

Abstract
The synthesis of new ribo and 2’-β-C-methyl ribo Janus type nucleosides J-AA, J-AG and J-AU is reported along with their ability to block HCV and HIV replication. Their toxicity was also assessed in Huh7, human lymphocytes, CEM and Vero cells
Keywords: HCV, Antiviral, prodrug, Janus
Hepatitis C virus (HCV), an enveloped single-stranded positive sense enveloped RNA virus discovered in 1989,1 is a leading cause of long term liver cirrhosis, resulting in liver transplantation, liver failure and hepatocellular carcinoma. 2 Globally, there are an estimated 170 million persons infected with the virus and 3 to 4 million persons are newly infected each year. Despite the existence of treatments involving pegylated interferon-α (IFN) and ribavirin (RBV), with or without protease inhibitors (PI) boceprevir (Victrelis) and telaprevir (Incivek),3 the limited efficacy and side effects of current therapies emphasize the need for additional improved therapeutic agents. Nucleoside inhibitors that target HCV NS5B polymerase have demonstrated clinical advantages of broader activity against various HCV genotypes and a higher barrier to the development of resistant viruses when compared to all other classes of HCV inhibitors.4 To date a number of 2’-modified nucleosides have shown potent activity against HCV (Figure 1).5,6 IDX-184 1, RO-5024048/RG-7128 2, and PSI-7977 (GS-7977) 3 are in advanced clinical trials as effective anti-HCV agents. Interestingly, highly base-modified tricyclic derivatives such as 47 and 58 have also shown potent anti-HCV activity.
Figure 1.
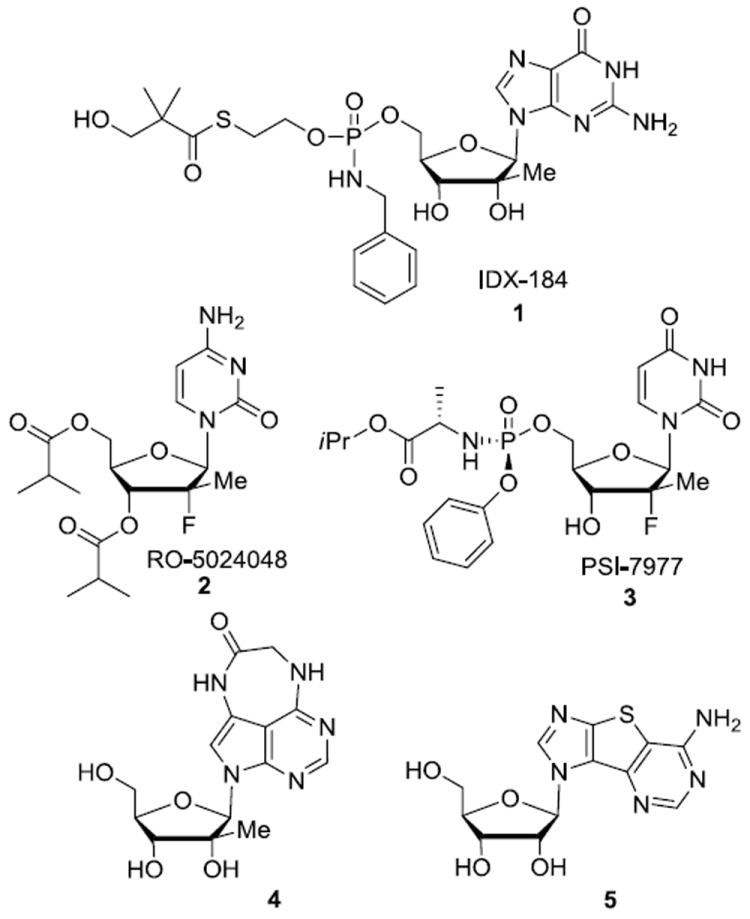
Nucleoside analogs exhibiting anti-HCV properties.
