

Abstract
Narcolepsy is characterized by excessive sleepiness and cataplexy, sudden episodes of muscle weakness during waking that are thought to be an intrusion of rapid eye movement sleep muscle atonia into wakefulness. One of the most striking aspects of cataplexy is that it is often triggered by strong, generally positive emotions, but little is known about the neural pathways through which positive emotions trigger muscle atonia. We hypothesized that the amygdala is functionally important for cataplexy because the amygdala has a role in processing emotional stimuli and it contains neurons that are active during cataplexy. Using anterograde and retrograde tracing in mice, we found that GABAergic neurons in the central nucleus of the amygdala heavily innervate neurons that maintain waking muscle tone such as those in the ventrolateral periaqueductal gray, lateral pontine tegmentum, locus ceruleus, and dorsal raphe. We then found that bilateral, excitotoxic lesions of the amygdala markedly reduced cataplexy in orexin knock-out mice, a model of narcolepsy. These lesions did not alter basic sleep–wake behavior but substantially reduced the triggering of cataplexy. Lesions also reduced the cataplexy events triggered by conditions associated with high arousal and positive emotions (i.e., wheel running and chocolate). These observations demonstrate that the amygdala is a functionally important part of the circuitry underlying cataplexy and suggest that increased amygdala activity in response to emotional stimuli could directly trigger cataplexy by inhibiting brainstem regions that suppress muscle atonia.
Introduction
Narcolepsy is caused by loss of the hypothalamic neurons that produce the orexin neuropeptides (Peyron et al., 2000; Thannickal et al., 2000). Loss of these cells or loss of just the orexin peptides results in severe sleepiness and cataplexy, sudden episodes of muscle weakness during waking. In people with narcolepsy, cataplexy is most often triggered by positive emotions such as those associated with laughter, joking, or delight (Overeem et al., 1999, 2011). Similarly, in narcoleptic dogs, cataplexy is usually triggered by palatable food or play (Siegel et al., 1986, 1991), and in mouse models of narcolepsy, cataplexy is increased by rewarding stimuli such as wheel running and palatable food (Espana et al., 2007; Clark et al., 2009). For >130 years, this striking and odd connection to positive emotions has been recognized as a key aspect of cataplexy (Gelineau, 1880; Schenck et al., 2007), but the neural mechanisms through which positive emotions trigger cataplexy remain unknown.
Several lines of evidence suggest that the amygdala could be a key site through which emotions trigger cataplexy. There is clear evidence that the amygdala is important for responses to positive stimuli across species; for example, in humans, amygdala activity increases in response to positive affective stimuli (Garavan et al., 2001; Straube et al., 2008; Davey et al., 2011), and in nonhuman primates, amygdala neurons encode the positive value of conditioned images (Nishijo et al., 1988). In rats, amygdala neurons encode positive stimulus associations (Paton et al., 2006), and amygdala-lesioned rats fail to approach stimuli of positive affective valence (Schoenbaum et al., 1998). Anatomically, the amygdala is well positioned to influence muscle tone and rapid eye movement (REM) sleep phenomena because the central nucleus of the amygdala (CeA) in rats innervates regions in the pons that regulate REM sleep (Boissard et al., 2003; Fung et al., 2011). In addition, Gulyani et al. (2002) recorded from the amygdala of freely behaving narcoleptic dogs and found many neurons with increased activity during cataplexy. These cells often showed a sharp increase in firing at the onset of cataplexy and then a return to baseline just as muscle tone recovered, suggesting that they may be part of the mechanism that triggers cataplexy.
We hypothesized that the amygdala is an important part of the circuitry that triggers cataplexy. We first examined whether the amygdala innervates key regions of the pons that regulate muscle tone in mice. Then, to test whether the amygdala is functionally important for cataplexy, we produced bilateral, excitotoxic lesions of the amygdala in orexin knock-out (KO) mice and examined their effect on cataplexy under baseline conditions and those likely to elicit positive emotions.
Materials and Methods
These studies were approved by the Institutional Animal Care and Use Committees of Beth Israel Deaconess Medical Center and Harvard Medical School and were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Animals.
For anatomic experiments, we used wild-type (WT) (n = 3) mice as well as mice expressing Cre only in neurons with the vesicular GABA transporter (VGAT–Cre mice) (n = 3) (Vong et al., 2011). For the lesion experiments, we used 32 male orexin KO mice, 12–22 weeks old and weighing 26–34 g. Founder orexin KO mice were a kind gift from M. Yanagisawa (University of Texas Southwestern, Dallas, TX) and were then backcrossed to C57BL/6J mice for over 10 generations (Mochizuki et al., 2004).
Surgery.
We anesthetized mice with ketamine–xylazine (100 and 10 mg/kg, i.p.) and placed them in a stereotaxic alignment system (model 1900; David Kopf Instruments). Using an air pressure injection system and glass micropipette (∼10 μm tip diameter), we bilaterally injected ibotenic acid (5% in PBS; 25–50 nl injected over 3–5 min) into the area of the CeA (1.35 mm posterior to bregma, ±2.75 mm lateral, and 4.5 mm ventral). We microinjected the amygdalae of controls with an equal volume of sterile PBS.
We then implanted mice with electrodes for recording the electroencephalogram (EEG) and electromyogram (EMG). In brief, stainless steel screws were implanted for frontoparietal EEG recordings (1.5 mm lateral and 1 mm anterior to bregma; 1.5 mm lateral and 3 mm posterior to bregma). EMG electrodes were made from fine, multi-stranded stainless steel wire (AS131; Cooner Wire), which were sutured into the neck extensor muscles. All electrodes were attached to a micro-strip connector affixed to the animal's head with dental cement. After surgery, mice were given 0.5 ml of 0.9% saline and meloxicam (5 mg/kg, i.p.).
Experimental protocol.
Two weeks after surgery, we transferred mice to recording cages in a sound-attenuated chamber with a 12 h light/dark cycle (30 lux daylight-type fluorescent tubes with lights on at 7:00 A.M.), constant temperature (23 ± 1°C), and food and water available ad libitum. The recording cable was attached to a low-torque electrical swivel, fixed above the cages that allowed free movement. Mice habituated to the cables for 4 d before the experiments began and remained connected throughout the study.
Mice were recorded during the 2.5–4 week period after excitotoxic or sham lesions. We first examined baseline sleep–wake behavior and cataplexy across 24 h using EEG, EMG, and infrared video recordings. We then studied mice under two conditions that should increase cataplexy: (1) access to a running wheel and (2) access to a running wheel plus chocolate. We placed a low-torque, polycarbonate running wheel (Fast-Trac; Bio-Serv) in each cage and recorded wheel rotations using a photodetector beneath each wheel. Running wheels increase cataplexy in orexin KO mice (Espana et al., 2007), and we chose this style of wheel because it does not interfere with the EEG recording cable. After 7 d habituation to the wheel, we recorded sleep–wake behavior and wheel rotations for 24 h. The next night, we gave mice 3 g of Hershey's milk chocolate at dark onset and recorded sleep–wake behavior and wheel running over the next 12 h (7:00 P.M. to 7:00 A.M.). We chose to use chocolate because it is used as a reward in rodent operant studies (Holahan et al., 2011; King et al., 2011), and cataplexy in mice and dogs is increased by palatable foods (Siegel et al., 1986; Clark et al., 2009).
Data acquisition and analyses.
EEG/EMG signals were acquired using Grass Instruments model 12 amplifiers and digitized at 256 Hz. Signals were digitally filtered (EEG, 0.3–30 Hz; EMG, 20–100 Hz) using SleepSign (Kissei Comtec). We manually scored behavior as wake, non-REM sleep, REM sleep, or cataplexy in 10 s epochs. Behavior was scored as cataplexy if the mouse had one or more epochs of muscle atonia accompanied by EEG theta that was preceded by at least 40 s of active wakefulness and was also followed by wakefulness (Scammell et al., 2009). Each potential episode of cataplexy was examined using infrared video recordings to ensure that the mouse was immobile.
