

Summary
Differences in chromatin-associated proteins allow the same genome to participate in multiple cell types and to respond to an array of stimuli in any given cell. To understand the fundamental properties of chromatin and to reveal its cell- and/or stimulus-specific behaviors, quantitative proteomics is an essential technology. This chapter details the methods for fractionation and quantitative mass spectrometric analysis of chromatin from hearts or isolated adult myocytes, detailing some of the considerations for applications to understanding heart disease. The state-of-the-art methodology for data interpretation and integration through bioinformatics is reviewed.
Keywords: Chromatin, cardiovascular disease, proteomics, mass spectrometry, bioinformatics
1. Introduction
How the same genome encodes multiple proteomes in the hundreds of cell types in a given multi-cellular eukaryote is one of the great unanswered questions of biology. The packaging of DNA in the nucleus occurs on the basis of the following structural hierarchy: a segment (~147 bp) of DNA wraps around a protein complex containing two copies each of four core histones (H2A, H2B, H3 and H4), constituting a nucleosome; this octameric protein complex in turn forms higher ordered structures of less well-defined architecture through interactions with linker histones (like H1) and other chromatin structural proteins. These higher order chromatin regions determine the overall shape and presentation of each region of the genome.
During mitosis, the structure of the chromosomes is well established—as are the highly orchestrated structural events during cytokinesis. The 3D structure of the non-mitotic genome, however, is poorly understood. The packing task of the nucleus is daunting: in the case of a cardiac cell, for example, the 2 copies of the ~3 billion base pair genome must be collapsed into ~350–400 μM3 of nuclear space; accommodation must also be made for RNA and protein (we have measured >1000 proteins in the cardiac nucleus,1 including >300 bound to chromatin1,2), while still maintaining conformational flexibility to enable large, rapid changes in gene expression. New insights into genomic structure provided by techniques like chromosomal conformation capture3,4 have demonstrated a non-random structure of the genome in an interphase nucleus, and suggest that coordinated packaging of chromosomal territories is a critical task to enable global gene expression programs in eukaryotes. Not addressed in these studies are the proteins contributing to the chromatin backbone and how quantitative changes in these chromatin structural proteins influence global gene expression in disease.
One of the most important problems in biology is explaining how genome-wide changes in transcription are facilitated by structural remodeling of chromatin. Thirty-seven years after the original nucleosome hypothesis was proposed,5 the field is still coming to understand how chromatin dynamics are structurally regulated in vivo with high resolution nucleosome mapping.6 The recent explosion of studies using chromatin immunoprecipitation (ChIP) and DNA sequencing to map the localization of select proteins across the genome7–10 has added new detail to the picture of gene regulation. Extensive work has been done to show how various classes of proteins (such as deacetylases, acetyltransferases and methyltransferases, among others) modulate chromatin accessibility and thereby gene expression;11–15 however, not revealed in these studies is how specific genes are targeted and how global remodeling is integrated across the genome. To initiate transcription, RNA polymerases themselves, as well as a cadre of chromatin-remodeling enzymes, reorganize and evict nucleosomes to free up first the promoter region, and subsequently downstream exons, for transcription.16–18
Because adult cardiac myocytes do not readily divide, it is well established that the injured heart will grow surviving myocytes to maintain cardiac output. This hypertrophic process is a common antecedent to heart failure in a variety of conditions,19 and involves ordered reprogramming of gene expression to convert the cell to a more primitive phenotype (the so called “fetal gene program”).20,21 To be transcribed, genes silenced during development must transition from a heterochromatic (tightly packed) to euchromatic (loosely packed) environment. This presents an intriguing scenario from the standpoint of chromatin regulation: cardiac transcriptional responses must somehow “find” the right genes to impinge upon through a combination of consensus motifs, associating proteins and—this part is a hypothesis— reception by a permissive global chromatin structure, poised for a given cellular phenotype.
Proteomics is a critical tool to answer questions of global chromatin regulation in a fully differentiated cell (the cardiomyocyte), with a complex gene expression profile and highly specialized physiological phenotype. This protocol describes methodologies for label-free quantitative analysis of subproteomes within the nucleus, and addresses unique challenges for analyzing heart tissue, including multiple cell types, abundant non-nuclear contractile proteins (Fig. 1) and bioinformatic and computational biology approaches (Fig. 2) for data interpretation.
Figure 1. Flow chart for fractionation and quantitative mass spectrometric analysis of chromatin.
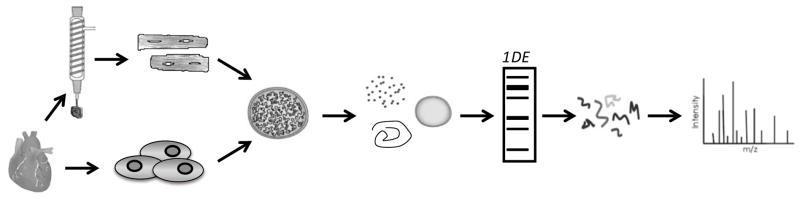
Whole mouse hearts are homogenized, or adult cardiac myocytes are lysed, and the lysate enriched for nuclei. Each nuclei sample can either be separated into nucleoplasm and chromatin fractions or into an acid-extracted fraction that enriches for histones. Proteins from each are separated by one-dimensional SDS-PAGE, and the bands trypsin digested and run by LC/MS/MS.
