AK, Froines JR, Kumagai Y. J Biol Chem. 2007;282:33396–33404. doi: 10.1074/jbc.M705224200. [DOI] [PubMed] [Google Scholar]
- 8.Gaur D, Thapliyal N, Kishore S, Pathak V. Efficacy of broncho-alveolar lavage and bronchial brush cytology in diagnosing lung cancers. 2007;24:73–77. [Google Scholar]
- 9.Romagnoli M, Vachier I, Vignola AM, Godard P, Bousquet J, Chanez P. Respir Med. 1999;93:461–466. doi: 10.1016/s0954-6111(99)90088-4. [DOI] [PubMed] [Google Scholar]
- 10.Yendamuri S, Vaporciyan AA, Zaidi T, Feng L, Fernandez R, Bekele NB, Hofstetter WL, Jiang F, Mehran RJ, Rice DC, Spitz MR, Swisher SG, Walsh GL, Roth JA, Katz RL. J Thorac Oncol. 2008;3:979–984. doi: 10.1097/JTO.0b013e3181834f3a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yang IV, Tomfohr J, Singh J, Foss CM, Marshall HE, Que LG, McElvania-Tekippe E, Florence S, Sundy JS, Schwartz DA. Am J Respir Crit Care Med. 2012;185:620–627. doi: 10.1164/rccm.201108-1503OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bewig B, Haacke TC, Tiroke A, Bastian A, Bottcher H, Hirt SW, Rautenberg P, Haverich A. Respiration. 2000;67:166–172. doi: 10.1159/000029481. [DOI] [PubMed] [Google Scholar]
- 13.Lu C. Chemical cytometry : ultrasensitive analysis of single cells. Wiley-VCH; Weinheim: 2010. p. xix.p. 247. [Google Scholar]
- 14.Kovarik ML, Allbritton NL. Trends Biotechnol. 2011;29:222–230. doi: 10.1016/j.tibtech.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lin Y, Trouillon R, Safina G, Ewing AG. Anal Chem. 2011;83:4369–4392. doi: 10.1021/ac2009838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Piccard H, Hu J, Fiten P, Proost P, Martens E, Van den Steen PE, Van Damme J, Opdenakker G. Electrophoresis. 2009;30:2366–2377. doi: 10.1002/elps.200800698. [DOI] [PubMed] [Google Scholar]
- 17.Keithley RB, Rosenthal AS, Essaka DC, Tanaka H, Yoshimura Y, Palcic MM, Hindsgaul O, Dovichi NJ. Analyst. 2013;138:164–170. doi: 10.1039/c2an36286d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Masui H, Castro L, Mendelsohn J. J Cell Biol. 1993;120:85–93. doi: 10.1083/jcb.120.1.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Proctor A, Wang Q, Lawrence DS, Allbritton NL. Analyst. 2012;137:3028–3038. doi: 10.1039/c2an16162a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nelson AR, Allbritton NL, Sims CE. Methods Cell Biol. 2007;82:709–722. doi: 10.1016/S0091-679X(06)82026-1. [DOI] [PubMed] [Google Scholar]
- 21.Sims CE, Meredith GD, Krasieva TB, Berns MW, Tromberg BJ, Allbritton NL. Anal Chem. 1998;70:4570–4577. doi: 10.1021/ac9802269. [DOI] [PubMed] [Google Scholar]
- 22.Efron B, Tibshirani R. An introduction to the bootstrap. Chapman & Hall; New York: 1993. p. xvi.p. 436. [Google Scholar]
- 23.Guyer CA, Woltjer RL, Coker KJ, Staros JV. Arch Biochem Biophys. 1994;312:573–578. doi: 10.1006/abbi.1994.1347. [DOI] [PubMed] [Google Scholar]
- 24.Wang Z, Wang M, Lazo JS, Carr BI. J Biol Chem. 2002;277:19470–19475. doi: 10.1074/jbc.M201097200. [DOI] [PubMed] [Google Scholar]
- 25.Tiganis T. IUBMB Life. 2002;53:3–14. doi: 10.1080/15216540210811. [DOI] [PubMed] [Google Scholar]
- 26.Weinberger R. Practical capillary electrophoresis. 2. Academic Press; San diego, CA: 2000. p. xvii.p. 462. [Google Scholar]
- 27.Perez-Rodriguez M, Prieto G, Rega C, Varela LM, Sarmiento F, Mosquera V. Langmuir. 1998;14:4422–4426. [Google Scholar]
- 28. [accessed July 17, 2012];Human Protein Reference Database. http://www.hprd.org/PhosphoMotif_finder.