Based on these compounds and inspired by Townsend’s work on linear tricyclic nucleosides, 9 we prepared a series of new nucleosides with potential anti-HCV activity that may be viewed as possessing the feature of two completely different bases simultaneously (Figure 2). These dual bases or Janus type 10 nucleosides, such as J-GA (Figure 2), presenting one face with a Watson-Crick donor/acceptor array of a guanine and the other face with an array of an adenine, could in principle, by rotation of the glycosidic bond, pair with either a cytosine or a uracil. Thus, we report herein the synthesis and the biological evaluation of ribo and new 2’-β-C-methyl ribo Janus type nucleosides J-AA, J-AG and J-AU (Figure 2; R = H or Me).
Figure 2.
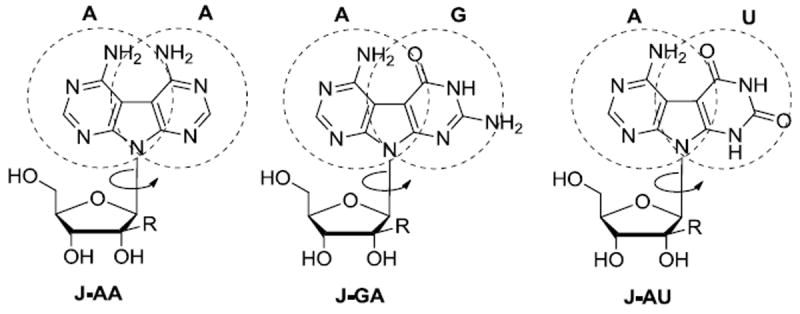
Janus type dual-base nucleosides
The Janus type nucleosides 12a, 14a and 19a were previously reported by Townsend,9 we have included our resynthesis of these nucleosides for comparison to the synthesis of the 2’-β-C-methyl ribo series and also to evaluate and compare both series for antiviral and cytotoxic activity. The synthesis of Janus type nucleosides 12a-b (J-AU) is summarized in Scheme 1. The Vorbr ggen coupling reaction between 4-amino-6-bromo-5-cyanopyrrolo[2,3-d]pyrimidine 611 and 1-O-acetyl-2,3,5-tri-O-benzoyl-β-D-ribofuranose 7a or 2’-β-C-methyl-1,2-di-O-benzoyl-3,5-di-O-toluyl-β-D-ribofuranose 7b12 using N,O-bis(trimethylsilyl)acetamide (BSA) as the silylation agent and TMSOTf as the Lewis acid provided compounds 8a and 8b in 71% and 65% respectively. 13 Compounds 8a-b were then deprotected in saturated methanolic ammonia to give 9a-b. Subsequent halogen displacement with liquid ammonia in a steel bomb was followed by treatment with hydroxylamine14 in ethanol which afforded compounds 11a-b in good yields. Finally, J-AU derivatives 12a-b15 were obtained in 51% and 46% yield, respectively, by treatment of amides 11a-b with diethyl carbonate in presence of sodium ethoxide. It is noteworthy that 1H NMR spectral data for Janus base nucleosides denote the presence of only one isomer (For compound 12b, 2’Me signal appears as a singlet), clearly indicating free rotation of the base.
Scheme 1.