Histology.
After recordings, we anesthetized mice with ketamine–xylazine (200 and 20 mg/kg, i.p.) and transcardially perfused them with 0.1 m PBS, followed by 10% Formalin. We postfixed brains in Formalin for 24 h and then cryoprotected them in 20% sucrose for 2 d. We coronally sectioned brains at 40 μm in a 1:3 series using a microtome. We stained one series with thionin and mapped the lesions on standard brain atlas maps (Paxinos and Franklin, 2001). Criteria for inclusion in the lesion group (n = 9) were symmetrical, bilateral lesions of the CeA. All lesions also produced some injury to the basolateral nucleus of the amygdala, and the largest lesions encompassed the entirety of the amygdala with slight injury to the piriform cortex and ventral regions of the caudate–putamen. There were no obvious behavioral differences between mice with small versus large lesions. We excluded 15 mice that received ibotenic acid injections because their lesions were off target. Six of the excluded mice had approximately unilateral lesions (i.e., a unilateral amygdala lesion and a unilateral miss, such that the overall lesioned area was approximately equivalent to bilaterally lesioned mice), but they had no change in sleep–wake behavior (data not shown) or cataplexy (baseline, 2.7 ± 0.2% of dark period; wheel running, 3.3 ± 0.6%; wheel running and chocolate, 6.6 ± 0.6%) compared with control mice.
Anterograde and retrograde tracing.
How amygdala circuitry regulates cataplexy is unknown. As a first step in identifying these pathways, we mapped connections between the amygdala and brainstem regions the regulate muscle tone. Under ketamine–xylazine anesthesia, we injected the retrograde tracer cholera toxin subunit B (CTB) (0.2%, 18 nl) into the ventrolateral periaqueductal gray/lateral pontine tegmentum (vlPAG/LPT) of WT mice to identify neurons projecting to the vlPAG/LPT. We also labeled another series for the VGAT mRNA using in situ hybridization to identify inhibitory neurons projecting to this atonia-suppressing region (Boissard et al., 2003). In addition, we microinjected an adeno-associated viral vector coding for channelrhodopsin-2 fused to mCherry (AAV–FLEX–ChR2–mCherry; 4 × 1012 particle/ml, 60 nl) into the amygdala of VGAT–Cre mice to anterogradely label GABAergic projections of the central nucleus.
Three weeks later, we perfused the mice and cut brain sections as above. For immunostaining, we placed sections in 0.3% hydrogen peroxide for 30 min and then incubated them overnight in primary antibody at room temperature. Primary antibodies included goat anti-CTB (1:15,000; List Biological), rabbit anti-DsRed (to label mCherry; 1:5000; Clontech), and rabbit anti-orexin-A (1:5000; Abcam). We then incubated sections with biotinylated secondary antibody (1:1000; Vector Laboratories) for 90 min, followed by horseradish peroxidase-conjugated avidin–biotin complex (1:1000; Vector Laboratories) for 1 h. We visualized immunoreactivity using 0.06% 3,3′-diaminobenzidine and 0.01% hydrogen peroxide. For double labeling of CTB and orexin, we used Alexa Fluor 555-conjugated anti-goat IgG antibody (1:1000; Invitrogen) and Alexa Fluor 488-conjugated anti-rabbit IgG antibody (1:1000; Invitrogen) as secondary antibodies.
For in situ hybridization, we generated a 919 bp riboprobe complementary to bases 876 to 1794 of the VGAT mRNA (GenBank accession number NM_009508). Using tail genomic DNA as a template, we amplified the probe fragment by PCR (forward, GCATGTTCGTGCTGGGCCTACC; reverse, CAGCGCAGCGTCAGCCCCCAG). Next, we added T7/SP6 promoter sites to the fragment, purified the DNA fragment (Min Elute PCR Purification Kit; Qiagen), and used T7 polymerase to generate a DIG-labeled antisense probe. We then incubated sections with 2 μg/ml VGAT riboprobe overnight at 50°C, incubated with 50 μg/ml RNase A for 30 min at 37°C, washed in decreasing concentrations of SSC buffer, incubated with peroxidase conjugated anti-DIG antibody (1:200; Roche) overnight, and reacted with tyramide-conjugated Cy3 (PerkinElmer Life and Analytical Sciences) for 30 min. For the combination of VGAT in situ hybridization and CTB immunostaining, we incubated sections with goat anti-CTB antibody overnight, followed by Alexa Fluor 488-conjugated anti-goat IgG antibody (1:1000; Invitrogen) for 1 h.
Statistical analyses.
We used paired t tests for comparisons within each group and unpaired t tests for comparisons between groups when data were normally distributed; Mann–Whitney U or Wilcoxon's signed-rank tests were used for non-normal data. We compared the frequency, duration, and total time spent in each behavioral state between treatments using unpaired t tests. All statistical analyses were performed using SigmaStat (SPSS) and applied a critical two-tailed α value of p < 0.05. Data are presented as mean ± SEM.
Results
The amygdala is anatomically well positioned to regulate cataplexy
The vlPAG and adjacent LPT are thought to be key sites for the suppression of atonia and REM sleep because lesions of this region increase REM sleep and may permit atonia to occur outside of REM sleep (Lu et al., 2006; Kaur et al., 2009). To identify brain regions that may regulate activity in the vlPAG/LPT, we microinjected CTB into the vlPAG/LPT of WT mice. CTB retrogradely labeled many brain regions, including approximately one-quarter of the orexin neurons (Fig. 1). Immunostaining for orexin-A showed that the orexin neurons heavily innervate the vlPAG/LPT, suggesting that, during wakefulness, the orexin neurons may help maintain activity in this atonia-suppressing region.
Figure 1.
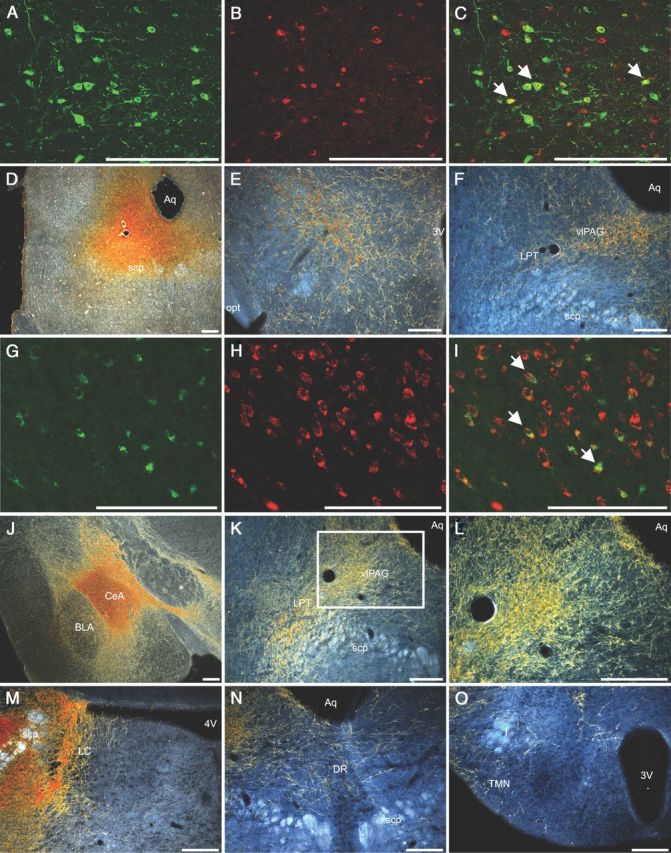
GABAergic neurons of the (CeA and the orexin neurons innervate brainstem regions that regulate REM atonia. A–C, Double-fluorescence immunolabeling for orexin-A (green) and CTB (red) demonstrates that a large number of orexin neurons are retrogradely labeled from the vlPAG and LPT. White arrows denote examples of double-labeled neurons. D, A representative injection of the retrograde tracer CTB into the vlPAG/LPT. E, F, Orexin neurons (brown) in the lateral hypothalamus innervate the vlPAG/LPT. G–I, After an injection of CTB into the vlPAG/LPT, nearly all neurons in the CeA retrogradely labeled with CTB (green) contain VGAT mRNA (red). White arrows denote examples of double-labeled neurons. J–L, Injection of AAV–ChR2–mCherry into the CeA of a VGAT–Cre mouse anterogradely labels a dense plexus of GABAergic nerve terminals in the vlPAG and LPT. L is a high-power view of the boxed vlPAG region in K. M–O, Injection of AAV–ChR2–mCherry into the CeA of a VGAT–Cre mouse also anterogradely labels nerve terminals in the dorsolateral LC and to a lesser extent the DR and TMN. 3V, Third ventricle; 4V, fourth ventricle; Aq, aqueduct; BLA, basolateral nucleus of the amygdala; f, fornix; opt, optic tract; scp, superior cerebellar peduncle. Scale bars, 250 μm.