Figure 2. Flow chart for bioinformatic analysis of proteomic data.

Raw MS spectra are obtained from LC/MS/MS, and the results searched against a protein database (UniProt) using the SEQUEST algorithm, which is embedded in Xcalibur. This list can be used directly to obtain global features of the proteome, such as physical-chemical properties and protein domains/families. After converting protein identifiers to gene identifiers, gene ontology-based annotation can be conducted. The location of genes within chromosomes can be extracted from Ensembl genome database and visualized by UCSC genome browser. Candidate regulatory elements, i.e. binding motifs of transcription factors can be found using MotifMap database or Clover algorithm. Refer to Table 1 for the availability of these tools.
2. Materials
Unless otherwise indicated, all solutions are made in deionized water, with the exception of the digestion and LC/MS/MS steps, for which all solutions should be HPLC grade and made in HPLC water unless specified otherwise.
2.1 Fractionation Components
Phosphate Buffered Saline (PBS).
Protease/Phosphatase Inhibitor Mix: 0.1mM phenylmethanesulfonylfluoride, protease inhibitor cocktail pellet (Roche, catalogue number 04 693 159 001), 0.2mM sodium orthovanadate, 0.1mM sodium fluoride and 10mM sodium butyrate. Rather than make a stock mix, these components should be added individually directly to the following buffers when they are made.
Whole Lysate Buffer: 20mM Tris pH 7.4, 150mM NaCl, 1mM ethylenediaminetetraacetic acid (EDTA), 1mM ethylene glycol tetraacetic acid (EGTA), 1% SDS, 2.5mM sodium pyrophosphate, 1mM glycerophosphate with protease/phosphatase inhibitor mix. Store at −20°C for up to one week.
100 μM strainer (catalogue number 352369) (BD Falcon).
Langendorff system.
Heart Perfusion Buffer: 100mM potassium glutamate, 10mM aspartic acid, 25mM KCl, 10mM KH2PO4, 2mM MgSO4, 20mM taurine, 5mM creatine, 0.5mM EGTA, 5mM 4-(2- hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) and 20mM glucose. Adjust pH to 7.2 with KOH. Add glucose just before use. Warm the perfusion buffer to 37°C before use.
Collagenase: Collagenase type II from Worthington is recommended. Dissolve the enzyme into perfusion buffer before use. We used a concentration of 1,000mg/L but this should be determined empirically.
Lysis Buffer: 0.15% Nonidet P-40 (NP-40), 10mM Tris pH 7.5, 15mM NaCl, with protease/phosphatase inhibitor mix. Store at −20°C for up to one week.
Sucrose Buffer: 10mM Tris pH 7.5, 15mM NaCl, 24% sucrose (w/v), with protease/phosphatase inhibitor mix. Make fresh.
Transfer pipette.
PBS/EDTA: 1mM EDTA in PBS. Store at room temperature for months.
TRIS, SDS, EDTA buffer: 50mM Tris pH 7.4, 10mM EDTA, 1% sodium dodecyl sulfate (SDS), with protease/phosphatase inhibitor mix. Store at −20°C for up to one week.
Detergent Extraction Buffer: 20mM HEPES pH 7.6, 7.5mM MgCl2, 0.2mM EDTA, 30mM NaCl, 1M Urea, 1% NP-40, with protease/phosphatase inhibitor mix. Store at −20°C for up to one week.
0.4N H2SO4.
Trichloroacetic acid.
Acetone.
1M Tris (pH unadjusted).
2.2 Protein Extraction and Electrophoresis Components
BCA protein assay (catalogue number 23227) (Thermo Scientific).
5x Laemmli Buffer: 60mM Tris pH 6.8, 25% glycerol, 5% β-mercaptoethanol, a few flakes bromophenol blue. Store extra at −20°C for months. Store working volume at room temperature for months.
Nitrocellulose membrane, 0.45 μM (catalogue number 162-0115) (BioRad).
Histone H2A antibody (catalogue number sc-8648) (Santa Cruz).
Adenine nucleotide transporter antibody (catalogue number sc-9299) (Santa Cruz).
Nitrile gloves (catalogue number 19-130-1597B) (Fisher).
Oriole (catalogue number 161-0496) (BioRad).
Stainless steel surgical blade (catalogue number 371211) (Bard-Parker).
2.3 Digestion and LC/MS/MS Components
Preparation of any solution that will be used in the digestion or LC/MS/MS steps should be prepared using a hairnet in addition to lab coat and gloves. Special care should be employed to prevent contamination using tips and tubes which have been sealed and when possible, using solvents dedicated for MS use that have been kept contaminant-free.
1.5ml Low Adhesion Microcentrifuge Tubes (catalogue number 1415-2600) (USA Scientific).