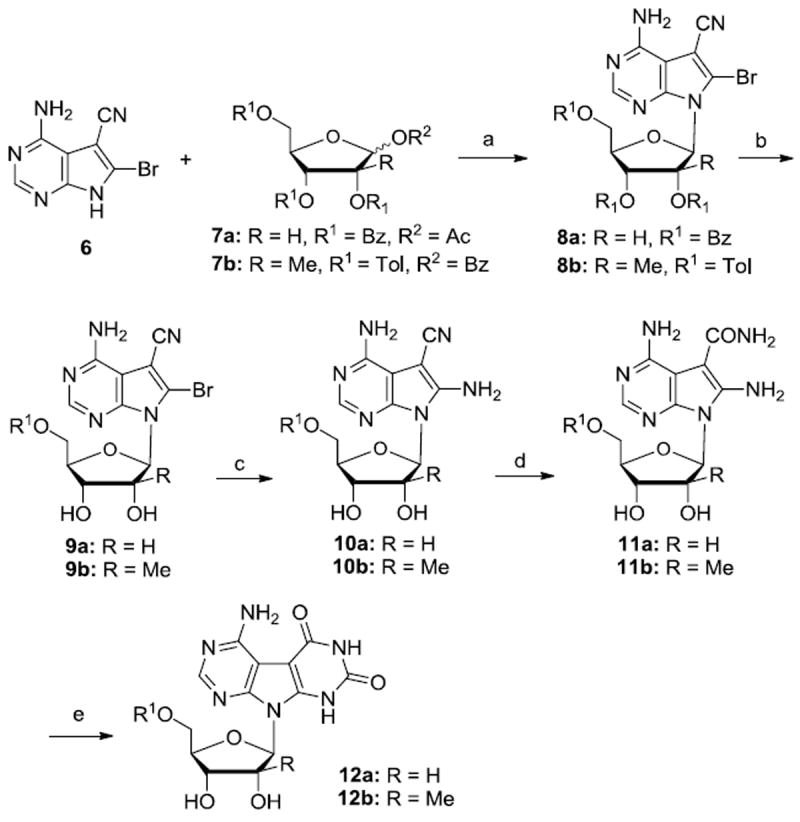
Reagents and conditions: (a) (i) 6, BSA, CH3CN, rt, 20 min; (ii) 7, TMSOTf, 80-85°C, 3 h, 8a: 71%, 8b: 65%; (b) NH3/MeOH, overnight, 9a:90%, 9b:92%; (c) NH3(l), 110 °C, 10a: 64%; 10b: 66%; (d) NH2OH, EtOH, reflux, 17 h, 11a: 68%; 11b: 81%; (e) (EtO)2CO, NaOEt, reflux, 30 h, 12a: 51%; 12b: 46%.
J-AA compounds 14a-b16 were easily prepared without isolation of intermediates by reaction of 10a-b with diethoxymethyl acetate followed by treatment with saturated methanolic ammonia solution followed by 25% aqueous acetic acid (Scheme 2).
Scheme 2.
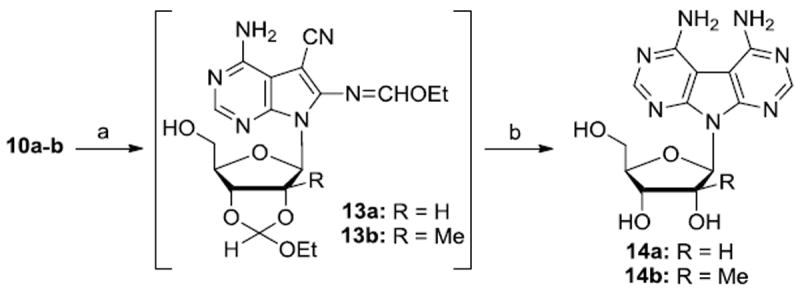
Reagents and conditions: (a) AcOCH(OEt)2, reflux, 2 h; (b) (i) NH3/MeOH, rt, 24 h; (ii) 25% HOAc, rt, 30 h for 13a, 72 h for 13b; (iii) 1N NaOH, 14a:33%; 14b:36%.
Finally, nucleosides J-AG 19a-b were prepared in 5 steps from intermediate 11a-b (Scheme 3). Thus, treatment of compounds 11a-b with carbon disulfide under basic conditions afforded, in quantitative manner, tricyclic sodium salts 15a-b, which were converted to their ammonium salt counterparts 17a-b after acidification and treatment with NH4OH. 17a-b were then oxidized using hydrogen peroxide and the resulting ammonium sulfonate intermediates 18a-b were subsequently reacted with ammonia in a steal vessel to give the desired J-AG nucleosides 19a-b.17
Scheme 3.
