

Abstract
Background
Kernel weight, controlled by quantitative trait loci (QTL), is an important component of grain yield in maize. Cytokinins (CKs) participate in determining grain morphology and final grain yield in crops. ZmIPT2, which is expressed mainly in the basal transfer cell layer, endosperm, and embryo during maize kernel development, encodes an isopentenyl transferase (IPT) that is involved in CK biosynthesis.
Results
The coding region of ZmIPT2 was sequenced across a panel of 175 maize inbred lines that are currently used in Chinese maize breeding programs. Only 16 single nucleotide polymorphisms (SNPs) and seven haplotypes were detected among these inbred lines. Nucleotide diversity (π) within the ZmIPT2 window and coding region were 0.347 and 0.0047, respectively, and they were significantly lower than the mean nucleotide diversity value of 0.372 for maize Chromosome 2 (P < 0.01). Association mapping revealed that a single nucleotide change from cytosine (C) to thymine (T) in the ZmIPT2 coding region, which converted a proline residue into a serine residue, was significantly associated with hundred kernel weight (HKW) in three environments (P <0.05), and explained 4.76% of the total phenotypic variation. In vitro characterization suggests that the dimethylallyl diphospate (DMAPP) IPT activity of ZmIPT2-T is higher than that of ZmIPT2-C, as the amounts of adenosine triphosphate (ATP), adenosine diphosphate (ADP), and adenosine monophosphate (AMP) consumed by ZmIPT2-T were 5.48-, 2.70-, and 1.87-fold, respectively, greater than those consumed by ZmIPT2-C. The effects of artificial selection on the ZmIPT2 coding region were evaluated using Tajima’s D tests across six subgroups of Chinese maize germplasm, with the most frequent favorable allele identified in subgroup PB (Partner B).
Conclusions
These results showed that ZmIPT2, which is associated with kernel weight, was subjected to artificial selection during the maize breeding process. ZmIPT2-T had higher IPT activity than ZmIPT2-C, and this favorable allele for kernel weight could be used in molecular marker-assisted selection for improvement of grain yield components in Chinese maize breeding programs.
Keywords: Maize, Isopentenyl transferase 2, Association mapping, Artificial selection
Background
Genetic improvement of grain yield is important for ensuring food security. Many important agronomic traits, including yield, show continuous phenotypic variation [1]. Grain yield is a complex quantitative trait, and is determined by several components including kernel number and kernel weight [2]. To date, more than 185 quantitative trait loci (QTL) underlying yield components including kernel row number (KRN), kernel number per row (KNPR), and hundred kernel weight (HKW) have been identified across the maize genome using various mapping populations (2012 November update to Gramene database), which has improved our understanding of the genetic basis of maize yield. Therefore, grain size and kernel weight are important targets for artificial selection for high grain yield. For example, QTL associated with GS3 [3] and GW5/SW5 [4,5] have been selected for grain size in rice. Also, QTL associated with TaGW2-6A Hap-6A-A for grain size [6] and TaCKX6-D1 [7] for grain weight have been identified and artificially selected from within Chinese wheat germplasm. Markers derived from QTL controlling similar components of grain yield in maize will be very useful for marker-assisted selection.
Several genes that control kernel weight or kernel number have been cloned in crops, and some of these genes were found to be involved in carbon and nitrogen metabolism [8-10], protein degradation [11,12], and hormone metabolism [13,14]. All of these processes affect the production and export of C- and N-assimilates to the seed, thereby increasing crop yield. C- and N-metabolism are essential to all processes in plants, including reproductive development, but are especially so during grain filling [8]. Among the genes involved in carbon metabolism, rice grain incomplete filling 1, which functions similarly to the invertase encoded by miniature 1 in maize [10], also encodes a cell-wall invertase required for carbon partitioning during early grain-filling [8]. A gene involved in nitrogen metabolism in rice, OsARG, encodes an arginine hydrolase that, when overexpressed, increases grain number per plant under nitrogen-limited conditions, due to increased nitrogen remobilization at the reproductive stage [9].
In addition, degradation of proteins via the ubiquitin/proteasome pathway negatively regulates cell division and grain yield. Another gene, Grain Weight 2 (GW2), which encodes a RING-type E3 ubiquitin ligase, functions in ubiquitin-mediated degradation, and the loss of GW2 function can enhance rice grain width, weight, and yield [11]. Furthermore, ZmGW2-CHR4, which functions in the same manner as GW2, is also associated with kernel width and kernel weight in maize [12]. Although they have potentially positive impacts on yield, alleles at these loci also should be monitored to avoid possible negative effects of introduced germplasm on yield.
As to the influence of plant hormones, brassinosteroids (BRs) and cytokinins (CKs) are useful for controlling grain yield in crops. BRs stimulate the transport of sucrose and other sugars to the endosperm and embryo. Expression of the maize, rice, or Arabidopsis thaliana C-22 hydroxylase that is involved in BR synthesis in stems, leaves, and roots had clear effects on seed weight [14]. In rice, the accumulation of CK in the inflorescence meristems increases the number of grains per panicle. The rice gene Gn1a encodes an oxidase/dehydrogenase (OsCKX2) that functions in the degradation of CKs. An 11-bp deletion in its coding region created a premature stop codon that reduced expression of OsCKX2 and resulted in increased grain number [13]. In wheat, TaCKX6-D1, an ortholog of rice OsCKX2, was significantly associated with 1000-grain weight by linkage mapping and association analysis [7]. Thus, the CK regulatory pathway likely plays a crucial role in grain yield.
There is also evidence for a role of CKs in balancing the source and sink relationship [15]. Accordingly, exogenous applications of CKs increased seed set and yield stability under heat stress in maize [16]. CKs are co-regulated by isopentenyl transferase (IPT) and oxidase/dehydrogenase in plants. The discovery of biosynthetic enzymes IPT1 through IPT10 [17-19] and degradative enzymes CKX1 through CKX12 [18,20-23] has led to a better understanding of the role of CKs in maize kernel development. Among the IPTs, IPT1 and IPT10 are highly abundant and are constitutively expressed in all organs, but other IPT transcripts show distinct spatial and temporal patterns of expression [18]. ZmIPT2 is specifically expressed in the pedicel, endosperm, and embryo [18,19]. ZmIPT2 consists of an intronless 966 bp coding region for dimethylallyl diphosphate (DMAPP): adenosine triphosphate (ATP)/adenosine diphosphate (ADP) IPT, which preferred ATP and ADP over adenosine monophosphate (AMP) as substrates for IPT activity [19].
To date, the allelic diversity of ZmIPT2 and the most favorable allele(s) affecting kernel weight have not yet been reported for maize. The objectives of this study were to (1) examine the sequence diversity of ZmIPT2 in 175 Chinese maize inbred lines; (2) test associations between nucleotide polymorphisms in ZmIPT2 and various yield components including KRN, KNPR, and HKW, and identify further favorable allele(s) for grain yield components; (3) characterize the IPT activity of the protein products of different alleles in vitro; and (4) investigate selection for favorable ZmIPT2 alleles for kernel weight during maize breeding in China.