0.65mL Prelubricated Microcentrifuge Tubes (catalogue number 3206) (Costar).
HPLC water.
Acetonitrile for LC-MS.
50mM NH4HCO3, 50% acetonitrile. Store at room temperature for a month.
10mM DTT/10mM TCEP: 10mM dithiothreitol, 10mM Tris(2-carboxyethyl)phosphine hydrochloride. Make fresh.
100mM Iodoacetamide. Make fresh.
0.02 μg/ μl trypsin (Promega, catalogue number V5111) in 50mM NH4HCO3. Make fresh on ice.
0.02 μg/ μl chymotrypsin (Roche, catalogue number 11 418 467 001) in 50mM NH4HCO3. Make fresh on ice.
5% formic acid. Store at room temperature for weeks.
0.1% formic acid, 50% acetonitrile. Store at room temperature for weeks.
HPLC vials and inserts (refer to your system for proper models).
Mobile phase A: 0.1% formic acid, 2% acetonitrile. Store at room temperature for weeks.
Mobile phase B: 0.1%formic acid, 80% acetonitrile. Store at room temperature for weeks.
Reversed phase column (catalogue number PFC7515-B14-10) (New Objective).
Xcalibur software (Thermo Scientific).
3. Methods
3.1 Heart Isolation for Whole Heart Lysate
Carry out all steps on ice unless otherwise indicated.
Fill a glass dounce with 2ml whole heart lysate buffer and place on ice along with a beaker of PBS. Anesthetize adult mouse (8–12wks) with isoflurane and sacrifice by cervical dislocation. Work quickly to remove the heart, rinse it in cold PBS and place in dounce. Homogenize on ice, being careful not to create bubbles, until there is no more visible tissue. Collect the lysate in a 2ml centrifuge tube.
Sonicate for 10–15 seconds, 3–6 times. Put sample on ice between sonications. Centrifuge at 4°C for 5 minutes at 16,200g. Remove the supernatant and store at −80°C. The supernatant is the whole heart lysate fraction.
3.2 Nuclear Isolation from Whole Heart
Carry out all steps on ice unless otherwise indicated.
Fill a glass dounce with 2ml lysis buffer and place on ice along with a beaker of PBS. Anesthetize adult mouse (8–12wks) with isoflurane and sacrifice by cervical dislocation. Work quickly to remove the heart, rinse it in cold PBS and place in dounce. Homogenize on ice, being careful not to create bubbles, until there is no more visible tissue. Pass lysate through a 100 μm strainer and collect the lysate in a 2ml centrifuge tube.
Centrifuge at 4°C for 5 minutes at 1,500g. Meanwhile, prepare a 2ml tube with 1ml of sucrose buffer on ice. After centrifugation, remove the supernatant and store at −80°C. This is the cytosolic fraction. Resuspend the pellet in 500 μl cold lysis buffer and gently place on top of the sucrose buffer using a transfer pipette. Be careful not to disturb the sucrose pad or mix the two layers.
Centrifuge at 4°C for 10 minutes at 2,400g. Remove the sucrose pad along with the lipid layer on top, and rinse the pellet in 200 μl cold PBS/EDTA. This is the nuclei pellet.
3.3 Nuclear Isolation from Adult Isolated Cardiomyocytes
Isolation of high quality cardiac myocytes is one of the most important factors for successful experimentation. The protocols for adult cardiac myocyte isolation vary between different laboratories and depend on the downstream experiments. Here we describe a succinct protocol using Langendorff perfusion apparatus. For more details on isolation of cardiac myocytes, please see these protocols 22–24.
Isolate whole heart from animal. The perfusion system should be prepared and warmed up before starting to remove heart from animal.
Connect aorta to the system and perfuse with perfusion buffer to remove blood from the heart, followed by collagenase to digest tissue. Maintain the temperature of perfusate at 36–37°C.
Cut the ventricles in a dish and mince into small pieces using a scalpel. Triturate with a plastic transfer pipette to dissect tissue.
Filter the cell suspension through a 100 μm Nylon cell strainer into 15ml tubes. Sediment cells by gravity for 8 to 10 minutes followed by gentle centrifugation for 1 minute at 180g. Wash pellet in PBS twice. Carry out the next steps on ice unless otherwise indicated.
Resuspend pellet in 1ml ice cold lysis buffer and incubate on ice for 10 minutes.
Centrifuge at 4°C for 5 minutes at 1,500g. Meanwhile, prepare a 2ml tube with 1ml of sucrose buffer on ice. After centrifugation, remove the supernatant and store at −80°C. This is the cytosolic fraction.
Resuspend the pellet in 500 μl cold lysis buffer and gently place on top of the sucrose buffer using a transfer pipette. Be careful not to disturb the sucrose pad or mix the two layers.
Centrifuge at 4°C for 10 minutes at 2,400g. Remove the sucrose pad along with the lipid layer on top, and rinse the pellet in 200 μl cold PBS/EDTA. This is the nuclei pellet.