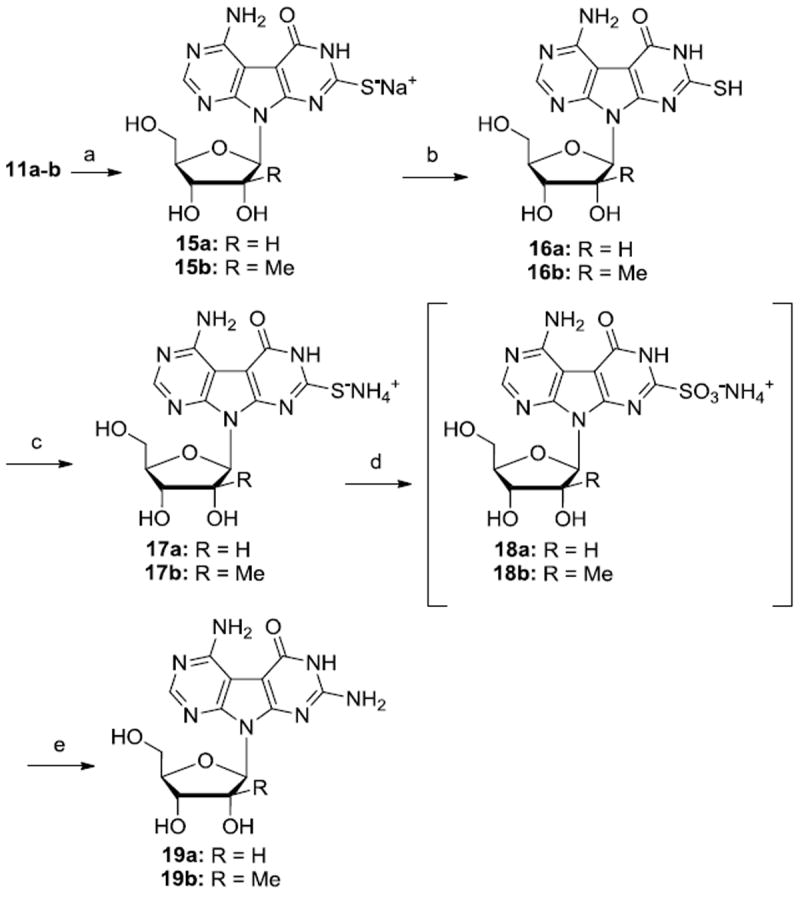
Reagents and conditions: (a) Carbon disulfide, NaOH, MeOH, 160-180 °C, 3 h, 15a: 100%; 15b: 100%; (b) 1N HCl, 16a: 62%; 16b: 51%; (c) NH4OH, rt; (d) H2O2, 0 °C;(e) NH3(l), 0 °C, then 120 °C, 3 h, over three steps, 19a: 40%; 19b: 35%.
Overall the presence of the 2’-C-Me group had little effect on the yield of the various reactions, however, in most cases there was a slight reduction in isolated yield. All synthesized tricyclic dual-base nucleosides 12a-b, 14a-b, 19a-b along with intermediates 9a-b, 10a-b, 11a-b were evaluated for inhibition of HCV RNA replication in Huh7 cells using a subgenomic HCV replicon system. 18 Cytotoxicity in Huh7 cells was determined simultaneously with anti-HCV activity by extraction and amplification of both HCV RNA and cellular ribosomal RNA (rRNA). 19 To determine the spectrum of activity of the compounds, anti-HIV activity was evaluated against HIV-1LAI in primary human peripheral blood mononuclear (PBM) cells and 3’-azido-3’-deoxythymidine (AZT) was used as a positive control. Cytotoxicity was determined in PBM, human lymphoblastoid CEM, and African Green monkey Vero cells (Table 1).20
Table 1.