Results
Phenotypic data
Phenotypic values for individual lines ranged from 7.81 to 18.62 g for KRN (mean = 12.80), from 8.26 to 34.67 g for KNRP (mean = 22.40), and from 12.35 to 47.60 g for HKW (mean = 28.36), as shown in Table 1. KRN was significantly positively correlated with KNPR (P <0.01), while HKW was negatively correlated with KNPR (P <0.05). Analysis of variance (ANOVA) indicated significant phenotypic variation for KRN, KNPR, and HKW among these 175 maize lines (P <0.01). Heritabilities estimated on a per-plot basis were 86.58% for KRN, 84.27% for KNRP, and 86.12% for HKW.
Table 1.
Mean squares from ANOVA and correlation coefficients for KRN, KNPR, and HKW
| Category | Source of variation | DF | KRN | KNPR | HKW (g) |
|---|---|---|---|---|---|
| ANOVA |
Year |
1 |
0.20 |
10.3** |
2.14 |
| |
Replication |
2 |
1.39 |
0.16 |
0.44 |
| |
Genotype |
172 |
9.81** |
10.14** |
12.12** |
| |
Year × Genotype |
171(169)a |
1.57** |
2.11** |
1.95** |
| Descriptive |
Range |
|
7.81-18.62 |
8.26-34.67 |
12.35-47.60 |
| Statistics |
Mean ± SD |
|
12.80 ± 1.80 |
22.40 ± 4.55 |
27.98 ± 5.46 |
| Correlation |
KNPR |
|
0.35** |
|
|
| coefficients |
HKW (g) |
|
−0.06 |
−0.16* |
|
| h2 | 86.58% | 84.27% | 86.12% |
DF degrees of freedom, HKW hundred kernel weight, KRN kernel row number, KNPR kernel number per row, h2 broad-sense heritability; **, P <0.01; *, P <0.05.
a Because the phenotypic values for some lines were missing for kernel weight, the DF differ. The numbers in parentheses indicate the DF for HKW and the numbers outside parentheses indicate the DF for KRN and KNPR.
Nucleotide diversity in the ZmIPT2 coding region
A total of 16 single nucleotide polymorphisms (SNPs) were identified in the ZmIPT2 coding region, with an average of one SNP every 60 bp, and six of these variants resulted in amino acid substitutions (Figure 1A). Seven haplotypes, named Hap_1 through Hap_7, were detected in our association panel based on these 16 SNPs (Figure 1B). A total of 75 (42.9%) inbred lines harbored Hap_1, while 10, 31, 22, 9, 10, and 18 inbred lines carried Hap_2 through 7, respectively. Hap_1 harbored the ZmIPT2-T allele at SNP28 (C-T), but other haplotypes bore the ZmIPT2-C allele. The window covering ZmIPT2 exhibited relatively lower diversity with a value of 0.347, and it was significantly lower than the average diversity value of 0.372 for Chromosome 2 across 175 maize inbred lines (P < 0.01) (Figure 2). Most polymorphisms in ZmIPT2 were found in the region from 360 to 590 bp, (P >0.005) (Figure 3). Among maize subgroups revealed by Xie et al. [24], subgroups Lancaster (Lan) and Lvda red cob (LRC) harbored higher levels of diversity in the ZmIPT2 coding region, compared with the average for all inbreds (Figure 3).
Figure 1.
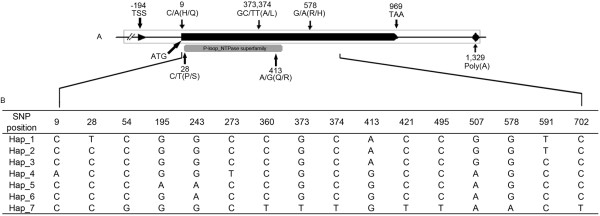
Nucleotide diversity and amino acid variants in the ZmIPT2 coding region. (A) The full sequence of ZmIPT2 gene. Sites are labeled according to the position of the corresponding polymorphisms in the aligned sequences, with amino acid alterations indicated in parentheses. The gray box indicates the IPT domain, which starts at position 28 and ends at position 480. (B) The ZmIPT2 genomic sequences grouped into seven haplotypes defined by 16 single nucleotide polymorphisms identified in the alignment.
Figure 2.
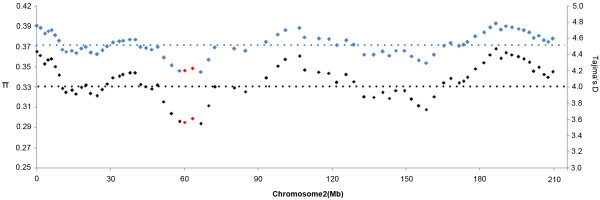
Sliding-Window analyses of nucleotide diversity π and Tajima’s D in the association panel, with a sliding window size of 500 SNP markers and step intervals of 50 SNP markers on maize chromosome 2. Blue diamonds, π; Black diamonds, Tajima’s D; Red diamonds, windows containing the ZmIPT2 gene locus. Blue dashes represent the mean π value; Black dashes represent the mean Tajima’s D value.
Figure 3.
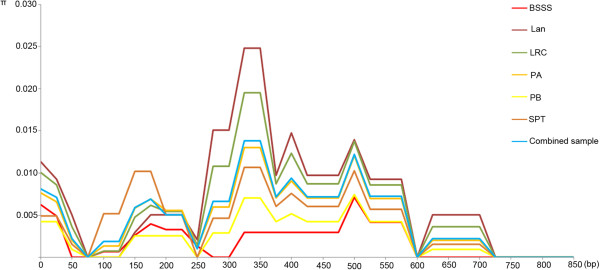
Sliding-Window analysis of nucleotide diversity in the ZmIPT2 coding region across six maize subgroups with 100 bp window and 25 bp step size. BSSS, derived from US BSSS and Reid germplasm; Lan, derived from US Lancaster Sure Crop germplasm; LRC, derived from Lvda Red Cob, a Chinese landrace; PA, derived from modern US hybrids in China; PB, derived from modern US hybrids in China; SPT, derived from Si-ping-tou, a Chinese landrace. Combined sample, 175 maize inbred lines.
Association analysis of KRN, KNPR, and HKW
Association analysis revealed that SNP28 was significantly associated with HKW across all three environments tested (P <0.05), and explained 5.42% of the phenotypic variation in HKW at Xinjiang in 2007, 3.41% of the phenotypic variation at Beijing in 2008, and 5.46% of the phenotypic variation at Xinjiang in 2008. Significant association of HKW with SNP195, SNP413, and SNP591 was detected in one or two environments (Table 2). However, only SNP413 was found to be associated with KNPR in Beijing (P <0.05), and no SNP was significantly associated with KRN (Table 2). The inbred lines containing the ZmIPT2-T allele had significantly (P <0.05) greater kernel weight than those carrying the ZmIPT2-C allele, as seen in the data from Xinjiang in 2007 and in Beijing in 2008 across this association panel (Figure 4). The P value obtained here supports the strength of the observed association between HKW and SNP28, and improves confidence in identification of the ZmIPT2-T allele in Hap_1 as the most favorable allele for kernel weight in maize.
Table 2.