In addition, we found that >90% of the retrogradely labeled neurons in the CeA contained VGAT mRNA. To examine this pathway with anterograde tracing, we injected an AAV coding for Cre-dependent channelrhodopsin-2 fused to mCherry into the CeA of VGAT–Cre mice. GABAergic neurons in the CeA strongly expressed mCherry and densely innervated the vlPAG/LPT. In addition, these neurons heavily innervated the dorsolateral locus ceruleus (LC), moderately innervated the dorsal raphe (DR) nucleus, and lightly innervated the tuberomammillary nucleus (TMN). These monoaminergic nuclei and the vlPAG/LPT are all hypothesized to suppress atonia, and inhibition of these sites by the CeA may promote muscle weakness or atonia.
Cataplexy is increased by stimuli associated with “positive emotions”
Under baseline conditions, control (unlesioned) orexin KO mice had 18 ± 4 episodes of cataplexy during the dark period (Table 1). Episodes had an average duration of 66 ± 5 s, resulting in 2.6% of the dark period spent in cataplexy. To examine whether stimuli associated with positive emotions increase cataplexy in orexin KO mice, we first gave orexin KO mice access to a running wheel. In agreement with previous results (Espana et al., 2007), we found that access to a running wheel increased the number of cataplexy bouts by 83% (n = 8, t(8) = −3.17, p = 0.01; Fig. 2). Cataplexy bouts became 40% shorter (t(8) = −4.27, p = 0.003), whereas the total amount of cataplexy was 3.1% of the dark period (t(8) = −0.88, p = 0.4).
Table 1.
Sleep–wake architecture and cataplexy during the dark period in control (n = 8) and amygdala-lesioned (n = 9) orexin KO mice
| Baseline |
Wheel running |
Wheel running and chocolate |
||||
|---|---|---|---|---|---|---|
| Control | Lesion | Control | Lesion | Control | Lesion | |
| Wake | ||||||
| Percentage | 65.2 ± 2.0 | 69.5 ± 2.4 | 78.4 ± 1.7 | 74.6 ± 3.0 | 90.1 ± 1.8 | 77.1 ± 3.1* |
| Bout number | 185 ± 16 | 173 ± 12 | 130 ± 11 | 140 ± 13 | 107 ± 15 | 156 ± 15* |
| Mean duration (s) | 166 ± 21 | 183 ± 21 | 281 ± 28 | 264 ± 47 | 443 ± 69 | 259 ± 59 |
| Non-REM | ||||||
| Percentage | 28.8 ± 2.0 | 26.0 ± 2.3 | 16.8 ± 1.8 | 21.5 ± 2.8 | 4.4 ± 1.6 | 17.2 ± 2.8* |
| Bout number | 167 ± 17 | 162 ± 14 | 96 ± 13 | 123 ± 13 | 39 ± 12 | 114 ± 15* |
| Mean Duration (s) | 80 ± 8 | 68 ± 3 | 79 ± 7 | 72 ± 6 | 44 ± 6 | 60 ± 6 |
| REM | ||||||
| Percentage | 3.5 ± 0.4 | 3.8 ± 0.4 | 1.6 ± 0.3 | 2.5 ± 0.4 | 0.4 ± 0.2 | 2.8 ± 0.5* |
| Bout number | 25 ± 3 | 30 ± 6 | 13 ± 2 | 17 ± 3 | 4 ± 2 | 19 ± 4* |
| Mean duration (s) | 61 ± 3 | 62 ± 7 | 55 ± 4 | 63 ± 5 | 38 ± 8 | 64 ± 5* |
| Cataplexy | ||||||
| Percentage | 2.6 ± 0.4 | 1.3 ± 0.3* | 3.1 ± 0.5 | 1.3 ± 0.4* | 5.0 ± 0.6 | 2.9 ± 0.6* |
| Bout number | 18 ± 4 | 13 ± 2* | 33 ± 6 | 16 ± 3 | 67 ± 10 | 40 ± 8* |
| Mean duration (s) | 66 ± 5 | 46 ± 5* | 40 ± 6 | 32 ± 4 | 34 ± 2 | 36 ± 2 |
*p < 0.05, significant difference from control mice (control vs lesion).
Figure 2.
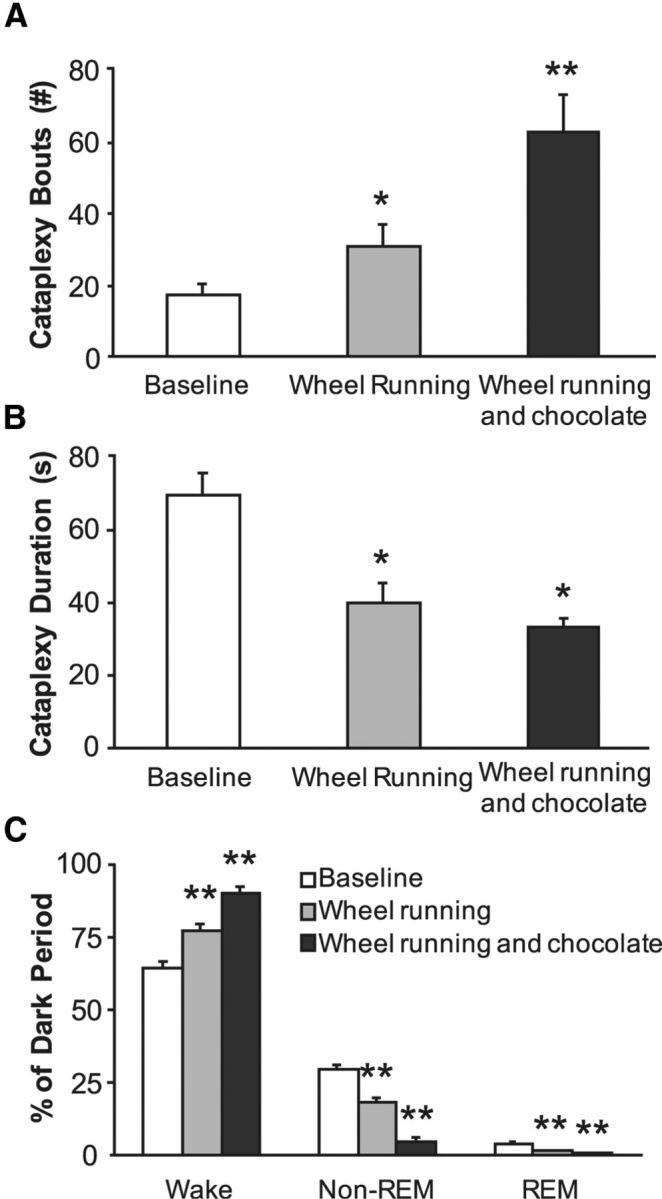
Wheel running and chocolate increase cataplexy in control mice. Wheel running and chocolate are rewarding stimuli that are likely associated with positive emotions. A, Both stimuli substantially increased the number of episodes of cataplexy in control orexin KO mice (n = 8). B, Both stimuli decreased the mean duration of cataplexy bouts. C, Wheel running increased waking at the expense of non-REM and REM sleep. This arousal response is even greater when mice have access to a running wheel plus chocolate. Data are from the baseline 12 h dark period. *p < 0.05; **p < 0.001 compared with baseline.