In vitro antiviral activity and cytotoxicity of compounds 12a-b, 14a-b, 19a-b, 9a-b, 10a-b, 11a-b and phosphoramidate prodrug 24.a
| Cmpd | Janus Type | Anti-HCV activity (μM)
|
rRNA (μM)
|
Anti-HIV-1 activity (μM)
|
Cytotoxicity, CC50 (μM)
|
||||
|---|---|---|---|---|---|---|---|---|---|
| EC50 | EC90 | CC50b | EC50 | EC90 | PBM | CEM | Vero | ||
| 12a | J-AU | 5.7 | 9.4 | 5.7 | 6.8 | 16.5 | 51.8 | 7.9 | 13.4 |
| 12b | J-AU | > 10 | > 10 | > 10 | > 100 | > 100 | > 100 | >100 | > 100 |
| 14a | J-AA | > 10 | > 10 | > 10 | > 100 | > 100 | 53.8 | 29.5 | 82.5 |
| 14b | J-AA | > 10 | > 10 | > 10 | ≥100 | ≥100 | > 100 | >100 | > 100 |
| 19a | J-GA | 3 | 22.5 | < 10 | 9.4 | 24.0 | 3.1 | 20.2 | 63.1 |
| 19b | J-GA | ~ 3 | 17.9 | ~ 3 | 3.3 | > 100 | 92.1 | 36.5 | > 100 |
| 24 | NA | > 10 | >10 | >10 | ND | ND | ND | ND | ND |
| 9a | NA | > 10 | > 10 | > 10 | 2.7 | 16.2 | 10.5 | 65.1 | 87.3 |
| 9b | NA | > 10 | > 10 | > 10 | >100 | > 100 | > 100 | > 100 | > 100 |
| 10a | NA | 0.2 | 0.3 | 0.15 | 0.38 | 1.1 | 0.10 | ND | 0.96 |
| 10b | NA | 6.8 | 21.8 | ~ 10 | 24.7 | 56.0 | 4.9 | 10.0 | > 100 |
| 11a | NA | 0.5 | 2.4 | ~ 10 | 0.49 | 1.5 | 3.1 | <1.0 | 7.6 |
| 11b | NA | > 10 | > 10 | > 10 | > 100 | > 100 | > 100 | > 100 | > 100 |
| 16a | NA | > 10 | > 10 | > 10 | 9.6 | 30.2 | 2.9 | 8.2 | 43.1 |
| 16b | NA | > 10 | > 10 | > 10 | > 100 | > 100 | > 100 | > 100 | > 100 |
| AZT | NA | > 10 | > 10 | > 10 | 0.0040 | 0.028 | > 100 | 14.3 | 53.0 |
All values are based on mean of replicate assays (n ≥ 2)
CC50: cytotoxic concentration that reduced the rRNA levels by 50% at 120 h.
ND: not determined.
From among the prepared compounds, targeted Janus nucleosides compounds, 12a (J-AU) and 19a-b (J-GA) along with intermediates 10a-b and 11a showed HCV activities that were not differentiable from the toxicity observed in the replicon Huh7 cell line. The remaining three Janus compounds 12b (J-AU) and 14a-b (J-AA) and intermediates 9a-b, 11b, and 16a-b did not display any anti-HCV activity in our Huh7 based replicon system. A similar result was seen in the HIV assay with all compounds that displayed anti-HIV activity were also found to have cytotoxicity toward PBM cells (compound 10a was the most toxic with a CC50 in PBM cells of 0.1 μM).
Since the lack of antiviral activity or cytotoxicity observed for 12b and 14b may be due to inefficient uptake and/or their inability to be intracellularly metabolized to the corresponding nucleoside triphosphates, a J-AA McGuigan type phosphoramidate prodrug 24 was prepared (Scheme 4). We chose 24 for prodrug synthesis as the adenine groups are presented in a manner consistent with adenosine while the J-AU nucleoside 12b presents the uracil group in a manner inconsistent with uridine.
Scheme 4.