ZmIPT2 polymorphisms associated with KRN, KNPR, and HKW in the association panel
| SNPs | Genotype | Frequency | Environment | KRN | KNPR | HKW |
|---|---|---|---|---|---|---|
| SNP9 |
C/A |
153/22 |
07XJ |
- |
- |
- |
| 08BJ |
- |
- |
- |
|||
| 08XJ |
- |
- |
- |
|||
| SNP28 |
T/C |
75/100 |
07XJ |
- |
- |
0.003 |
| 08BJ |
- |
- |
0.0252 |
|||
| 08XJ |
- |
- |
0.0027 |
|||
| SNP54 |
C/G |
157/18 |
07XJ |
- |
- |
- |
| 08BJ |
- |
- |
- |
|||
| 08XJ |
- |
- |
- |
|||
| SNP195 |
G/A |
166/9 |
07XJ |
- |
- |
0.0386 |
| 08BJ |
- |
- |
0.0031 |
|||
| 08XJ |
- |
- |
- |
|||
| SNP243 |
G/A |
156/19 |
07XJ |
- |
- |
- |
| 08BJ |
- |
- |
- |
|||
| 08XJ |
- |
- |
- |
|||
| SNP413 |
A/G |
115/6 |
07XJ |
- |
- |
0.0302 |
| 08BJ |
- |
0.029 |
- |
|||
| 08XJ |
- |
- |
0.0351 |
|||
| SNP591 | T/C | 85/90 | 07XJ |
- |
- |
- |
| 08BJ |
- |
- |
- |
|||
| 08XJ | - | - | 0.0187 |
HKW hundred kernel weight, KRN kernel row number, KNPR kernel number per row, 07XJ Xinjiang in 2007, 08BJ Beijing in 2008, 08XJ Xinjiang in 2008; -, not significant at P <0.05.
Figure 4.
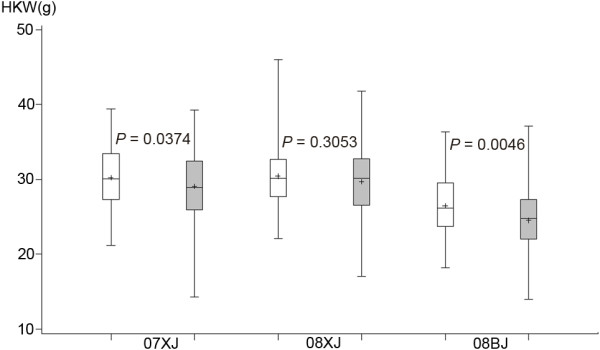
Hundred-kernel weight (HKW) comparison of groups harboring either ZmIPT2-T or ZmIPT2-C.P, P value for t-test comparing two groups carrying different alleles in each of three environments. 07XJ, Xinjiang in 2007; 08XJ, Xinjiang in 2008; 08BJ, Beijing in 2008. White boxes represent the ZmIPT2-T group; grey boxes represent the ZmIPT2-C group.
Characterization of ZmIPT2
Both the ZmIPT2-T and ZmIPT2-C protein alleles have the same pI and aliphatic index. However, the transition at SNP28 from cytosine (C) to thymine (T) converts proline into serine, resulting in a decrease in the hydropathicity of the entire ZmIPT2 protein from −0.006 to −0.008, as analyzed using GRAVY. The IPT coding region aligned with the P-loop NTPase superfamily that is characteristic of all NTP-binding proteins from position 28 to 480 with an E-value, or probability of a protein sequence of a given bit score occurring by chance, of 1.3e-06 (NCBI blastp database) (Figure 1A). The 3-D structure predicted by SWISS-MODEL (changed to http://swissmodel.expasy.org/) showed that the spatial structure of ZmIPT2-T and ZmIPT2-C differed due to variation in hydrophobicity between the two proteins (Additional file 1). These kinds of changes could affect the binding of substrates to the enzyme, which is mediated by hydrogen bonds [25]. This mutation and resultant differences in physicochemical properties imply that IPT activity could differ between the protein products of the ZmIPT2-T and ZmIPT2-C alleles.
To further confirm the CK biosynthetic function of these two ZmIPT2 products in vitro, Escherichia coli prokaryotic expression vectors were constructed to express and purify ZmIPT2 as C-terminal His-tagged recombinant proteins. ZmIPT2-T and ZmIPT2-C purified on Talon® columns reached 95-100% purity, as quantified by Coomassie staining after PAGE (Additional file 2). Purified proteins were then used to assay DMAPP:ATP, DMAPP:ADP, and DMAPP:AMP transferase activity. Figure 5 shows the profile obtained from the reaction mixture of purified ZmIPT2 protein incubated with DMAPP and substrates, including ATP, ADP, and AMP. ZmIPT2-T converted AMP to isopentenyl adenosine monophosphate (iPMP) with higher efficiency than did ZmIPT2-C, as indicated by the amount of iPMP after reaction (Figure 5C). The mean AMP consumption by ZmIPT2-T and ZmIPT2-C were 0.20 μmol and 0.07 μmol, respectively (Additional file 3). Similar results were obtained with ATP or ADP as the substrate (Figure 5A, B). ZmIPT2-T consumed more ADP and ATP, with means of 270.2% and 548.4%, respectively, than consumed by ZmIPT2-C. Additionally, ZmIPT2-T consumed much more ATP and ADP than it did AMP, with means of 7.38- and 13.61-fold the amount of AMP consumed, respectively (Additional file 3). These results indicate that the protein product of the ZmIPT2-T allele has higher IPT enzyme activity and prefers ADP as substrate, and so could have a more favorable impact on yield.
Figure 5.
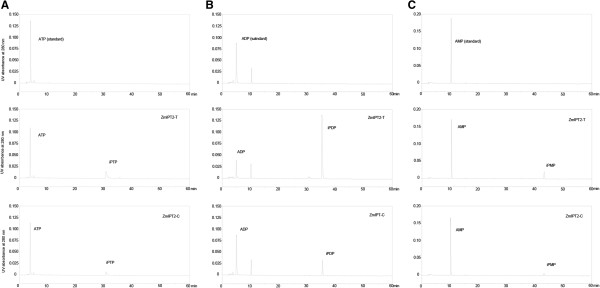
Determination of isopentenyl transferase activity of ZmIPT2. A, B, and C depict determination of DMAPP:ATP, DMAPP:ADP, and DMAPP:AMP isopentenyl transferase activity of recombinant ZmIPT2 protein. iPTP = isopentenyl adenosine triphosphate, iPDP = isopentenyl adenosine diphosphate, and iPMP = isopentenyl adenosine monophosphate.
Selection in the region of the ZmIPT2 locus
A total of 4282 SNPs [26] were used to analyze the effects of selection occurring in this region of Chromosome 2 during selection and breeding. The estimates for π and Tajima’s D in the ZmIPT2 window showed lower values than the mean estimates for those parameters for all of maize Chromosome 2 (Figure 2). Tajima’s D tests of the ZmIPT2 coding region and genomic window identified different artificial selection effects across these six maize subgroups, with the largest effect identified in subgroup PB (Figure 6). Notably, 81% of inbred lines in subgroup PB harbored the favorable allele ZmIPT2-T (Table 3). These results indicate that artificial selection occurred in the genomic region harboring ZmIPT2 during maize breeding.
Figure 6.

Tajima’s D values for the ZmIPT2 coding region and hundred kernel weights across six maize subgroups. PB, derived from modern US hybrids in China; BSSS, derived from US BSSS and Reid germplasm; LRC, derived from Lvda Red Cob, a Chinese landrace; PA, derived from modern US hybrids in China; Lan, derived from US Lancaster Sure Crop germplasm; SPT, derived from Si-ping-tou, a Chinese landrace. HKW, hundred-kernel weight. 07XJ, Xinjiang in 2007; 08XJ, Xinjiang in 2008; 08BJ, Beijing in 2008.