3.4 Whole Nuclei Preparation
Carry out all steps on ice unless otherwise indicated.
Resuspend the nuclei pellet from section 3.2 (whole heart) or 3.3 (isolated myocytes) in 300 μl TRIS, SDS, EDTA buffer.
Sonicate for 10–15 seconds, 3–6 times. Put sample on ice between sonications. Centrifuge at 4°C for 5 minutes at 16,200g. The pellet should be small. If it is not, additional sonication may be necessary. Remove the supernatant and store at −80°C. The supernatant is the whole nuclei fraction.
3.5 Nucleoplasm and Chromatin Fractionation
Carry out all steps on ice unless otherwise indicated.
Resuspend the nuclei pellet from section 3.2 (whole heart) or 3.3 (isolated myocytes) in 200 μl detergent extraction buffer, and vortex for 10 seconds twice, then place on ice for 10 minutes.
Centrifuge at 4°C for 5 minutes at 16,200g. Remove the supernatant and store at −80°C. This is the nucleoplasm. Rinse the pellet with 200 μl cold PBS/EDTA, and resuspend it in 300 μl TRIS, SDS, EDTA buffer.
Sonicate for 10–15 seconds, 3–6 times. Put sample on ice between sonications. Centrifuge at 4°C for 5 minutes at 16,200g. The pellet should be small. If it is not, additional sonication may be necessary. Remove the supernatant and store at −80°C. The supernatant is the chromatin fraction.
3.6 Acid Extraction (see Note 1)
Carry out all steps on ice unless otherwise indicated.
Resuspend the nuclei pellet from section 3.2 (whole heart) or 3.3 (isolated myocytes) in 200 μl detergent extraction buffer, and vortex for 10 seconds twice, then place on ice for 10 minutes.
Centrifuge at 4°C for 5 minutes at 16,200g. Remove the supernatant and store at −80°C. This is the nucleoplasm. Rinse the pellet with 200 μl cold PBS/EDTA, and resuspend it in 400 μl 0.4N H2SO4. Vortex sample until clumps are dissolved and rotate at 4°C for 30 minutes or overnight.
Centrifuge at 4°C for 10 minutes at 16,000g. Remove the supernatant to a new tube and add to it 132 μl of trichloroacetic acid, drop-by-drop. Invert between drops and place on ice for 30 minutes.
Centrifuge at 4°C for 10 minutes at 16,000g. Discard the supernatant. The supernatant is the acid insoluble fraction. Rinse the pellet with 200 μl of ice-cold acetone. Centrifuge at 4°C for 10 minutes at 16,000g. Repeat the wash by rinsing the pellet with 200 μl of ice-cold acetone and again centrifuging at 4°C for 10 minutes at 16,000g. Discard the supernatant and air-dry the pellet.
Resuspend the pellet in 100 μl of TRIS, SDS, EDTA buffer. Use 1M Tris stock (pH unadjusted) to set the pH to 8. Sonicate for 15 minutes in a water bath (partially filled with ice) to resuspend the protein. Store at −80°C. This is the acid extracted fraction.
3.7 Protein Extraction, Electrophoresis and Digestion
The following steps can be performed at room temperature unless otherwise indicated. When working with samples that will be run on the mass spectrometer, it is important to prevent keratin contamination. The experimenter should wear a lab coat, gloves and hairnet and work in an area free of dust. Similarly, use tips and tubes that have been kept covered, and wash with soap and water the gel running apparatus and box the gel will be stained in. Spray down any surface the gel will touch with 75% ethanol, and wipe with a Kimwipe. Finally use HPLC grade solvents that have been kept clean of contamination.
Remove samples from −80°C and thaw on ice. Using the bicinchoninic acid (BCA) protein assay, determine the protein concentration of each sample. Dilute samples using 5x Laemmli buffer. (Buffer must be at least 1/5 of the final volume, but can be more.) Depending on the fraction, we usually dilute our samples to 0.5–5 μg/ μl. Boil the samples at 100°C for 10 minutes and then return to ice. Once diluted in Laemmli buffer, samples can be stored at −20°C.
Run equal amounts of each sample on a one-dimensional gel. Depending on your intentions, 20 μg of sample is a good starting amount. After running, the gel can be transferred to a nitrocellulose membrane and blotted to verify success of the fractionation (see Note 2), or used for mass spectrometric analysis (subsequent steps).
Transfer gel to a clean, light tight box. Avoid touching the gel, and instead use a squirt bottle with deionized water to loosen the gel off of the glass plate and into the box. Add enough Oriole stain to cover (use nitrile gloves when handling Oriole), and leave on shaker for at least 90 minutes. The gel can also be stained over night at 4°C; however a small amount may precipitate out leaving tiny overexposed flecks on the gel. After staining, the Oriole should be disposed of properly and the gel can be kept at 4°C in deionized water.