Chocolate is often used as a reward in rodent studies (Hsu et al., 2010; Martin-Garcia et al., 2011; Burokas et al., 2012), so we then gave orexin KO mice chocolate with continued access to a running wheel. This combination of stimuli increased the number of cataplexy bouts 272% above baseline (n = 8, t(8) = −6.06, p < 0.001). As with running wheels alone, cataplexy bouts were 48% shorter (t(8) = 2.68, p = 0.004), but the total amount of cataplexy in the dark period increased to 5% (t(8) = −3.32, p = 0.01). These observations demonstrate that stimuli associated with positive emotions increase murine cataplexy, just as in people and dogs with narcolepsy.
Running wheels and chocolate also affected sleep–wake behavior. The amount of wake during the dark period increased by 20% with running wheels alone (t(8) = −5.42, p < 0.001) and by 38% with running wheels plus chocolate (t(8) = −11.52, p < 0.001). In fact, the arousing influence of running wheels plus chocolate was so strong that orexin KO mice were awake for 90% of the dark period, with proportional reductions in non-REM and REM sleep.
Amygdala lesions decreased cataplexy under baseline conditions
We microinjected ibotenic acid into the right and left amygdalae to produce excitotoxic lesions. Most lesions encompassed both the central and basolateral nuclei, but lesions varied, with a few affecting only the CeA and several involving much of the amygdaloid complex (Fig. 3). Nine mice met criteria for acceptable bilateral lesions. Control mice injected with PBS (n = 8) showed no evidence of amygdala injury.
Figure 3.

Excitotoxic lesions of the amygdala. Ibotenic acid injections successfully lesioned the CeA and often extended into the basolateral nucleus (BLA). Different colored lines represent the nine different mice from the lesion group. Drawings are adapted from a mouse brain atlas (Paxinos and Franklin, 2001), and anteroposterior coordinates are relative to bregma. BLP, Posterior basolateral amygdala; CPu, caudate–putamen; LH, lateral hypothalamus.
Bilateral amygdala lesions significantly reduced the amount of time spent in cataplexy during the dark period (t(16) = 2.58, p = 0.02; Fig. 4), with 28% fewer bouts of cataplexy (t(16) = −6.95, p < 0.001) and a 30% decrease in the average duration of cataplexy bouts (U(8) = 81, p < 0.001). The lesions did not affect wake and non-REM or REM sleep (Table 1). Amygdala lesions did not appear to disrupt muscle atonia during REM sleep.
Figure 4.

Amygdala lesions reduce cataplexy in orexin KO mice. A, Under baseline conditions, bilateral amygdala lesions decreased the total amount of cataplexy compared with controls. This decrease was attributable to a reduction in the number of cataplexy bouts and a shortening of bouts. B, Amygdala lesions also decreased the amount of cataplexy when mice had access to running wheels. C, Amygdala lesions reduced the amount of cataplexy when mice had access to running wheels and chocolate. Data are from the 12 h dark period. *p < 0.05; **p < 0.001 compared with control orexin KO mice.
Amygdala lesions decreased cataplexy triggered by a positive stimulus
To determine whether the amygdala contributes to the cataplexy elicited by a positive stimulus, we examined cataplexy and sleep–wake behavior in amygdala-lesioned orexin KO mice with access to a running wheel. Lesioned mice spent 58% less time in cataplexy than control mice (t(16) = 2.52, p = 0.02). The lesioned mice also had 52% fewer cataplexy bouts and a 20% decrease in the average duration of cataplexy bouts, but these changes did not reach statistical significance. Amygdala lesions had no effect on sleep–wake architecture in the presence of a running wheel. Lesions had no significant effect on total wheel rotations (7375 ± 2603 vs 10,877 ± 2058 rotations, lesioned vs control, U(16) = 18, p = 0.16).
Amygdala lesions decreased cataplexy triggered by strong positive stimuli
The combination of chocolate plus a running wheel dramatically increased cataplexy in control orexin KO mice, but lesioned mice had 42% less cataplexy than controls (t(16) = 2.57, p = 0.02). The lesioned mice also had 40% fewer cataplexy bouts (t(16) = 2.04, p = 0.04), with little change in the duration of cataplexy bouts. The lesions did not alter the amount of chocolate consumed (2.2 ± 0.2 vs 1.9 ± 0.4 g, lesioned vs control, t(16) = 0.94, p = 0.36). Although lesioned mice seemed to run less, this reduction did not reach statistical significance (9846 ± 3850 vs 18,627 ± 2769 rotations, lesioned vs control, t(16) = 1.85, p = 0.08).
Control mice had a very strong arousal response to chocolate plus a running wheel, but this response was less intense in the lesioned mice. Control mice were awake for 90% of the dark period, but lesioned mice were awake for 77% of the period, with proportionately greater amounts of non-REM and REM sleep.
Discussion
For >130 years, researchers have appreciated that cataplexy is triggered by positive emotional stimuli, but very little has been understood about the mechanisms linking emotions to muscle atonia. We found that, in mice, the CeA has robust GABAergic projections to key brainstem regions for suppressing atonia, including the vlPAG/LPT, LC, and DR (Lu et al., 2006). Furthermore, bilateral lesions of the amygdala selectively reduced cataplexy in orexin KO mice without altering the very similar state of REM sleep. These results provide strong evidence that the amygdala has a functional role in triggering cataplexy.
Amygdala lesions reduce cataplexy
Researchers have hypothesized that the limbic system may trigger cataplexy (Overeem et al., 1999, 2011; Gulyani et al., 2002; Espana et al., 2007), but no studies have tested this directly. We found that amygdala lesions substantially reduced cataplexy across a variety of conditions. Specifically, under both baseline and stimulating, cataplexy-inducing conditions, lesions centered on the CeA reduced the number of bouts of cataplexy and decreased the total amount of cataplexy but had minimal effects on cataplexy duration. Thus, it appears likely that the amygdala is important for emotionally stimulated cataplexy and perhaps spontaneous cataplexy as well. Most likely, the amygdala helps generate emotional responses to salient positive stimuli and then relays this information to brainstem regions that regulate muscle tone (Brinley-Reed et al., 1995; Balleine and Killcross, 2006; Paton et al., 2006; Straube et al., 2008; Ball et al., 2009).
However, it is possible that the amygdala lesions reduced cataplexy through nonspecific changes in emotional state, locomotion, or motivated behavior. Although we cannot rule out indirect effects of the amygdala lesions, direct effects on cataplexy seem more likely because the amygdala heavily innervates brainstem regions that regulate motor tone and amygdala neurons increase their firing at the onset of cataplexy (Gulyani et al., 2002).
Importantly, amygdala lesions did not eliminate cataplexy, suggesting that additional forebrain regions may contribute. The largest lesions reduced baseline cataplexy by ∼65%, yet even in these mice, running wheels and chocolate still increased cataplexy. This makes it likely that, although the amygdala is important, additional neural mechanisms can still relay signals related to emotion to brainstem regions that regulate atonia. In the future, it will be helpful to examine the roles of the bed nucleus, basal forebrain, lateral hypothalamus, and limbic cortex because these regions also project to pontine regions that regulate muscle atonia and may contribute to the triggering of cataplexy (Boissard et al., 2003; Clément et al., 2011; Hsieh et al., 2011).