Table 3.
Distribution of the ZmIPT2-T allele among six subgroups in 175 Chinese inbred lines
| PB | BSSS | LRC | PA | Lan | SPT | |
|---|---|---|---|---|---|---|
|
ZmIPT2-T |
17 |
18 |
11 |
19 |
5 |
5 |
| No.a |
21 |
29 |
27 |
55 |
18 |
25 |
| Frequencyb | 81% | 62% | 41% | 35% | 28% | 20% |
a The number of inbred lines in each subgroup.
b The frequency of the ZmIPT2-T allele in each subgroup.
Discussion
Grain yield is a complex trait controlled by minor genes
Grain yield is one of the most important and complex traits in maize. Although many studies on the genetic basis of maize yield-related traits have been reported, few favorable alleles for yield candidate genes have been identified because single genes identified in association studies explain only a small proportion of phenotypic variation. In this study, SNP28 in the ZmIPT2 coding region was significantly associated with kernel weight across three environments, explaining 4.76% of phenotypic variation. Relatively small allelic effects were also found for one polymorphism in the ZmGS3 region, which explained 6.29% of variation in Beijing and 7.73% of that in Hainan [27], and another SNP, S40 in the ZmGW2-CHR4 region, which accounted for less than 10% of variation across three environments [12]. Thus, the identification and pyramiding of a large number of favorable alleles will be the key to molecular marker-assisted breeding of high-yielding maize.
Association mapping of complex traits
As an effective way to dissect complex traits and identify favorable alleles or haplotypes, association mapping has been widely used for many maize traits, including flowering time [28], leaf architecture [29], plant height [26], and disease resistance [30-32]. With candidate-gene association mapping, excellent allelic variants of many genes have been identified, such as crtRB1 for β-carotenoid synthesis in maize kernels [33]; Dwarf8 for maize flowering time [6,34,35]; ZmGW2-CHR4[36] and ZmGS3[27], which are related to maize kernel size and weight; TaGW2, which is associated with wheat kernel weight and maturity [37]; and other genes related to starch synthesis in rice [38].
Contribution of CKs to grain yield
Plant hormones control many aspects of development, and like other hormones, CKs affect many plant physiological and biochemical processes, including those controlling delayed senescence [39], suppression of auxin-induced apical dominance [40], signaling of nitrogen availability [41], cell division and differentiation [42], and sink strength [15]. IPT proteins, which are involved in synthesis of CKs, are encoded by a multi-gene family in maize [17-19], rice [43], soybean [44], Arabidopsis [45,46], petunia [47], hop [48] and pea [49]. A favorable IPT allele that resulted in increased grain weight has now been identified in maize using an association mapping approach (Figure 3).
CK content is co-regulated by two families of genes, including isopentenyl transferase and an oxidase/dehydrogenase in plants. In rice, reduced expression of OsCKX2, which encodes the oxidase/dehydrogenase, caused an increase in grain number per panicle, due to CK accumulation in inflorescence meristems. OsCKX2 is expressed strongly in pistils, at an intermediate level in inflorescences and seeds, and at low levels in other organs [13]. TaCKX6-D1, a wheat ortholog of rice OsCKX2, was related to HGW by linkage mapping and association analysis. TaCKX6_D1 was expressed at a high level in seeds and pistils, but at a low level in other organs [7]. One study showed that ZmIPT2 is specifically expressed in pedicel, endosperm, and embryo, and plays a major role in CK biosynthesis for maize kernel development [18,19]. In this study, however, only SNP413 was found to be significantly associated with KNPR in one of the three experimental environments (Beijing), and no associated SNP was found for KRN (Table 2). CKs coordinately induce extracellular invertase and hexose transporter activity, which are functionally coupled to supply carbohydrates to sink tissues [15]. In this study, a single nucleotide substitution from C to T in the ZmIPT2 coding region was significantly associated with kernel weight. This suggests that manipulation of CK metabolism in reproductive organs could be an effective approach to further increase crop grain yields, by increasing the flow of assimilates from source to sink, or by increasing sink capacity.
In vitro characterization of the ZmIPT2 enzyme showed that it prefers ADP and ATP over AMP as substrates for DMAPP IPT activity (Figure 5, Additional file 3), consistent with the results of a previous study [19]. Six amino acid substitutions, none of which were conservative, occurred in ZmIPT2, compared with 10 other maize ZmIPTs [18], indicating that ZmIPT2, and its specific expression pattern, is conserved and essential for kernel development. Interestingly, the mutation associated with the favorable allele, ZmIPT2-T, was detected in the ZmIPT2 coding region, and found to result in a serine to proline substitution in the conserved IPT domain that caused the ZmIPT2-T enzyme to consume more ATP, ADP, and AMP than the ZmIPT2-C enzyme (Figure 5, Additional file 3). Thus, selection for the molecular marker SNP28 should improve yield by positively influencing maize kernel development via an increase in IPT enzyme activity.
Nucleotide diversity in ZmIPT2 across six subgroups
In this study, the coding region of ZmIPT2 was sequenced across 175 Chinese maize inbreds. The coding regions of functional genes tend to be relatively conserved, due to their specificity for and affinity with other types of molecules. Thus, under natural selection, beneficial variants tend to become gradually fixed, while detrimental variants tend to be eliminated [37]. Lower levels of diversity within the ZmIPT2 locus were estimated in subgroups PB and BSSS, compared to the combined group (P < 0.01) (Figure 3), suggesting either that the germplasm base remained remarkably narrow or that strong positive selection at that locus occurred in these subgroups. According to pedigrees, 17 inbreds in subgroup PB were mainly derived from US hybrid P78599. However, subgroup Lan harbored the most diversity in the ZmIPT2 coding region across all these inbreds (Table 1; Figure 3). This subgroup has been identified as having the most genetic diversity in one study using 145 SSR loci randomly distributed across the genome [50].
Selection for grain yield during the breeding process
It is hypothesized that natural selection favored smaller-seeded wild ancestors with a larger number of seeds per plant, earlier maturity, and wider geographic distribution [51]. However, high crop yield associated with larger grain size and weight has been the objective of artificial selection during most breeding programs. For example, the genes GS3 [52] and GW5/SW5 [3,53] have been proven to be involved in the evolution of grain size in rice. Unlike rice, in wheat and maize, artificial selection is associated with an almost uniform increase in seed or grain size. The allele TaGW2-6A Hap-6A-A, which was associated with larger grain size, increased in frequency under positive selection from 50.0% in the 1950s to current levels of 77.42% [37]. Under strong selection at a given locus, domestication has the potential to drastically increase LD and reduce diversity [4].