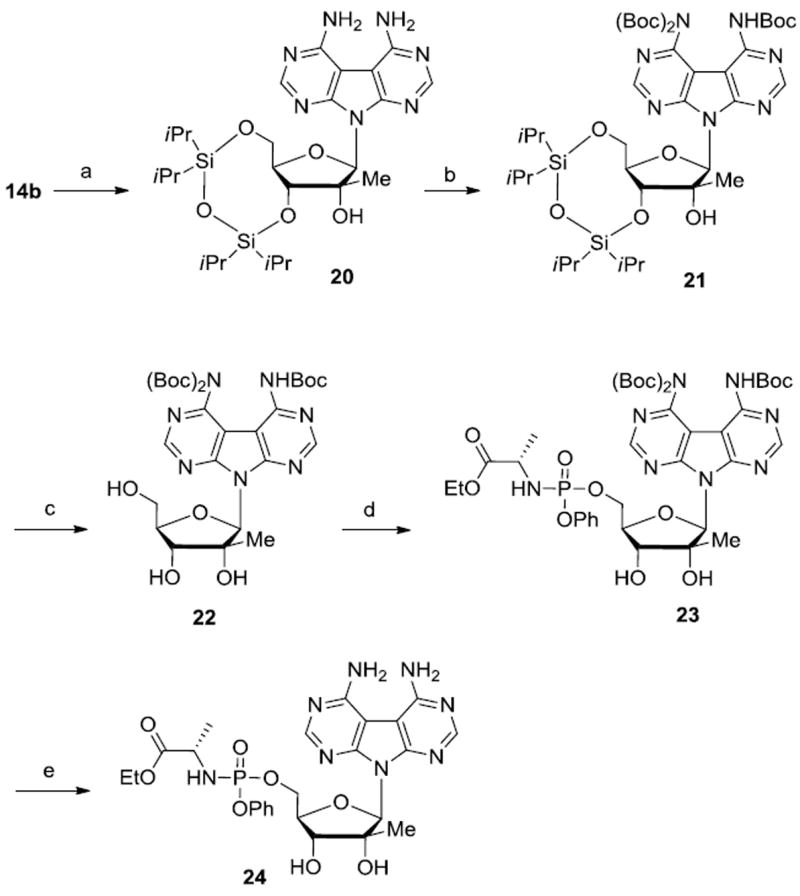
Reagents and conditions: (a) 1,3-dichloro-1,1,3,3-tetraisopropyldisiloxane, pyridine, rt, 72%; (b) (Boc)2O, DMAP, THF, rt, 30 h, 51%; (c) Et3N.3HF, THF, rt, 15 h, 55%; (d) phenyl(ethoxy-l-alaninyl)phosphorochloridate, t-BuMgCl, THF, rt, overnight; (e) 80% aq. TFA, rt, 2 h, 33% over two steps.
Attempts to synthesize prodrug 24 by direct coupling of nucleoside 14b with phenyl(ethoxy-l-alaninyl)phosphorochloridate in presence of either t-BuMgCl or NMI afforded only trace amount of the desired product. These difficulties lead us to envisage a temporarily protection of the exocyclic amino groups before coupling with the chlorophosphoramidate. Thus, compound 14b was reacted with 1,3-dichloro-1,1,3,3-tetraisopropyldisiloxane in pyridine to form 3’,5’-protected intermediate 20. Subsequently, the amino groups were protected with tert-butoxycarbonyl anhydride ((Boc)2O) to give the corresponding tri-N-Boc protected derivative 21. Interestingly, despite the use of a large excess of (Boc)2O, we were unable to introduce four Boc groups on compound 20. Treatment of 21 with Et3N.3HF provided N-Boc-protected nucleoside 22, which was then reacted with phenyl(ethoxy-l-alaninyl)phosphorochloridate in presence of t-BuMgCl to give phosphoramidate 23. Deprotection with 80% aqueous TFA at room temperature provided target monophosphate prodrug 24 in 33% yield over 2 steps. Unfortunately, monophosphate prodrug 24 did not display any inhibition of HCV RNA replication in the replicon system.