The amygdala may trigger cataplexy through direct projections to the pons
Cataplexy and the atonia of REM sleep share many similarities, and it is likely that these phenomena are mediated by the same brainstem pathways. The sublaterodorsal nucleus (SLD) is a key site that indirectly inhibits motor neurons during REM sleep, through spinal and medullary GABAergic and glycinergic premotor neurons (Boissard et al., 2002; Lu et al., 2006; Brooks and Peever, 2012; Luppi et al., 2012). During wakefulness, the vlPAG/LPT and monoaminergic neurons inhibit the SLD and suppress atonia, but during REM sleep, these neurons are inactive, disinhibiting the SLD and permitting atonia to occur (Lu et al., 2006). Monaminergic neurons can also increase muscle tone through direct projections to motor neurons, but monoamine neurons fall silent during REM sleep (Wu et al., 1999, 2004; Fenik et al., 2005; Chan et al., 2006; Takahashi et al., 2010). Much less is known about the patterns of neuronal activity during cataplexy. It is clear that neurons of the LC and DR reduce firing (Wu et al., 1999, 2004), and although it has not yet been studied directly, cataplexy is likely associated with reduced activity in the vlPAG/LPT and increased activity in the SLD.
Several lines of evidence now suggest that the amygdala could trigger cataplexy by relaying signals associated with positive emotions to these same brainstem regions. The amygdala helps generate emotional responses to positive affective stimuli, reward, and social play (Balleine and Killcross, 2006; Paton et al., 2006; Straube et al., 2008; Ball et al., 2009; Bermudez and Schultz, 2010; Davey et al., 2011; Trezza et al., 2012). Brain regions that mediate reward and motivation [e.g., prefrontal cortex (PFC) or dopaminergic neurons of the ventral tegmental area (VTA)] innervate the amygdala in which they could excite brainstem-projecting CeA neurons that trigger cataplexy (Asan, 1998). Importantly, in narcoleptic dogs, neurons in the amygdala increase their activity just before and during cataplexy (Gulyani et al., 2002); there is increased dopamine release in the amygdala in response to pleasurable stimuli (Miller et al., 1990), and manipulation of VTA dopamine neuron activity modulates cataplexy (Reid et al., 1996). Although amygdala activity during cataplexy has not yet been examined in humans, two studies have reported abnormal amygdala responses to emotional stimuli in people with narcolepsy, with increased amygdala and VTA responses to positive rewards and decreased amygdala responses to aversive stimuli (Ponz et al., 2010a,b). Here we demonstrate GABAergic projections from the CeA to the vlPAG/LPT and to brainstem monoaminergic nuclei. Activation of these projections, perhaps by PFC or VTA dopamine neurons, could trigger cataplexy as well as the momentary muscle weakness associated with laughter even in healthy individuals (Overeem et al., 1999).
In healthy animals, these inhibitory signals from the CeA are likely counterbalanced by excitatory signals from the orexin neurons. The orexin neurons are active under conditions of high arousal, positive emotions, and social interaction (Lee et al., 2005; Mileykovskiy et al., 2005; Blouin et al., 2013), and we and others have shown that they innervate the vlPAG/LPT (Lu et al., 2006; Kaur et al., 2009), as well as the LC and DR (Peyron et al., 1998). Under normal conditions, any inhibition of the vlPAG/LPT by the CeA during periods of positive emotion would be counterbalanced by the orexin neurons and subsequent monoamine activation, thus maintaining muscle tone via inhibition of the SLD and direct excitation of motor neurons (Fig. 5) (Lu et al., 2006; Hsieh et al., 2011; Burgess and Scammell, 2012; Clément et al., 2012). However, with loss of orexin signaling as in narcolepsy, the inhibitory projections from the CeA would be unopposed, resulting in cataplexy. This model helps explain how positive emotions may trigger cataplexy, and although some elements of the model still need to be confirmed, all are testable with current techniques.
Figure 5.
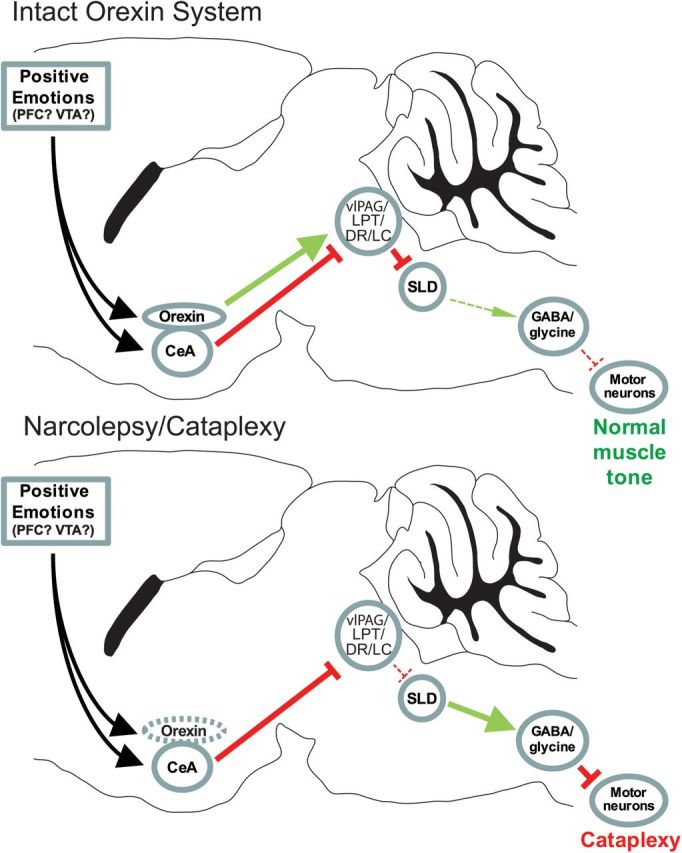
A model of the neural pathways through which positive emotions may trigger cataplexy. During wakefulness, neurons in the LC, DR, vlPAG, and adjacent LPT inhibit neurons in the SLD that generate atonia. With strong, positive emotions, neurons in the CeA may receive excitatory signals from PFC or VTA dopamine neurons and inhibit these brainstem regions. Normally, this CeA inhibition is offset by excitatory signals from the orexin neurons. Loss of orexin signaling in narcolepsy upsets this balance, so that the CeA can inhibit the vlPAG/LPT and monoaminergic neurons, resulting in cataplexy. Direct excitatory signals from monoaminergic nuclei and orexin neurons to motor neurons, not depicted here, could also increase muscle tone.
Limitations
A few limitations warrant discussion. Although our lesions were centered on the CeA, many extended into adjacent parts of the amygdala, and we cannot conclude that the CeA per se is important for triggering cataplexy. However, the CeA was the only area consistently ablated in our mice, and the majority of brainstem-projecting neurons in the amygdala are located in the CeA (Hopkins and Holstege, 1978; Wallace et al., 1989, 1992; Sah and Lopez De Armentia, 2003). Another concern is that, although chocolate is rewarding for mice, it also increases wakefulness and locomotor activity, and some of the increase in cataplexy may have been related to these behavioral effects or psychoactive compounds in chocolate, such as caffeine. However, the increase in cataplexy with chocolate was much larger than the increase in wakefulness, and previous studies have shown that caffeine does not increase cataplexy (Willie et al., 2003, Okuro et al., 2010). Considering the rewarding effects of chocolate in mice (Hsu et al., 2010; Martin-Garcia et al., 2011), we think that the increase in cataplexy with chocolate is mainly attributable to increases in positive affect. A final limitation is that we cannot be certain that this reduction was attributable to a disruption of positive affect because cataplexy was reduced in all three conditions, but a growing literature demonstrates many roles for the amygdala in mediating positive affect (Garavan et al., 2001; Baxter and Murray, 2002; Balleine and Killcross, 2006; Paton et al., 2006; Straube et al., 2008; Trezza and Campolongo, 2009). In narcoleptic dogs and people, cataplexy is clearly triggered by rewarding stimuli and positive emotional states (Gelineau, 1880; Siegel et al., 1989; Overeem et al., 2011), and in narcoleptic mice, cataplexy is increased by stimuli that presumably trigger positive affect (Espana et al., 2007; Clark et al., 2009). This convergent evidence suggests that disruption of signals related to positive affect is the main cause of the reduction in cataplexy.