Estimates of the parameters π and Tajima’s D in this study showed that artificial selection took place at the ZmIPT2 locus (Figure 3). However, only Hap_1, among seven haplotypes examined here, harbored the ZmIPT2-T allele (Figure 1), and this favorable allele was associated with increased grain weight in two of the three experimental environments (Figure 4), suggesting positive selection for higher grain weight. Similarly, a 95.7% reduction in nucleotide diversity across the GS3 locus occurred among rice accessions carrying the A allele for GS3 [52]. The current study found that the ZmIPT2-T allele was not evenly distributed among these six maize subgroups, occurring with the highest frequency in subgroup PB, in which 81% of the tested lines harbor the favorable allele (Table 3). The lowest Tajima’s D value was also found for subgroup PB (Figure 6) indicating that distinct positive selection pressure has taken place at the ZmIPT2 locus in this subgroup. Selection for the favorable allele ZmIPT2-T within each subgroup should be continued in order to improve maize yield.
Conclusions
A favorable allele, ZmIPT2-T, was associated with kernel weight in Chinese maize germplasm. ZmIPT2-T was shown in vitro to have higher IPT activity than ZmIPT2-C with ADP, ATP, or AMP as substrate. Artificial selection at the ZmIPT2 locus was detected with Tajima’s D tests across six subgroups of Chinese maize germplasm, with the most frequent favorable allele identified in subgroup PB. This favorable allele could be used in molecular marker-assisted selection for improvement of grain yield components in Chinese maize breeding programs.
Methods
Experimental design and statistical analyses
In this study, a total of 175 maize inbred lines were used, which were subdivided into six subgroups [24], including 29 BSSS lines derived from US BSSS and Reid germplasm; 55 PA (Partner A) lines derived from modern US hybrids in China; 21 PB (Partner B, harboring the distinct heterosis reaction of PA) lines derived from modern US hybrids in China; 18 Lan lines derived from US Lancaster Sure Crop germplasm; 27 LRC lines derived from Lvda Red Cob, a Chinese landrace; and 25 SPT lines derived from Si-ping-tou, a Chinese landrace. All lines were planted at Xinjiang in 2007 and 2008, and at Beijing in 2008 to measure grain yield components, including KRN, KNPR, and HKW. A randomized complete-block design was employed with three replications of 20 plants of each line per location planted in 4.5-m rows, 0.6 m apart. Normal maize agricultural practices were carried out. At maturity, all ears were harvested manually and dried to grain moisture of 13%. KRN was scored as number of rows per ear, KNPR was scored as total kernels in a row per ear, and HKW was measured on 100 randomly selected kernels per ear. The average phenotypic values of each plot over three replications in each environment were used for final analysis.
Descriptive statistics and analysis of variance (ANOVA) for KRN, KNPR, and HKW were obtained using the program PROC GLM in SAS software version 9.13 (Copyright © 2009 SAS Institute Inc., 2009. SAS and all other SAS Institute Inc. product or service names are registered trademarks or trademarks of SAS Institute Inc., Cary, NC, USA). The SAS program PROC CORR was used to quantify the relationship among the three traits. Broad-sense heritability was estimated based on plot as h2 = σg2/ (σg2 + σgl2/n + σe2/nr), where σg2 is genetic variance, σgl2 is genotype-by-environment interaction, σe2 is error variance, r is the number of replications, and n is the number of locations. The estimates for σg2, σgl2, and σe2 were acquired from ANOVA [5].
DNA isolation, PCR amplification and DNA sequencing
Genomic DNA was extracted from maize leaves using the CTAB method [54]. Primers ZmIPT2-5′ (5′-ATCATCAAGACAATGGAGCACGGTG-3′) and ZmIPT2-3′ (5′-CGTCCGCTAGCTACTTATGCATCAG-3′) were designed based on the published ZmIPT2 cDNA sequence (Accession number EU263126). ZmIPT2 genomic sequences were amplified from each of 175 inbred lines in 25 μL reaction mixtures containing 20 ng of genomic DNA, 0.2 mM of each dNTP, 1 μM of each primer, 2 × GC buffer (2 mM Mg2+), and 2.5 U TransTaq High Fidelity DNA polymerase (Transgen Biotechnology Corporation, Beijing, China). Touchdown PCR was applied as follows: 94°C for 2 min (one cycle);94°C for 30 s, 65°C for 45 s, and 72°C for 90 s (5 cycles, annealing temperature reduced 1°C per cycle);94°C for 30 s, 60°C for 45 s and 72°C for 1 min 30 s (30 cycles); 72°C for 7 min, then the reaction was then held at 4°C. The products were sequenced (ABI3730) at the public laboratory of the National Key Facility of Crop Gene Resources and Genetic Improvement, Institute of Crop Science, Chinese Academy of Agricultural Science.
Nucleotide diversity and selection in breeding process
DNA sequences of all lines were analyzed using DnaSP Version 4.00 [55]. The Sliding-Window analysis of nucleotide diversity was performed with window size set at 100 bp and steps of 25 bp. The parameter π was estimated as the average proportion of nucleotide differences between all possible pairs of sequences in the sample [56]. A total of 4282 SNPs on maize Chromosome 2 from the MaizeSNP50 BeadChip (Illumina, Inc.) were used to estimate π and Tajima’s D using TASSEL 3.0 software [26,57].
Association mapping of KRN, KNPR and HKW
To exclude the effect of population structure on association mapping, population structure and kinship information for 175 lines were analyzed using STRUCTURE version 2.3 and SPAGeDi software with SNPs selected from a collection of 5000 SNPs identified in a previous study and selected based on physical position and minor allele frequency [26]. Association mapping between three yield-related traits and the nucleotide diversity of the ZmIPT2 coding region was performed using TASSEL 3.0 [57] with a mixed linear model (MLM) controlling both population structure (Q) and relative kinship (K).
Characterization of ZmIPT2
The 3D protein model of ZmIPT2 was predicted using the online process SWISS-MODEL Automated Mode (http://swissmodel.expasy.org/). Physicochemical properties of ZmIPT2 proteins, including isoelectric point (pI), aliphatic index, and grand average of hydropathicity (GRAVY) were analyzed using ProtParam (http://web.expasy.org/protparam/). These kinds of changes could affect the binding of substrates to the enzyme, which is mediated by hydrogen bonds [25]. The conserved domain was used to query the smart web program (http://smart.embl-heidelberg.de/).
To characterize the enzyme activity of the products of these two ZmIPT2 alleles (the T variant and the C variant), prokaryotic expression and enzyme activity determination of ZmIPT2 were carried out in vitro. The gene fragments harboring ZmIPT2-T or ZmIPT2-C were amplified using gene-specific primers with appropriate NdeI and NotI restriction site extensions, (5′-GGCATATGGAGCACGGTGCCGTCGC-3′ and 5′-CCGCGGCCGCTCATCATGCATCAGCCACGGCGGTGA-3′) from the ‘Ye478’ (Hap_1) and ‘Guan17’ (Hap_2) inbred lines, respectively. The PCR products were digested with NdeI and NotI, and then cloned into pET30b (Novagen, Darmstadt, Germany) at these same restriction enzyme sites. The recombinant plasmids were integrated into BL21 (DE3) E. coli competent cells (Tiangen Biotech Co., Ltd, Beijing, China). The cultures were incubated at 37°C with shaking until log phase at OD600 = ~ 0.4. Expression of the IPT protein was induced by incubation with 1 mM IPTG at 37°C for 3 h, followed by purification using the HisTALON™ Gravity column Purification Kit (Clontech Laboratories, Inc., CN) according to the manufacturer’s guidelines. Sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis analysis (PAGE) was performed to ensure purity, and protein density was measured using the Macro-BCA Assay Kit (Gragen Life Science Inc., CN).