Image the gel using a UV light box. Spray off the surface with 75% ethanol and wipe down with a Kimwipe several times before placing your gel on it. It is best to take images at multiple exposures. Choose an exposure that best reflects what you will see when you cut the gel, to mark and label the bands that will be cut. For studies of the total proteome, we cut each lane into 25 2-mm bands. Cut out each band using a small razor, and further section it into 3 equal pieces, width-wise. Collect the three pieces into a labeled 1.5ml tube. (It is best to use “low bind” tubes for digestion.) Store pieces at −20°C.
Digest the gel pieces (see Note 4). Wash gel plugs twice in 50mM NH4HCO3/50% acetonitrile (ACN), dehydrate with ACN, and dry in a Speedvac. Reduce with 10mM DTT/10mM TCEP at 56°C for 30 minutes. Wash and dehydrate the gel plugs as before and alkylate in 100mM iodoacetamide for 22 minutes in the dark. Wash, dehydrate and Speedvac as before. Digest in 0.02 μg/ μl trypsin solution in 50mM NH4HCO3 overnight at 37°C (see Note 3 for enzymatic considerations). Halt the digestion with 5% formic acid solution and collect the supernatant in a 0.65ml low bind tube. Further extract peptides in 0.1% formic acid/50% ACN twice, collecting the supernatant after each, and Speedvac the combined supernatant down to approximately 30 μl. For quantitative analysis, be careful to concentrate each sample equally. Transfer the samples to HPLC vials. Store the samples and the left over gel plugs at −20°C. For an extended protocol see our Jove publication (PMID: 19455095).
3.8 LC/MS/MS
Inject 10 μl of sample for each run using a nano-flow method optimized for a range of peptides. We use a linear gradient from 5%B to 50%B over 60 minutes, to 95%B over the next 15 minutes, and then we hold at 95%B for 10 minutes. We use a 200nL/min flow rate through a New Objective, reversed phase, C18 column (75 μm i.d.). For MS/MS, use a high mass accuracy mass spectrometer and acquire date in a data-dependent mode. We use a Thermo Orbitrap to fragment the six most abundant parent ions.
We recommend repeating this process for at least three biological replicates (samples from different animals) and two technical replicates (the same sample analyzed an additional time by LC/MS/MS). Gross changes in peptide abundance between experimental conditions can be observed in the total ion chromatogram (TIC) which displays each peptide detected as a three-dimensional peak Fig. 3.
Figure 3. 3D rendering of total ion chromatogram.
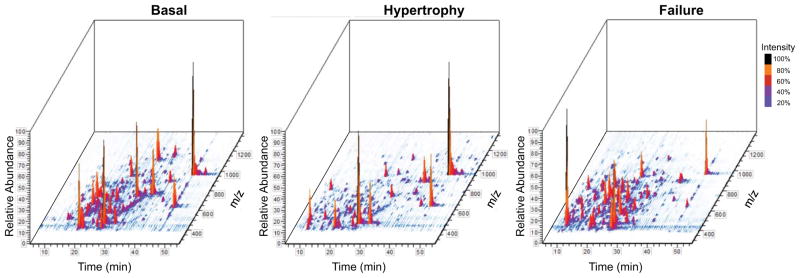
The total ion chromatogram (TIC) from an LC/MS run can be viewed in 3-dimensions to display all peptides detected as individual peaks. The 3D-TIC of acid-extracted chromatin proteins from basal (left panel), hypertrophic (middle panel) and failing (right panel) mouse myocardium illustrate the changes in protein abundance that occur during disease progression. Figures generated using Qual Browser embedded in Xcalibur software. X-axis (time), Y-axis (intensity), Z-axis (m/z).
3.9 Database Searching
Identify peptides by searching the spectra against the Uniprot protein database of choice via a search algorithm (such as SEQUEST or MASCOT) integrated into commercially available software such as BioWorks and Xcalibur, or publically available software such as PROWL, X!Tandem and SpectraST. More details on database searching algorithms can be found in another protocol article25.
Consider modifying search parameters to allow for cysteine carbamidomethylation and methionine oxidation, two common modifications created during the sample processing.
Calculate a false positive rate using reverse database searching. A reverse database is created by reversing protein sequences in the original database.
Filter protein identifications to only accept matches of a threshold confidence. We recommend the following parameters to start with: Xcorr >3 (+2), >4 (+3), >5 (+4); deltaCN >0.1, consensus score ≥20, mass tolerance 2Da for parent ion, mass tolerance of 0.5Da for product ion, at least 2 unique peptides per protein and no more than 3 missed cleavages. Manually inspect all spectra used for protein identification and reject identifications when less than two peptides are identified (see Note 5).
3.10 Label-free Quantitation
Label-free quantitation requires consistent sample prep and LC/MS/MS conditions as well as the use of a high mass accuracy mass spectrometer. While quantitative comparisons using metabolic labeling combine samples into a single run and compare spectra from that same run, label-free quantitation requires samples be run individually and spectra compared across runs. Thus, the following quantitation relies on having taken effort to ensure high reproducibility in the previous steps (see Note 6).