In conclusion, as a part of our drug discovery program, we have synthesized some ribo and novel 2’-C-Me tricyclic dual base nucleosides J-AU, J-AG and J-AA and a McGuigan type phosphoramidate prodrug of J-AA. Many of these Janus type nucleosides were found to be cytotoxic in multiple cell lines with only 2’-C-Me J-AA 12b and 2’-C-Me J-AG 14b being devoid of cytotoxicity in the four cell lines tested. Our studies did not reveal any anti-HCV or -HIV activity that was free from cytotoxicity indicating that our changes to the nucleoside base might have been too drastic for recognition by phosphorylation kinases and/or HCV NS5B polymerase.
Acknowledgments
This work was supported in part by NIH grant 5P30-AI-50409 (CFAR), 1R01-MH-100999 and by the Department of Veterans Affairs. Dr. Schinazi is the founder and a major shareholder of RFS Pharma, LLC. Emory received no funding from RFS Pharma, LLC to perform this work and vice versa.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Choo QL, Kuo G, Weiner AJ, Overby LR, Bradley DW, Houghton M. Science. 1989;244:359–362. doi: 10.1016/s0168-8278(02)00051-x. [DOI] [PubMed] [Google Scholar]
- 2.Kanwal F, Hoang T, Kramer JR, Asch SM, Goetz MB, Zeringue A, Richardson P, El-Serag HB. BMC Gastroenterol. 2011;140:1182. doi: 10.1053/j.gastro.2010.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sheridan C. Nature Biotech. 2011;29:553. doi: 10.1038/nbt0711-553. [DOI] [PubMed] [Google Scholar]
- 4.McCown MF, Rajyaguru S, Le Pogam S, Ali S, Jiang WR, Kang H, Symons J, Cammack N, Najera I. Antimicrob Agents Chemother. 2008;52:1604. doi: 10.1128/AAC.01317-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(a) Eldrup AB, Prhavc M, Brooks J, Bhat B, Prakash TP, Song Q, Bera S, Bhat N, Dande P, Cook PD, Bennett CF, Carroll SS, Ball RG, Bosserman M, Burlein C, Colwell LF, Fay JF, Flores OA, Getty K, LaFemina RL, Leone J, MacCoss M, McMasters DR, Tomassini JE, Von Langen D, Wolanski B, Olsen DB. J Med Chem. 2004;47:5284. doi: 10.1021/jm040068f. [DOI] [PubMed] [Google Scholar]; (b) Clark JL, Hollecker L, Mason JC, Stuyver LJ, Tharnish PM, Lostia S, McBrayer TR, Schinazi RF, Watanabe KA, Otto MJ, Furman PA, Stec WJ, Patterson SE, Pankiewicz KW. J Med Chem. 2005;48:5504. doi: 10.1021/jm0502788. [DOI] [PubMed] [Google Scholar]
- 6.For a recent review of NS5B anti-HCV agents, see: Sofia MJ, Chang W, Furman PA, Mosley RT, Ross BS. J Med Chem. 2012;55:2481. doi: 10.1021/jm201384j.
- 7.Keicher JD, Roberts CD, Liehr SJR, Zheng X, Prhavc M, Rajwanshi VK, Grifftth RC, Kim CU. WO2006093987 PCT.
- 8.Seley-Radtke KL, Zhang Z, Wauchope OR, Zimmermann S, Ivanov A, Korba BE. Nucleic Acids Symp. 2008;52:635–636. doi: 10.1093/nass/nrn321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chung F-L, Schram KH, Panzica RP, Earl RA, Wotring LL, Townsend LB. J Med Chem. 1980;23:1158. doi: 10.1021/jm00185a002. [DOI] [PubMed] [Google Scholar]
- 10.Inspired by Janus, the Roman god of gates and doors, beginnings and endings, and hence represented with a double-faced head, each looking in opposite directions.