Regulation of arousal and REM sleep by the amygdala
In addition to a key role in cataplexy, the amygdala has additional effects on sleep–wake behavior. We found that, under the very arousing condition of running wheels plus chocolate, unlesioned mice had clear increases in wakefulness and locomotion, but this response was blunted in mice with amygdala lesions. Similarly, rhesus monkeys with amygdala lesions have reduced arousal compared with controls under gentle restraint (Benca et al., 2000), and pharmacological inactivation of the amygdala can decrease arousal in rats (Sanford et al., 2006a). In addition, pharmacology studies in rats suggest that the amygdala can increase or decrease REM sleep (Sanford et al., 2002, 2006a,b; Pawlyk et al., 2006; Dong et al., 2012). Thus, in addition to promoting cataplexy, neurons of the amygdala can regulate arousal and REM sleep, perhaps through pathways distinct from those that trigger cataplexy.
Conclusions
These results demonstrate that the amygdala is functionally important for the production of cataplexy in narcoleptic mice, and, in response to positive emotions, it may trigger muscle atonia by inhibiting brainstem nuclei that suppress atonia. Our findings suggest several avenues of additional research, including investigating the roles of other forebrain or limbic structures in triggering cataplexy and, more generally, their roles in sleep and muscle atonia. In particular, optogenetic and pharmacogenetic tools could be used to determine which neuronal populations in the amygdala or other regions are sufficient to trigger cataplexy. The recent finding that orexin levels in the amygdala are highest during positive emotions and social interactions in humans suggests that direct lateral hypothalamic–amygdala interactions could be an important area of future study as well (Blouin et al., 2013). Our data and the resulting model of how emotional stimuli trigger cataplexy could suggest a mechanism for treating cataplexy based on its most common triggers.
Footnotes
This work was supported by National Institutes of Health Grant NS055367 (T.E.S.) and Canadian Institutes of Health Research, Natural Sciences and Engineering Research Council of Canada, and Ontario Graduate Scholarship Program (J.H.P. and C.R.B.). We thank Dr. C. Alexandre, M. Yamamoto, and L. J. Agostinelli for their advice and technical assistance, as well as Drs. B. Lowell and L. Vong for VGAT–cre mice.
References
- Asan E. The catecholaminergic innervation of the rat amygdala. Adv Anat Embryol Cell Biol. 1998;142:1–118. doi: 10.1007/978-3-642-72085-7_1. [DOI] [PubMed] [Google Scholar]
- Ball T, Derix J, Wentlandt J, Wieckhorst B, Speck O, Schulze-Bonhage A, Mutschler I. Anatomical specificity of functional amygdala imaging of responses to stimuli with positive and negative emotional valence. J Neurosci Methods. 2009;180:57–70. doi: 10.1016/j.jneumeth.2009.02.022. [DOI] [PubMed] [Google Scholar]
- Balleine BW, Killcross S. Parallel incentive processing: an integrated view of amygdala function. Trends Neurosci. 2006;29:272–279. doi: 10.1016/j.tins.2006.03.002. [DOI] [PubMed] [Google Scholar]
- Baxter MG, Murray EA. The amygdala and reward. Nat Rev Neurosci. 2002;3:563–573. doi: 10.1038/nrn875. [DOI] [PubMed] [Google Scholar]
- Benca RM, Obermeyer WH, Shelton SE, Droster J, Kalin NH. Effects of amygdala lesions on sleep in rhesus monkeys. Brain Res. 2000;879:130–138. doi: 10.1016/S0006-8993(00)02761-X. [DOI] [PubMed] [Google Scholar]
- Bermudez MA, Schultz W. Responses of amygdala neurons to positive reward-predicting stimuli depend on background reward (contingency) rather than stimulus-reward pairing (contiguity) J Neurophysiol. 2010;103:1158–1170. doi: 10.1152/jn.00933.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blouin AM, Fried I, Wilson CL, Staba RJ, Behnke EJ, Lam HA, Maidment NT, Karlsson KAE, Lapierre JL, Siegel JM. Human hypocretin and melanin-concentrating hormone levels are linked to emotion and social interaction. Nat Commun. 2013;4:1547. doi: 10.1038/ncomms2461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boissard R, Gervasoni D, Schmidt MH, Barbagli B, Fort P, Luppi PH. The rat ponto-medullary network responsible for paradoxical sleep onset and maintenance: a combined microinjection and functional neuroanatomical study. Eur J Neurosci. 2002;16:1959–1973. doi: 10.1046/j.1460-9568.2002.02257.x. [DOI] [PubMed] [Google Scholar]
- Boissard R, Fort P, Gervasoni D, Barbagli B, Luppi PH. Localization of the GABAergic and non-GABAergic neurons projecting to the sublaterodorsal nucleus and potentially gating paradoxical sleep onset. Eur J Neurosci. 2003;18:1627–1639. doi: 10.1046/j.1460-9568.2003.02861.x. [DOI] [PubMed] [Google Scholar]
- Brinley-Reed M, Mascagni F, McDonald AJ. Synaptology of prefrontal cortical projections to the basolateral amygdala: an electron microscopic study in the rat. Neurosci Lett. 1995;202:45–48. doi: 10.1016/0304-3940(95)12212-5. [DOI] [PubMed] [Google Scholar]
- Brooks PL, Peever JH. Identification of the transmitter and receptor mechanisms responsible for REM sleep paralysis. J Neurosci. 2012;32:9785–9795. doi: 10.1523/JNEUROSCI.0482-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess CR, Scammell TE. Narcolepsy: neural mechanisms of sleepiness and cataplexy. J Neurosci. 2012;32:12305–12311. doi: 10.1523/JNEUROSCI.2630-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burokas A, Gutiérrez-Cuesta J, Martín-García E, Maldonado R. Operant model of frustrated expected reward in mice. Addict Biol. 2012;17:770–782. doi: 10.1111/j.1369-1600.2011.00423.x. [DOI] [PubMed] [Google Scholar]
- Chan E, Steenland HW, Liu H, Horner RL. Endogenous excitatory drive modulating respiratory muscle activity across sleep-wake states. Am J Respir Crit Care Med. 2006;174:1264–1273. doi: 10.1164/rccm.200605-597OC. [DOI] [PubMed] [Google Scholar]
- Clark EL, Baumann CR, Cano G, Scammell TE, Mochizuki T. Feeding-elicited cataplexy in orexin knockout mice. Neuroscience. 2009;161:970–977. doi: 10.1016/j.neuroscience.2009.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clément O, Sapin E, Bérod A, Fort P, Luppi PH. Evidence that neurons of the sublaterodorsal tegmental nucleus triggering paradoxical (REM) sleep are glutamatergic. Sleep. 2011;34:419–423. doi: 10.1093/sleep/34.4.419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clément O, Sapin E, Libourel PA, Arthaud S, Brischoux F, Fort P, Luppi PH. The lateral hypothalamic area controls paradoxical (REM) sleep by means of descending projections to brainstem GABAergic neurons. J Neurosci. 2012;32:16763–16774. doi: 10.1523/JNEUROSCI.1885-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davey CG, Allen NB, Harrison BJ, Yücel M. Increased amygdala response to positive social feedback in young people with major depressive disorder. Biol Psychiatry. 2011;69:734–741. doi: 10.1016/j.biopsych.2010.12.004. [DOI] [PubMed] [Google Scholar]