Use individual peptide signals to determine the relative abundance of specific peptides between samples (i.e. healthy and diseased, or treated and untreated) using label-free quantitation. Available programs include Census (Prof. Yates’ group),26 Elucidator (Microsoft),27 SIEVE (Thermo Scientific),28 Scaffold (Proteome Software).29 Briefly, these programs will align data across runs, normalize signal intensity and quantify peptide peaks through spectral counting or by measuring peak area to generate peptide abundance ratios between samples.
Connect peptide abundance to protein identification by employing search algorithms (Mascot, Sequest, X!Tandem) in the above programs.
Assess accuracy and reproducibility through ANOVA analysis of the data. We recommend generating a PCA plot to compare different biological replicates (different experimental samples) and technical replicates (running the same sample on the mass spectrometer multiple times). As already mentioned, these replicates are crucial to label-free quantitation, and a PCA plot will allow you to confirm reproducible grouping of data, with technical replicates most closely clustered. Additionally, close clustering of biological replicates not only confirms accuracy, but also determines the presence of proteomic differences between treatment groups. One representative result is shown in Fig. 4.
Figure 4. Analysis of variance and principal component analysis.

Cardiac chromatin fractions from 3 basal mice (blue), 3 mice in cardiac hypertrophy (red) and 3 mice in cardiac failure (green) were run on a one-dimensional protein gel. Proteins of 75–150kDa were cut and digested. Each sample was analyzed twice by LC/MS/MS for a technical replicate. The data was searched and protein abundance determined by label-free quantitation. ANOVA analysis was used to generate a PCA plot where axes represent peptide intensity and each dot represents a single replicate. Clustering by disease state reveals distinct proteomic changes occuring during cardiac hypertrophy and failure, and confirms the ability of this methodology to reproducibly detect these changes.
3.11 Bioinformatic Analysis
Depending on the different purposes of the project, many bioinformatics tools are available for various analyses. One interesting question to ask is where these genes coding for identified proteins are located in the genome. In this section we will map the proteins identified by MS to chromosomes and visualize the location of these genes within genome. We will then conduct gene ontology analysis for these proteins to build a gene ontology tree with enriched terms. Most of the procedures are straightforward and do not require program coding. Refer to Table 1 for the description of these tools.
Table 1.
Publicly available tools and databases for bioinformatic analysis
| Name URL | Function (Refer to the manual of each tool for more details) |
|---|---|
| Algorithms | |
| ID Mapping www.uniprot.org/?tab=mapping | Map identifiers to or from UniProtKB. |
| PICR www.ebi.ac.uk/Tools/picr/ | Map identifiers based on sequence identity. |
| UCSC Genome Browser genome.ucsc.edu | Visualize the location of genes within genome. |
| ProtParam web.expasy.org/protparam/ | Compute various physiochemical parameters for a given protein. |
| PROSITE prosite.expasy.org | Determine protein domains and families. |
| DAVID Bioinformatics Resources david.abcc.ncifcrf.gov | Annotate large list of genes based on gene ontology. |
| GENE ONTOLOGY (GO) Tools go.princeton.edu | GO Term Finder and GO Term Mapper. Find significant GO terms shared by a list of genes. |
| Clover zlab.bu.edu/clover/ | Identify overrepresented functional sites within a set of DNA sequences. |
| Cytoscape www.cytoscape.org | Analyze and visualize complex protein-protein interaction networks. |
| Databases | |
| Database of Interacting Proteins dip.doe-mbi.ucla.edu/ | Database for experimentally determined interactions between proteins. |
| Ensembl www.ensembl.org | Genome database for vertebrates and other eukaryotic species. |
| InterPro www.ebi.ac.uk/interpro/ | Database for protein families, domains, regions and sites. |
| KEGG PATHWAY www.genome.jp/kegg/pathway.html | Database of molecular interaction and reaction networks. |
| MotifMap motifmap.ics.uci.edu | Comprehensive maps of candidate regulatory elements encoded in the genomes of model species. |
Convert UniProt identifiers to Ensembl Genomes or RefSeq nucleotide identifiers using ID Mapping (see Note 7).
Extract gene annotation information from UCSC Genome Browser. Go to UCSC Genome Browser and select mouse as species. Upload gene identifiers from previous step to the Table Browser and download the gene structure information in BED format.
Visualize the location of these genes in chromosomes. Submit the BED file generated from the previous step to UCSC Genome Browser as a custom track.
Build gene ontology tree with enriched gene ontology terms. Convert UniProt identifiers into Mouse Genome Informatics (MGI) identifiers using ID Mapping. Submit MGI IDs to GO Term Finder and choose “MGI (Generic GO slim)” for GO Slim, if mouse sample is used. Genes can be clustered by GO terms using GO Term Mapper.
Acknowledgments
The Vondriska lab is supported by grants from the National Heart, Lung and Blood Institute of the NIH and the Laubisch Endowment at UCLA. EM is recipient of the Jennifer S. Buchwald Graduate Fellowship in Physiology at UCLA; HC is the recipient of an American Heart Association Pre-doctoral Fellowship; and SF is the recipient of an NIH K99 Award.