Purified protein was used to determine DMAPP:ATP, DMAPP:ADP, and DMAPP:AMP IPT activities. Each purified protein (~0.13 mg/ml) was incubated in a reaction mixture containing 12.5 mM Tris–HCl (pH 7.5), 37.5 mM KCl, 5 mM MgCl2, 1 mM DMAPP (Sigma-Aldrich Co., St. Louis, USA), and 1 mM ATP (Sigma-Aldrich Co., St. Louis, USA), ADP (Sigma-Aldrich Co., St. Louis, USA) or AMP (Sigma-Aldrich Co., St. Louis, USA) for 2 h at 30°C. The reaction was stopped by boiling for 5 min. The reaction products were separated by reversed-phase HPLC (Shimadzu LC-10AT series HPLC system with a SPD-10AVP UV–vis detector) with a C18-ODS2 column (Kromasil), according to the following program: 20 mM KH2PO4 for 15 min, linear gradient of 0% acetonitrile, 20 mM KH2PO4 to 20% acetonitrile, and 4 mM KH2PO4 over 30 min. UV absorbance was monitored at 280 nm.
Abbreviations
ANOVA: Analysis of variance; BRs: Brassinosteroids; CKs: Cytokinins; DMAPP: Dimethylallyl diphospate; GLM: General linear model; HKW: Hundred kernel weight; IPT: Isopentenyl transferase; KRN: Kernel row number; KNPR: Kernel number per row; LD: Linkage disequilibrium; MLM: Mixed linear model; QTL: Quantitative trait loci; SNP: Single nucleotide polymorphisms; 07XJ: Xinjiang location in 2007; 08BJ: Beijing location in 2008; 08XJ: Xinjiang location in 2008.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
SZ and XL conceived and designed the experiments. JW and BL performed the experiments. JW, BL, CL, XY, HW, ZH, ML, DZ, and XC contributed reagents, materials, and analysis tools. JW, BL, and XL wrote the manuscript. SZ, XL, and JW coordinated the research. All authors read and approved the final manuscript.
Supplementary Material
3-D structure of ZmIPT2 predicted with the program Swiss-PdbViewer. Ser and Pro represent amino acids in ZmIPT2-T and ZmIPT2-C, respectively.
Purified recombinant proteins for enzyme activity determination.
The consumption of ATP, ADP, and AMP by ZmIPT2-C and ZmIPT2-T during in vitro enzyme activity determination.
Contributor Information
Jianfeng Weng, Email: jfweng@126.com.
Bo Li, Email: libosunny@163.com.
Changlin Liu, Email: sichuanlcl@126.com.
Xiaoyan Yang, Email: yangxiaoyan6600@126.com.
Hongwei Wang, Email: whw741119@163.com.
Zhuanfang Hao, Email: haozhuanfang@yahoo.com.cn.
Mingshun Li, Email: mshunli@yahoo.com.cn.
Degui Zhang, Email: zdgzdg2010@yahoo.com.cn.
Xiaoke Ci, Email: xkc_99@yahoo.com.cn.
Xinhai Li, Email: lixh2008caas@yahoo.cn.
Shihuang Zhang, Email: cshzhang2000@yahoo.com.cn.
Acknowledgements
This study was supported by National Basic Research Program (2011CB100100) and the National High Technology Research and Development Program (2012AA10A306) from the Ministry of Science and Technology of China.
References
- Yano M. Genetic and molecular dissection of naturally occurring variation. Curr Opin Plant Biol. 2001;4(2):130–135. doi: 10.1016/S1369-5266(00)00148-5. [DOI] [PubMed] [Google Scholar]
- Austin DF, Lee M. Comparative mapping in F2∶3 and F6∶7 generations of quantitative trait loci for grain yield and yield components in maize. TAG Theor Appl Genet. 1996;92(7):817–826. doi: 10.1007/BF00221893. [DOI] [PubMed] [Google Scholar]
- Shomura A, Izawa T, Ebana K, Ebitani T, Kanegae H, Konishi S, Yano M. Deletion in a gene associated with grain size increased yields during rice domestication. Nat Genet. 2008;40(8):1023–1028. doi: 10.1038/ng.169. [DOI] [PubMed] [Google Scholar]
- Palaisa KA, Morgante M, Williams M, Rafalski A. Contrasting Effects of Selection on Sequence Diversity and Linkage Disequilibrium at Two Phytoene Synthase Loci. Plant Cell. 2003;15(8):1795–1806. doi: 10.1105/tpc.012526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knapp SJ, Stroup WW, Ross WM. Exact confidence intervals for heritability on a progeny mean basis. Crop Sci. 1985;25(1):192–194. doi: 10.2135/cropsci1985.0011183X002500010046x. [DOI] [Google Scholar]
- Camus-Kulandaivelu L, Veyrieras J-B, Madur D, Combes V, Fourmann M, Barraud S, Dubreuil P, Gouesnard B, Manicacci D, Charcosset A. Maize adaptation to temperate climate: relationship between population structure and polymorphism in the Dwarf8 Gene. Genetics. 2006;172(4):2449–2463. doi: 10.1534/genetics.105.048603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Zhao YL, Gao LF, Zhao GY, Zhou RH, Zhang BS, Jia JZ. TaCKX6-D1, the ortholog of rice OsCKX2, is associated with grain weight in hexaploid wheat. New Phytol. 2012;195(3):574–584. doi: 10.1111/j.1469-8137.2012.04194.x. [DOI] [PubMed] [Google Scholar]
- Wang E, Wang J, Zhu X, Hao W, Wang L, Li Q, Zhang L, He W, Lu B, Lin H. et al. Control of rice grain-filling and yield by a gene with a potential signature of domestication. Nat Genet. 2008;40(11):1370–1374. doi: 10.1038/ng.220. [DOI] [PubMed] [Google Scholar]
- Ma X, Cheng Z, Qin R, Qiu Y, Heng Y, Yang H, Ren Y, Wang X, Bi J, Ma X. et al. OsARG encodes an arginase that plays critical roles in panicle development and grain production in rice. Plant J. 2013;73(2):190–200. doi: 10.1111/j.1365-313x.2012.05122.x. [DOI] [PubMed] [Google Scholar]
- Cheng WH, Taliercio EW, Chourey PS. The miniature1 seed locus of maize encodes a cell wall invertase required for normal development of endosperm and maternal cells in the pedicel. Plant Cell. 1996;8(6):971–983. doi: 10.1105/tpc.8.6.971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song XJ, Huang W, Shi M, Zhu MZ, Lin HX. A QTL for rice grain width and weight encodes a previously unknown RING-type E3 ubiquitin ligase. Nat Genet. 2007;39(5):623–630. doi: 10.1038/ng2014. [DOI] [PubMed] [Google Scholar]
- Li Q, Li L, Yang X, Warburton M, Bai G, Dai J, Li J, Yan J. Relationship, evolutionary fate and function of two maize co-orthologs of rice GW2 associated with kernel size and weight. BMC Plant Biol. 2010;10(1):143. doi: 10.1186/1471-2229-10-143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashikari M, Sakakibara H, Lin S, Yamamoto T, Takashi T, Nishimura A, Angeles ER, Qian Q, Kitano H, Matsuoka M. Cytokinin oxidase regulates rice grain production. Science. 2005;309(5735):741–745. doi: 10.1126/science.1113373. [DOI] [PubMed] [Google Scholar]