- Dong E, Wellman LL, Yang L, Sanford LD. Effects of microinjections of Group II metabotropic glutamate agents into the amygdala on sleep. Brain Res. 2012;1452:85–95. doi: 10.1016/j.brainres.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- España RA, McCormack SL, Mochizuki T, Scammell TE. Running promotes wakefulness and increases cataplexy in orexin knockout mice. Sleep. 2007;30:1417–1425. doi: 10.1093/sleep/30.11.1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenik VB, Davies RO, Kubin L. REM sleep-like atonia of hypoglossal (XII) motoneurons is caused by loss of noradrenergic and serotonergic inputs. Am J Respir Crit Care Med. 2005;172:1322–1330. doi: 10.1164/rccm.200412-1750OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fung SJ, Xi M, Zhang J, Torterolo P, Sampogna S, Morales FR, Chase MH. Projection neurons from the central nucleus of the amygdala to the nucleus pontis oralis. J Neurosci Res. 2011;89:429–436. doi: 10.1002/jnr.22554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garavan H, Pendergrass JC, Ross TJ, Stein EA, Risinger RC. Amygdala response to both positively and negatively valenced stimuli. Neuroreport. 2001;12:2779–2783. doi: 10.1097/00001756-200108280-00036. [DOI] [PubMed] [Google Scholar]
- Gelineau J. De la narcolepsie. Gazette des Hôpitaux. 1880;53:626–628. [Google Scholar]
- Gulyani S, Wu MF, Nienhuis R, John J, Siegel JM. Cataplexy-related neurons in the amygdala of the narcoleptic dog. Neuroscience. 2002;112:355–365. doi: 10.1016/S0306-4522(02)00089-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holahan MR, Madularu D, McConnell EM, Walsh R, DeRosa MC. Intra-accumbens injection of a dopamine aptamer abates MK-801-induced cognitive dysfunction in a model of schizophrenia. PLoS One. 2011;6:e22239. doi: 10.1371/journal.pone.0022239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopkins DA, Holstege G. Amygdaloid projections to the mesencephalon, pons and medulla oblongata in the cat. Exp Brain Res. 1978;32:529–547. doi: 10.1007/BF00239551. [DOI] [PubMed] [Google Scholar]
- Hsieh KC, Gvilia I, Kumar S, Uschakov A, McGinty D, Alam MN, Szymusiak R. c-Fos expression in neurons projecting from the preoptic and lateral hypothalamic areas to the ventrolateral periaqueductal gray in relation to sleep states. Neuroscience. 2011;188:55–67. doi: 10.1016/j.neuroscience.2011.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu CT, Patton DF, Mistlberger RE, Steele AD. Palatable meal anticipation in mice. PLoS One. 2010;5:e12903. doi: 10.1371/journal.pone.0012903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur S, Thankachan S, Begum S, Liu M, Blanco-Centurion C, Shiromani PJ. Hypocretin-2 saporin lesions of the ventrolateral periaquaductal gray (vlPAG) increase REM sleep in hypocretin knockout mice. PLoS One. 2009;4:e6346. doi: 10.1371/journal.pone.0006346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King SJ, Isaacs AM, O'Farrell E, Abizaid A. Motivation to obtain preferred foods is enhanced by ghrelin in the ventral tegmental area. Horm Behav. 2011;60:572–580. doi: 10.1016/j.yhbeh.2011.08.006. [DOI] [PubMed] [Google Scholar]
- Lee MG, Hassani OK, Jones BE. Discharge of identified orexin/hypocretin neurons across the sleep-waking cycle. J Neurosci. 2005;25:6716–6720. doi: 10.1523/JNEUROSCI.1887-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Sherman D, Devor M, Saper CB. A putative flip-flop switch for control of REM sleep. Nature. 2006;441:589–594. doi: 10.1038/nature04767. [DOI] [PubMed] [Google Scholar]
- Luppi PH, Clement O, Sapin E, Peyron C, Gervasoni D, Léger L, Fort P. Brainstem mechanisms of paradoxical (REM) sleep generation. Pflugers Arch. 2012;463:43–52. doi: 10.1007/s00424-011-1054-y. [DOI] [PubMed] [Google Scholar]
- Martin-Garcia E, Burokas A, Kostrzewa E, Gieryk A, Korostynski M, Ziolkowska B, Przewlocka B, Przewlocki R, Maldonado R. New operant model of reinstatement of food-seeking behavior in mice. Psychopharmacology (Berl) 2011;215:49–70. doi: 10.1007/s00213-010-2110-6. [DOI] [PubMed] [Google Scholar]
- Mileykovskiy BY, Kiyashchenko LI, Siegel JM. Behavioral correlates of activity in identified hypocretin/orexin neurons. Neuron. 2005;46:787–798. doi: 10.1016/j.neuron.2005.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JD, Faull KF, Bowersox SS, Dement WC. CNS monoamines and their metabolites in canine narcolepsy: a replication study. Brain Res. 1990;509:169–171. doi: 10.1016/0006-8993(90)90328-9. [DOI] [PubMed] [Google Scholar]
- Mochizuki T, Crocker A, McCormack S, Yanagisawa M, Sakurai T, Scammell TE. Behavioral state instability in orexin knock-out mice. J Neurosci. 2004;24:6291–6300. doi: 10.1523/JNEUROSCI.0586-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishijo H, Ono T, Nishino H. Single neuron responses in amygdala of alert monkey during complex sensory stimulation with affective significance. J Neurosci. 1988;8:3570–3583. doi: 10.1523/JNEUROSCI.08-10-03570.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuro M, Fujiki N, Kotorii N, Ishimaru Y, Sokoloff P, Nishino S. Effects of paraxanthine and caffeine on sleep, locomotor activity, and body temperature in orexin/ataxin-3 transgenic narcoleptic mice. Sleep. 2010;33:930–942. doi: 10.1093/sleep/33.7.930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overeem S, Lammers GJ, van Dijk JG. Weak with laughter. Lancet. 1999;354:838. doi: 10.1016/S0140-6736(99)80023-3. [DOI] [PubMed] [Google Scholar]
- Overeem S, van Nues SJ, van der Zande WL, Donjacour CE, van Mierlo P, Lammers GJ. The clinical features of cataplexy: a questionnaire study in narcolepsy patients with and without hypocretin-1 deficiency. Sleep Med. 2011;12:12–18. doi: 10.1016/j.sleep.2010.05.010. [DOI] [PubMed] [Google Scholar]
- Paton JJ, Belova MA, Morrison SE, Salzman CD. The primate amygdala represents the positive and negative value of visual stimuli during learning. Nature. 2006;439:865–870. doi: 10.1038/nature04490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawlyk AC, Sanford LD, Brennan FX, Morrison AR, Ross RJ. Corticotropin-releasing factor microinjection into the central nucleus of the amygdala alters REM sleep. Pharmacol Rep. 2006;58:125–130. [PubMed] [Google Scholar]
- Paxinos G, Franklin K. The mouse brain in stereotaxic coordinates. New York: Academic; 2001. [Google Scholar]