Footnotes
Acid extraction allows a greater enrichment for histone proteins as compared to chromatin fractionation, as it selects for proteins that are tightly bound to the DNA. Performing analysis using both fractions allows for a more complete characterization.
Before carrying out mass spectrometric analysis, assess the purity of your fractionation by running a gel and transferring to a nitrocellulose membrane. Probe for a nuclear protein (we use H2A) and a mitochondrial protein (we use adenine nucleotide transporter). Electron microscopy of the nuclei pellet (last steps of section 3.2 and 3.3) can also verify enrichment of intact nuclei.
We typically digest in trypsin 18–24 hours at 37°C. A digestion markedly shorter than this could be incomplete, likewise going longer than 24 hours runs the risk of non-specific cleavage at other residues. Because trypsin cleaves at the C-terminal end of lysines and arginines, the acidic tails of histones are cut into peptides that are often too small to be detected by the mass spectrometer. Substituting chymotrypsin for trypsin for the low-molecular weight bands is an option to get around this without sacrificing the reproducibility of trypsin. Digest in 0.02 μg/ μl chymotrypsin solution in 50mM NH4HCO3 overnight at room temperature for approximately 20 hours. The specificity of cleavage, and thus the peptides that will be generated, will vary with timing, and can be adjusted depending on the specific goals of the experiment.
The protocol included is for in gel digestion as the fractions we are analyzing are very complex. One-dimensional separation by mass via electrophoresis and subsequent cutting of bands greatly reduces the complexity of the sample by distributing the proteins of one fractionation across multiple samples (bands), thereby increasing the ability of the mass spectrometer to detect lower abundance peptides. However, for less complex fractionations (such as acid extractions from a homogenous cell population) or for samples which have undergone subsequent purification (via HPLC for example) the one-dimensional separation may not be necessary and the sample could be directly digested in solution without running a gel or cutting bands. When working with a new protein sample, assess the complexity by running a gel and staining with Oriole. Determine if in-solution digestion is possible based off of the number and intensity of bands. For samples with a large number of bands of varying intensity, in gel digestion would be preferable. However in-solution digestion could be an option for samples with only a few bands of equal intensity, including an equally or more intense band at the molecular weight of your protein of interest. Note that among other adjustments for in- solution digestion, the concentration of enzyme and the length of digestion will vary.
With trypsin, and even chymotrypsin, digestion (see Note 4), histone proteins will often be cleaved into only one or a few detectable peptides (due to their small size and amino acid composition). Thus we recommend searching low molecular weight bands twice to increase the ability to identify histones in these samples. First, search using the above recommended protocol. Then, re-search the data removing the requirements for 2 unique peptides and for the Consensus Score, since this score also takes into account the number of detected peptides and may be low as a result. This will generate a list of possible histone proteins which must be confirmed by manual inspection of their spectra. Briefly, the identified peptide(s) for each should be examined for three key things. First, you should confirm the presence of the parent ion. Second, you should confirm that the majority of the high abundant and medium abundant daughter ions are assigned to your peptide. Third, you should look individually at the B and Y ions by charge. Working across a peptide from N terminal to C terminal there are a series of peptide bonds that could have broken, each one generating a B and Y ion of a different mass. As a rule of thumb, a good fragmentation will give at least five ions of the same charge (for example B2+) in a row. A final consideration for histones is the potential for the single identified peptide to match to more than one variant. In this case, knowledge of the sample and what other histone peptides were identified, as well as other methods may be necessary.
Even when samples are prepared together and run using the same buffers on the same day, small variations in injection, ionization and fragmentation must be accounted for. Several means of normalizing data between runs exist. Peptide elution profiles can be normalized through alignment algorithms. Additionally, signal intensity can be normalized to either background noise, a known analyte in your sample, or a known standard that was added during sample preparation. In all cases, biological and technical replicates are still necessary for determining statistical significance.
Identifier conversion between protein database and gene database is used extensively in this section. However, it should be noted that one gene identifier may be mapped to multiple protein identifiers, and vice versa. PICR tool provided by EBI maps identifiers according to the similarity between protein and gene sequences, and is useful when only sequence is available. However, it is slower than ID Mapping and may take several hours or days to map if a long list of identifiers are provided.