- Wu CY, Trieu A, Radhakrishnan P, Kwok SF, Harris S, Zhang K, Wang J, Wan J, Zhai H, Takatsuto S. et al. Brassinosteroids regulate grain filling in rice. Plant Cell. 2008;20(8):2130–2145. doi: 10.1105/tpc.107.055087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roitsch T, Ehneß R. Regulation of source/sink relations by cytokinins. Plant Growth Regul. 2000;32(2):359–367. [Google Scholar]
- Cheikh N, Jones RJ. Disruption of maize kernel growth and development by heat stress (role of cytokinin/abscisic acid balance) Plant Physiol. 1994;106(1):45–51. doi: 10.1104/pp.106.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brugiere N, Jiao S, Hantke S, Zinselmeier C, Roessler JA, Niu X, Jones RJ, Habben JE. Cytokinin oxidase gene expression in maize is localized to the vasculature, and is induced by cytokinins, abscisic acid, and abiotic stress. Plant Physiol. 2003;132(3):1228–1240. doi: 10.1104/pp.102.017707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vyroubalová Š, Vaclaviková K, Turečková V, Novák O, Smehilová M, Hluska T, Ohnoutková L, Frébort I, Galuszka P. Characterization of new maize genes putatively involved in cytokinin metabolism and their expression during osmotic stress in relation to cytokinin levels. Plant Physiol. 2009;151(1):433–447. doi: 10.1104/pp.109.142489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brugière N, Humbert S, Rizzo N, Bohn J, Habben J. A member of the maize isopentenyl transferase gene family, Zea mays isopentenyl transferase 2(ZmIPT2), encodes a cytokinin biosynthetic enzyme expressed during kernel development. Plant Mol Biol. 2008;67(3):215–229. doi: 10.1007/s11103-008-9312-x. [DOI] [PubMed] [Google Scholar]
- Morris RO, Bilyeu KD, Laskey JG, Cheikh NN. Isolation of a gene encoding a glycosylated cytokinin oxidase from maize. Biochem Biophys Res Commun. 1999;255(2):328–333. doi: 10.1006/bbrc.1999.0199. [DOI] [PubMed] [Google Scholar]
- Houba-Hérin N, Pethe C, D’Alayer J, Laloue M. Cytokinin oxidase from Zea mays: purification, cDNA cloning and expression in moss protoplasts. Plant J. 1999;17(6):615–626. doi: 10.1046/j.1365-313X.1999.00408.x. [DOI] [PubMed] [Google Scholar]
- Massonneau A, Houba-Hérin N, Pethe C, Madzak C, Falque M, Mercy M, Kopecny D, Majira A, Rogowsky P, Laloue M. Maize cytokinin oxidase genes: differential expression and cloning of two new cDNAs. J Exp Bot. 2004;55(408):2549–2557. doi: 10.1093/jxb/erh274. [DOI] [PubMed] [Google Scholar]
- Šmehilová M, Galuszka P, Bilyeu KD, Jaworek P, Kowalska M, Šebela M, Sedlářová M, English JT, Frébort I. Subcellular localization and biochemical comparison of cytosolic and secreted cytokinin dehydrogenase enzymes from maize. J Exp Bot. 2009;60(9):2701–2712. doi: 10.1093/jxb/erp126. [DOI] [PubMed] [Google Scholar]
- Xie C, Zhang S, Li M, Li X, Hao Z, Bai L, Zhang D, Liang Y. Inferring genome ancestry and estimating molecular relatedness among 187 Chinese maize inbred lines. J Genet Genomics. 2007;34(8):738–748. doi: 10.1016/S1673-8527(07)60083-6. [DOI] [PubMed] [Google Scholar]
- Kamada-Nobusada T, Sakakibara H. Molecular basis for cytokinin biosynthesis. Phytochemistry. 2009;70(4):444–449. doi: 10.1016/j.phytochem.2009.02.007. [DOI] [PubMed] [Google Scholar]
- Weng J, Xie C, Hao Z, Wang J, Liu C, Li M, Zhang D, Bai L, Zhang S, Li X. Genome-wide association study identifies candidate genes that affect plant height in Chinese elite maize (Zea mays L.) Inbred Lines. PLoS ONE. 2011;6(12):e29229. doi: 10.1371/journal.pone.0029229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Yang X, Bai G, Warburton M, Mahuku G, Gore M, Dai J, Li J, Yan J. Cloning and characterization of a putative GS3 ortholog involved in maize kernel development. TAG Theor Appl Genet. 2010;120(4):753–763. doi: 10.1007/s00122-009-1196-x. [DOI] [PubMed] [Google Scholar]
- Buckler ES, Holland JB, Bradbury PJ, Acharya CB, Brown PJ, Browne C, Ersoz E, Flint-Garcia S, Garcia A, Glaubitz JC. et al. The genetic architecture of maize flowering time. Science. 2009;325:714–718. doi: 10.1126/science.1174276. [DOI] [PubMed] [Google Scholar]
- Tian F, Bradbury PJ, Brown PJ, Hung H, Sun Q, Flint-Garcia S, Rocheford TR, McMullen MD, Holland JB, Buckler ES. Genome-wide association study of leaf architecture in the maize nested association mapping population. Nat Genet. 2011;43:159–162. doi: 10.1038/ng.746. [DOI] [PubMed] [Google Scholar]
- Poland JA, Bradbury PJ, Buckler ES, Nelson RJ. Genome-wide nested association mapping of quantitative resistance to northern leaf blight in maize. Proc Natl Acad Sci USA. 2011;108:6893–6898. doi: 10.1073/pnas.1010894108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng J, Liu X, Wang Z, Wang J, Zhang L, Hao Z, Xie C, Li M, Zhang D, Bai L. et al. Molecular mapping of the major resistance quantitative trait locus qHS2.09 with simple sequence repeat and single nucleotide polymorphism markers in maize. Phytopathology. 2012;102(7):692–699. doi: 10.1094/PHYTO-12-11-0330. [DOI] [PubMed] [Google Scholar]
- Kump KL, Bradbury PJ, Wisser RJ, Buckler ES, Belcher AR, Oropeza-Rosas MA, Zwonitzer JC, Kresovich S, McMullen MD, Ware D. et al. Genome-wide association study of quantitative resistance to southern leaf blight in the maize nested association mapping population. Nat Genet. 2011;43:163–168. doi: 10.1038/ng.747. [DOI] [PubMed] [Google Scholar]
- Yan J, Kandianis CB, Harjes CE, Bai L, Kim E-H, Yang X, Skinner DJ, Fu Z, Mitchell S, Li Q. et al. Rare genetic variation at Zea mays crtRB1 increases β-carotene in maize grain. Nat Genet. 2010;42(4):322–327. doi: 10.1038/ng.551. [DOI] [PubMed] [Google Scholar]
- Thornsberry J, Goodman M, Doebley J, Kresovich S, Nielsen D, Buckler E. Dwarf8 polymorphisms associate with variation in flowering time. Nat Genet. 2001;28(3):286–289. doi: 10.1038/90135. [DOI] [PubMed] [Google Scholar]