- 29.Jia Z, Barford D, Flint AJ, Tonks NK. Science. 1995;268:1754–1758. doi: 10.1126/science.7540771. [DOI] [PubMed] [Google Scholar]
- 30.Pouliot P, Bergeron S, Marette A, Olivier M. Immunology. 2009;128:534–542. doi: 10.1111/j.1365-2567.2009.03139.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Salmeen A, Andersen JN, Myers MP, Tonks NK, Barford D. Mol Cell. 2000;6:1401–1412. doi: 10.1016/s1097-2765(00)00137-4. [DOI] [PubMed] [Google Scholar]
- 32.Zhang ZY, Walsh AB, Wu L, McNamara DJ, Dobrusin EM, Miller WT. J Biol Chem. 1996;271:5386–5392. doi: 10.1074/jbc.271.10.5386. [DOI] [PubMed] [Google Scholar]
- 33.Zhao Z, Zander NF, Malencik DA, Anderson SR, Fischer EH. Anal Biochem. 1992;202:361–366. doi: 10.1016/0003-2697(92)90119-r. [DOI] [PubMed] [Google Scholar]
- 34.Samet JM, Tal TL. Annu Rev Pharmacol Toxicol. 2010;50:215–235. doi: 10.1146/annurev.pharmtox.010909.105841. [DOI] [PubMed] [Google Scholar]
- 35.Chen F, Shi X. Environ Health Perspect. 2002;110(Suppl 5):807–811. doi: 10.1289/ehp.02110s5807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huyer G, Liu S, Kelly J, Moffat J, Payette P, Kennedy B, Tsaprailis G, Gresser MJ, Ramachandran C. J Biol Chem. 1997;272:843–851. doi: 10.1074/jbc.272.2.843. [DOI] [PubMed] [Google Scholar]
- 37.Haase H, Maret W. Exp Cell Res. 2003;291:289–298. doi: 10.1016/s0014-4827(03)00406-3. [DOI] [PubMed] [Google Scholar]
- 38.Ahn JH, Cho SY, Ha JD, Chu SY, Jung SH, Jung YS, Baek JY, Choi IK, Shin EY, Kang SK, Kim SS, Cheon HG, Yang SD, Choi JK. Bioorg Med Chem Lett. 2002;12:1941–1946. doi: 10.1016/s0960-894x(02)00331-1. [DOI] [PubMed] [Google Scholar]
- 39.Zander NF, Lorenzen JA, Cool DE, Tonks NK, Daum G, Krebs EG, Fischer EH. Biochemistry. 1991;30:6964–6970. doi: 10.1021/bi00242a022. [DOI] [PubMed] [Google Scholar]
- 40.Mo XY, Cascio P, Lemerise K, Goldberg AL, Rock K. J Immunol. 1999;163:5851–5859. [PubMed] [Google Scholar]
- 41.Beninga J, Rock KL, Goldberg AL. J Biol Chem. 1998;273:18734–18742. doi: 10.1074/jbc.273.30.18734. [DOI] [PubMed] [Google Scholar]
- 42.Reits E, Griekspoor A, Neijssen J, Groothuis T, Jalink K, van Veelen P, Janssen H, Calafat J, Drijfhout JW, Neefjes J. Immunity. 2003;18:97–108. doi: 10.1016/s1074-7613(02)00511-3. [DOI] [PubMed] [Google Scholar]
- 43.Schmid A, Kortmann H, Dittrich PS, Blank LM. Curr Opin Biotechnol. 2010;21:12–20. doi: 10.1016/j.copbio.2010.01.007. [DOI] [PubMed] [Google Scholar]
- 44.Li H, Sims CE, Wu HY, Allbritton NL. Anal Chem. 2001;73:4625–4631. doi: 10.1021/ac0105235. [DOI] [PubMed] [Google Scholar]
- 45.Stockholm D, Benchaouir R, Picot J, Rameau P, Neildez TM, Landini G, Laplace-Builhe C, Paldi A. PLoS One. 2007;2:e394. doi: 10.1371/journal.pone.0000394. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.