- Peyron C, Tighe DK, van den Pol AN, de Lecea L, Heller HC, Sutcliffe JG, Kilduff TS. Neurons containing hypocretin (orexin) project to multiple neuronal systems. J Neurosci. 1998;18:9996–10015. doi: 10.1523/JNEUROSCI.18-23-09996.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peyron C, Faraco J, Rogers W, Ripley B, Overeem S, Charnay Y, Nevsimalova S, Aldrich M, Reynolds D, Albin R, Li R, Hungs M, Pedrazzoli M, Padigaru M, Kucherlapati M, Fan J, Maki R, Lammers GJ, Bouras C, Kucherlapati R, Nishino S, Mignot E. A mutation in a case of early onset narcolepsy and a generalized absence of hypocretin peptides in human narcoleptic brains. Nat Med. 2000;6:991–997. doi: 10.1038/79690. [DOI] [PubMed] [Google Scholar]
- Ponz A, Khatami R, Poryazova R, Werth E, Boesiger P, Bassetti CL, Schwartz S. Abnormal activity in reward brain circuits in human narcolepsy with cataplexy. Ann Neurol. 2010a;67:190–200. doi: 10.1002/ana.21825. [DOI] [PubMed] [Google Scholar]
- Ponz A, Khatami R, Poryazova R, Werth E, Boesiger P, Schwartz S, Bassetti CL. Reduced amygdala activity during aversive conditioning in human narcolepsy. Ann Neurol. 2010b;67:394–398. doi: 10.1002/ana.21881. [DOI] [PubMed] [Google Scholar]
- Reid MS, Tafti M, Nishino S, Sampathkumaran R, Siegel JM, Mignot E. Local administration of dopaminergic drugs into the ventral tegmental area modulates cataplexy in the narcoleptic canine. Brain Res. 1996;733:83–100. doi: 10.1016/0006-8993(96)00541-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sah P, Lopez De Armentia M. Excitatory synaptic transmission in the lateral and central amygdala. Ann N Y Acad Sci. 2003;985:67–77. doi: 10.1111/j.1749-6632.2003.tb07072.x. [DOI] [PubMed] [Google Scholar]
- Sanford LD, Parris B, Tang X. GABAergic regulation of the central nucleus of the amygdala: implications for sleep control. Brain Res. 2002;956:276–284. doi: 10.1016/S0006-8993(02)03552-7. [DOI] [PubMed] [Google Scholar]
- Sanford LD, Yang L, Liu X, Tang X. Effects of tetrodotoxin (TTX) inactivation of the central nucleus of the amygdala (CNA) on dark period sleep and activity. Brain Res. 2006a;1084:80–88. doi: 10.1016/j.brainres.2006.02.020. [DOI] [PubMed] [Google Scholar]
- Sanford LD, Yang L, Tang X, Dong E, Ross RJ, Morrison AR. Cholinergic regulation of the central nucleus of the amygdala in rats: effects of local microinjections of cholinomimetics and cholinergic antagonists on arousal and sleep. Neuroscience. 2006b;141:2167–2176. doi: 10.1016/j.neuroscience.2006.05.064. [DOI] [PubMed] [Google Scholar]
- Scammell TE, Willie JT, Guilleminault C, Siegel JM. A consensus definition of cataplexy in mouse models of narcolepsy. Sleep. 2009;32:111–116. [PMC free article] [PubMed] [Google Scholar]
- Schenck CH, Bassetti CL, Arnulf I, Mignot E. English translations of the first clinical reports on narcolepsy and cataplexy by Westphal and Gelineau in the late 19th century, with commentary. J Clin Sleep Med. 2007;3:301–311. [PMC free article] [PubMed] [Google Scholar]
- Schoenbaum G, Chiba AA, Gallagher M. Orbitofrontal cortex and basolateral amygdala encode expected outcomes during learning. Nat Neurosci. 1998;1:155–159. doi: 10.1038/407. [DOI] [PubMed] [Google Scholar]
- Siegel JM, Fahringer H, Tomaszewski KS, Kaitin K, Kilduff T, Dement WC. Heart rate and blood pressure changes associated with cataplexy in canine narcolepsy. Sleep. 1986;9:216–221. doi: 10.1093/sleep/9.1.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel JM, Tomaszewski KS, Fahringer H, Cave G, Kilduff T, Dement WC. Heart rate and blood pressure changes during sleep-waking cycles and cataplexy in narcoleptic dogs. Am J Physiol. 1989;256:H111–H119. doi: 10.1152/ajpheart.1989.256.1.H111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel JM, Nienhuis R, Fahringer HM, Paul R, Shiromani P, Dement WC, Mignot E, Chiu C. Neuronal activity in narcolepsy: identification of cataplexy-related cells in the medial medulla. Science. 1991;252:1315–1318. doi: 10.1126/science.1925546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straube T, Pohlack S, Mentzel HJ, Miltner WH. Differential amygdala activation to negative and positive emotional pictures during an indirect task. Behav Brain Res. 2008;191:285–288. doi: 10.1016/j.bbr.2008.03.040. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Kayama Y, Lin JS, Sakai K. Locus coeruleus neuronal activity during the sleep-waking cycle in mice. Neuroscience. 2010;169:1115–1126. doi: 10.1016/j.neuroscience.2010.06.009. [DOI] [PubMed] [Google Scholar]
- Thannickal TC, Moore RY, Nienhuis R, Ramanathan L, Gulyani S, Aldrich M, Cornford M, Siegel JM. Reduced number of hypocretin neurons in human narcolepsy. Neuron. 2000;27:469–474. doi: 10.1016/S0896-6273(00)00058-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trezza V, Campolongo P. Toward understanding the neurobiology of social attachment: role of estrogen receptors in the medial amygdala. J Neurosci. 2009;29:1–2. doi: 10.1523/JNEUROSCI.5193-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trezza V, Damsteegt R, Manduca A, Petrosino S, Van Kerkhof LW, Pasterkamp RJ, Zhou Y, Campolongo P, Cuomo V, Di Marzo V, Vanderschuren LJ. Endocannabinoids in amygdala and nucleus accumbens mediate social play reward in adolescent rats. J Neurosci. 2012;32:14899–14908. doi: 10.1523/JNEUROSCI.0114-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vong L, Ye C, Yang Z, Choi B, Chua S, Jr, Lowell BB. Leptin action on GABAergic neurons prevents obesity and reduces inhibitory tone to POMC neurons. Neuron. 2011;71:142–154. doi: 10.1016/j.neuron.2011.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace DM, Magnuson DJ, Gray TS. The amygdalo-brainstem pathway: selective innervation of dopaminergic, noradrenergic and adrenergic cells in the rat. Neurosci Lett. 1989;97:252–258. doi: 10.1016/0304-3940(89)90606-X. [DOI] [PubMed] [Google Scholar]
- Wallace DM, Magnuson DJ, Gray TS. Organization of amygdaloid projections to brainstem dopaminergic, noradrenergic, and adrenergic cell groups in the rat. Brain Res Bull. 1992;28:447–454. doi: 10.1016/0361-9230(92)90046-Z. [DOI] [PubMed] [Google Scholar]
- Willie JT, Chemelli RM, Sinton CM, Tokita S, Williams SC, Kisanuki YY, Marcus JN, Lee C, Elmquist JK, Kohlmeier KA, Leonard CS, Richardson JA, Hammer RE, Yanagisawa M. Distinct narcolepsy syndromes in Orexin receptor-2 and Orexin null mice: molecular genetic dissection of Non-REM and REM sleep regulatory processes. Neuron. 2003;38:715–730. doi: 10.1016/S0896-6273(03)00330-1. [DOI] [PubMed] [Google Scholar]
- Wu MF, Gulyani SA, Yau E, Mignot E, Phan B, Siegel JM. Locus coeruleus neurons: cessation of activity during cataplexy. Neuroscience. 1999;91:1389–1399. doi: 10.1016/S0306-4522(98)00600-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu MF, John J, Boehmer LN, Yau D, Nguyen GB, Siegel JM. Activity of dorsal raphe cells across the sleep-waking cycle and during cataplexy in narcoleptic dogs. J Physiol. 2004;554:202–215. doi: 10.1113/jphysiol.2003.052134. [DOI] [PMC free article] [PubMed] [Google Scholar]