- Andersen JR, Schrag T, Melchinger AE, Zein I, Lübberstedt T. Validation of Dwarf8 polymorphisms associated with flowering time in elite European inbred lines of maize (Zea mays L.) TAG Theor Appl Genet. 2005;111(2):206–217. doi: 10.1007/s00122-005-1996-6. [DOI] [PubMed] [Google Scholar]
- Li J, Zhang Z, Li Y, Wang Q, Zhou Y. QTL consistency and meta-analysis for grain yield components in three generations in maize. TAG Theor Appl Genet. 2011;122(4):771–782. doi: 10.1007/s00122-010-1485-4. [DOI] [PubMed] [Google Scholar]
- Su Z, Hao C, Wang L, Dong Y, Zhang X. Identification and development of a functional marker of TaGW2 associated with grain weight in bread wheat (Triticum aestivum L.) TAG Theor Appl Genet. 2011;122(1):211–223. doi: 10.1007/s00122-010-1437-z. [DOI] [PubMed] [Google Scholar]
- Tian Z, Qian Q, Liu Q, Yan M, Liu X, Yan C, Liu G, Gao Z, Tang S, Zeng D. et al. Allelic diversities in rice starch biosynthesis lead to a diverse array of rice eating and cooking qualities. Proc Natl Acad Sci USA. 2009;106(51):21760–21765. doi: 10.1073/pnas.0912396106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liang C, Xu Y, Gianfagna T, Huang B. Effects of ipt gene expression on leaf senescence induced by nitrogen or phosphorus seficiency in creeping bentgrass. J Am Soc Hortic Sci. 2010;135(2):108–115. [Google Scholar]
- Wang G, Romheld V, Li C, Bangerth F. Involvement of auxin and CKs in boron deficiency induced changes in apical dominance of pea plants (Pisum sativum L.) J Plant Physiol. 2006;163(6):591–600. doi: 10.1016/j.jplph.2005.09.014. [DOI] [PubMed] [Google Scholar]
- Kiba T, Kudo T, Kojima M, Sakakibara H. Hormonal control of nitrogen acquisition: roles of auxin, abscisic acid, and cytokinin. J Exp Bot. 2011;62(4):1399–1409. doi: 10.1093/jxb/erq410. [DOI] [PubMed] [Google Scholar]
- Mok DW, Mok MC. Cytokinin metabolism and action. Annu Rev Plant Physiol Plant Mol Biol. 2001;52(1):89–118. doi: 10.1146/annurev.arplant.52.1.89. [DOI] [PubMed] [Google Scholar]
- Sakamoto T, Sakakibara H, Kojima M, Yamamoto Y, Nagasaki H, Inukai Y, Sato Y, Matsuoka M. Ectopic expression of KNOTTED1-like homeobox protein induces expression of cytokinin biosynthesis genes in rice. Plant Physiol. 2006;142(1):54–62. doi: 10.1104/pp.106.085811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye C, Wu S, Kong F, Zhou C, Yang Q, Sun Y, Wang B. Identification and characterization of an isopentenyltransferase (IPT) gene in soybean (Glycine max L.) Plant Sci. 2006;170(3):542–550. doi: 10.1016/j.plantsci.2005.10.008. [DOI] [Google Scholar]
- Kakimoto T. Identification of plant cytokinin biosynthetic enzymes as dimethylallyl diphosphate:ATP/ADP isopentenyltransferases. Plant Cell Physiol. 2001;42(7):677–685. doi: 10.1093/pcp/pce112. [DOI] [PubMed] [Google Scholar]
- Takei K, Sakakibara H, Sugiyama T. Identification of genes encoding adenylate isopentenyltransferase, a cytokinin biosynthesis enzyme, in Arabidopsis thaliana. J Biol Chem. 2001;276(28):26405–26410. doi: 10.1074/jbc.M102130200. [DOI] [PubMed] [Google Scholar]
- Zubko E, Adams CJ, Macháèková I, Malbeck J, Scollan C, Meyer P. Activation tagging identifies a gene from Petunia hybrida responsible for the production of active cytokinins in plants. Plant J. 2002;29(6):797–808. doi: 10.1046/j.1365-313X.2002.01256.x. [DOI] [PubMed] [Google Scholar]
- Sakano Y, Okada Y, Matsunaga A, Suwama T, Kaneko T, Ito K, Noguchi H, Abe I. Molecular cloning, expression, and characterization of adenylate isopentenyltransferase from hop (Humulus lupulus L.) Phytochemistry. 2004;65(17):2439–2446. doi: 10.1016/j.phytochem.2004.08.006. [DOI] [PubMed] [Google Scholar]
- Tanaka M, Takei K, Kojima M, Sakakibara H, Mori H. Auxin controls local cytokinin biosynthesis in the nodal stem in apical dominance. Plant J. 2006;45(6):1028–1036. doi: 10.1111/j.1365-313X.2006.02656.x. [DOI] [PubMed] [Google Scholar]
- Wang R, Yu Y, Zhao J, Shi Y, Song Y, Wang T, Li Y. Population structure and linkage disequilibrium of a mini core set of maize inbred lines in China. TAG Theor Appl Genet. 2008;117(7):1141–1153. doi: 10.1007/s00122-008-0852-x. [DOI] [PubMed] [Google Scholar]
- Salamini F, Ozkan H, Brandolini A, Schafer-Pregl R, Martin W. Genetics and geography of wild cereal domestication in the near east. Nat Rev Genet. 2002;3(6):429–441. doi: 10.1038/nrg817. [DOI] [PubMed] [Google Scholar]
- Takano-Kai N, Jiang H, Kubo T, Sweeney M, Matsumoto T, Kanamori H, Padhukasahasram B, Bustamante C, Yoshimura A, Doi K. et al. Evolutionary history of GS3, a gene conferring grain length in rice. Genetics. 2009;182(4):1323–1334. doi: 10.1534/genetics.109.103002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng J, Gu S, Wan X, Gao H, Guo T, Su N, Lei C, Zhang X, Cheng Z, Guo X. et al. Isolation and initial characterization of GW5, a major QTL associated with rice grain width and weight. Cell Res. 2008;18(12):1199–1209. doi: 10.1038/cr.2008.307. [DOI] [PubMed] [Google Scholar]
- Murray MG, Thompson WF. Rapid isolation of high molecular weight plant DNA. Nucleic Acids Res. 1980;8:4321–4326. doi: 10.1093/nar/8.19.4321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozas J, Rozas R. DnaSP version 3: an integrated program for molecular population genetics and molecular evolution analysis. Bioinformatics. 1999;15(2):174–175. doi: 10.1093/bioinformatics/15.2.174. [DOI] [PubMed] [Google Scholar]
- Nei M. Molecular evolutionary genetics. New York: Columbia University Press; 1987. [Google Scholar]
- Bradbury PJ, Zhang Z, Kroon DE, Casstevens TM, Ramdoss Y, Buckler ES. TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics. 2007;23(19):2633–2635. doi: 10.1093/bioinformatics/btm308. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
3-D structure of ZmIPT2 predicted with the program Swiss-PdbViewer. Ser and Pro represent amino acids in ZmIPT2-T and ZmIPT2-C, respectively.
Purified recombinant proteins for enzyme activity determination.
The consumption of ATP, ADP, and AMP by ZmIPT2-C and ZmIPT2-T during in vitro enzyme activity determination.