- 11.Tolman RL, Robins RK, Townsend LB. J Am Chem Soc. 1969;91:2102. doi: 10.1021/ja01036a040. [DOI] [PubMed] [Google Scholar]
- 12.Franchetti P, Cappellacci L, Marchetti S, Trincavelli L, Martini C, Mazzoni MR, Lucacchini A, Grfantini M. J Med Chem. 1998;41:1708. doi: 10.1021/jm9707737. [DOI] [PubMed] [Google Scholar]
- 13.(a) Wang G, Tam RC, Gunic E, Du J, Bard J, Pai B. J Med Chem. 2000;43:2566. doi: 10.1021/jm000035+. [DOI] [PubMed] [Google Scholar]; (b) Ding Y, An H, Hong Z, Girardet J-L. Bioorg Med Chem Lett. 2005;15:725. doi: 10.1016/j.bmcl.2004.11.019. [DOI] [PubMed] [Google Scholar]
- 14.Hurd CD. Inorg Synth. 1939;1:87. [Google Scholar]
- 15.Selected spectral data for compound 12b: 1H-NMR (400 MHz, DMSO-d6) δ: 0.74 (s, 3H), 3.16 (d, 2H, J = 5.2 Hz), 3.71-3.90 (m, 3H), 4.09-4.13 (m, 1H), 5.19 (br s, 1H), 5.43 (br s, 1H), 6.33 (s, 1H), 7.43 (br s, 1H), 7.51 (br s, 1H), 8.15 (s, 1H), 11.29 (br s, 1H), 12.03 (br s, 1H). LC/MS (m/z), calcd for C14H16N6O6 (M++H), 364.1; found, 365.2.
- 16.Selected spectral data for compound 14b: 1H-NMR (400 MHz, DMSO-d6) δ: 0.82 (s, 3H, CH3), 3.76-3.78 (m, 2H, H5’), 3.90-3.93 (m, 1H, H4’), 4.61-4.65 (m, 1H, H3’), 4.99 (br s, 2H, 2× OH), 5.20 (d, 1H, J = 7.2 Hz, OH), 6.55 (s, 1H, H1’), 6.88 (br s, 4H, 2× NH2), 8.32 (s, 2H, ArH). LC/MS (m/z), calcd for C14H17N7O4 (M++H), 348.1; found, 348.2.
- 17.Selected spectral data for compound 19b: 1H-NMR (400 MHz, CD3OD) δ: 0.98 (s, 3H), 3.99-4.08 (m, 3H), 4.61 (d, 1H, J = 8.8 Hz), 6.47 (s, 1H), 8.19 (s, 1H). LC/MS (m/z), calcd for C14H17N7O5 (M++H), 363.1; found, 364.1.
- 18.Rondla R, Coats SJ, McBrayer TR, Grier J, Johns M, Tharnish PM, Whitaker T, Zhou L-H, Schinazi RF. Antivir Chem Chemother. 2009;20:99. doi: 10.3851/IMP1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stuyver LJ, Whitaker T, McBrayer TR, Hernandez-Santiago BI, Lostia S, Tharnish PM, Ramesh M, Chu CK, Jordan R, Shi J, Rachakonda S, Watanabe KA, Otto MJ, Schinazi RF. Antimicrob Agents Chemother. 2003;47:244. doi: 10.1128/AAC.47.1.244-254.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.(a) Schinazi RF, Sommadossi JP, Saalmann V, Cannon DL, Xie M-W, Hart GC, Smith GA, Hahn EF. Antimicrob Agents Chemother. 1990;34:1061. doi: 10.1128/aac.34.6.1061. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Stuyver LJ, Lostia S, Adams M, Mathew J, Pai BS, Grier J, Tharnish P, Choi Y, Chong Y, Choo H, Chu CK, Otto MJ, Schinazi RF. Antimicrob Agents Chemother. 2002;46:3854. doi: 10.1128/AAC.46.12.3854-3860.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]