References
- 1.Franklin S, et al. Specialized compartments of cardiac nuclei exhibit distinct proteomic anatomy. Mol Cell Proteomics. 2011;10:703. doi: 10.1074/mcp.M110.000703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Franklin S, et al. Quantitative analysis of chromatin proteome reveals remodeling principles and identifies HMGB2 as a regulator of hypertrophic growth. Mol Cell Proteomics. 2012 doi: 10.1074/mcp.M111.014258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lieberman-Aiden E, et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009;326:289–293. doi: 10.1126/science.1181369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.van Steensel B, Dekker J. Genomics tools for unraveling chromosome architecture. Nat Biotechnol. 2010;28:1089–1095. doi: 10.1038/nbt.1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kornberg RD. Chromatin structure: a repeating unit of histones and DNA. Science. 1974;184:868–871. doi: 10.1126/science.184.4139.868. [DOI] [PubMed] [Google Scholar]
- 6.Zhang Z, Pugh BF. High-resolution genome-wide mapping of the primary structure of chromatin. Cell. 2011;144:175–186. doi: 10.1016/j.cell.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rada-Iglesias A, et al. A unique chromatin signature uncovers early developmental enhancers in humans. Nature. 2010;470:279–283. doi: 10.1038/nature09692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barski A, et al. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–837. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 9.Cuddapah S, et al. Genomic profiling of HMGN1 reveals an association with chromatin at regulatory regions. Mol Cell Biol. 2011;31:700–709. doi: 10.1128/MCB.00740-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schones DE, Zhao K. Genome-wide approaches to studying chromatin modifications. Nat Rev Genet. 2008;9:179–191. doi: 10.1038/nrg2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 12.Schreiber SL, Bernstein BE. Signaling network model of chromatin. Cell. 2002;111:771–778. doi: 10.1016/s0092-8674(02)01196-0. [DOI] [PubMed] [Google Scholar]
- 13.Matouk CC, Marsden PA. Epigenetic regulation of vascular endothelial gene expression. Circ Res. 2008;102:873–887. doi: 10.1161/CIRCRESAHA.107.171025. [DOI] [PubMed] [Google Scholar]
- 14.Ho L, Crabtree GR. Chromatin remodelling during development. Nature. 2010;463:474–484. doi: 10.1038/nature08911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Haberland M, Montgomery RL, Olson EN. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat Rev Genet. 2009;10:32–42. doi: 10.1038/nrg2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Narlikar GJ, Fan HY, Kingston RE. Cooperation between complexes that regulate chromatin structure and transcription. Cell. 2002;108:475–487. doi: 10.1016/s0092-8674(02)00654-2. [DOI] [PubMed] [Google Scholar]
- 17.Bai L, Santangelo TJ, Wang MD. Single-molecule analysis of RNA polymerase transcription. Annu Rev Biophys Biomol Struct. 2006;35:343–360. doi: 10.1146/annurev.biophys.35.010406.150153. [DOI] [PubMed] [Google Scholar]
- 18.Cairns BR. The logic of chromatin architecture and remodelling at promoters. Nature. 2009;461:193–198. doi: 10.1038/nature08450. [DOI] [PubMed] [Google Scholar]
- 19.Heineke J, Molkentin JD. Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat Rev Mol Cell Biol. 2006;7:589–600. doi: 10.1038/nrm1983. [DOI] [PubMed] [Google Scholar]
- 20.Rajabi M, Kassiotis C, Razeghi P, Taegtmeyer H. Return to the fetal gene program protects the stressed heart: a strong hypothesis. Heart Fail Rev. 2007;12:331–343. doi: 10.1007/s10741-007-9034-1. [DOI] [PubMed] [Google Scholar]
- 21.Razeghi P, et al. Metabolic gene expression in fetal and failing human heart. Circulation. 2001;104:2923–2931. doi: 10.1161/hc4901.100526. [DOI] [PubMed] [Google Scholar]
- 22.Louch WE, Sheehan KA, Wolska BM. Methods in cardiomyocyte isolation, culture, and gene transfer. J Mol Cell Cardiol. 2011;51:288–298. doi: 10.1016/j.yjmcc.2011.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schluter KD, Schreiber D. Adult ventricular cardiomyocytes: isolation and culture. Methods Mol Biol. 2005;290:305–314. doi: 10.1385/1-59259-838-2:305. [DOI] [PubMed] [Google Scholar]
- 24.O’Connell TD, Rodrigo MC, Simpson PC. Isolation and culture of adult mouse cardiac myocytes. Methods Mol Biol. 2007;357:271–296. doi: 10.1385/1-59745-214-9:271. [DOI] [PubMed] [Google Scholar]
- 25.Nesvizhskii AI, Vitek O, Aebersold R. Analysis and validation of proteomic data generated by tandem mass spectrometry. Nat Methods. 2007;4:787–797. doi: 10.1038/nmeth1088. [DOI] [PubMed] [Google Scholar]
- 26.Park SK, Venable JD, Xu T, Yates JR., 3rd A quantitative analysis software tool for mass spectrometry-based proteomics. Nat Methods. 2008;5:319–322. doi: 10.1038/nmeth.1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lomenick B, et al. Target identification using drug affinity responsive target stability (DARTS) Proc Natl Acad Sci U S A. 2009;106:21984–21989. doi: 10.1073/pnas.0910040106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kamleh A, et al. Metabolomic profiling using Orbitrap Fourier transform mass spectrometry with hydrophilic interaction chromatography: a method with wide applicability to analysis of biomolecules. Rapid Commun Mass Spectrom. 2008;22:1912–1918. doi: 10.1002/rcm.3564. [DOI] [PubMed] [Google Scholar]
- 29.Searle BC. Scaffold: a bioinformatic tool for validating MS/MS-based proteomic studies. Proteomics. 2010;10:1265–1269. doi: 10.1002/pmic.200900437. [DOI] [PubMed] [Google Scholar]