

Abstract
It has been appreciated for some time that cardiovascular disease involves large-scale transcriptional changes in various cell types. What has become increasingly clear only in the last few years, however, is the role of chromatin remodeling in cardiovascular phenotypes in normal physiology as well as in development and disease. This review summarizes the state of the chromatin field in terms of distinct mechanisms to regulate chromatin structure in vivo, identifying when these modes of regulation have been demonstrated in cardiovascular tissues. We describe areas in which a better understanding of chromatin structure is leading to new insights into the fundamental biology of cardiovascular disease.
Introduction
The ordered packing in the nucleus of six billion base pairs of DNA, along with thousands of proteins and RNAs, is one of the most remarkable feats of molecular organization in biology. Genomic structure is highly dynamic in vivo: mitotic reorganization of the chromosomes is exquisitely choreographed and, when not replicating itself, the genome can scaffold rapid and large-scale changes in transcription. Genomic function is inherently dependent on non-DNA factors and is likewise quite plastic: the same DNA holds the information to create every cell in a multicellular eukaryote which, when read by the appropriate protein and RNA machinery, can lead to normal differentiation, multipotency, or disease.
The functional unit of chromatin is the nucleosome, a heteromultimeric protein complex comprised of 8 proteins and bound by ~150 bp of DNA, the structure of which is known at atomic resolution.(1) The precise structural features of the endogenous genome are unknown but there are several key properties that influence the likelihood of a region of DNA to be available for transcription, including: histone variants (and their post-translational modification, PTM), histone modifying enzymes and ATP-dependent chromatin remodeling proteins, non-nucleosomal chromatin structural proteins, and non-protein-coding RNAs. The DNA sequence itself also has a major role in genomic structure, and modifications to this sequence (e.g. cytosine methylation) participate in local accessibility and transcription. These DNA and non-DNA factors together endow global features alternatively associated with gene expression, referred to as euchromatin, or transcriptional repression, labeled heterochromatin (which can further be divided into the constitutive or facultative variety, depending on whether it is universal or cell type-specific, respectively; Figures 1 and 3). To understand transitions in genomic structure that underlie changes in phenotype of the cell, the logic through which these multiple factors influence genomic structure must be revealed.
Figure 1. Mechanisms of chromatin remodeling.

(Top Panel) DNA can be packaged as heterochromatin (tightly packaged and generally inaccessible to proteins such as transcriptional machinery) or as euchromatin (accessible to transcription). (Middle Panel) Chromatin remodeling proteins can alter DNA methylation or histone PTMs, the combination of which can be recognized by “reader proteins” (purple) to confer chromatin state. (Bottom Panel) Non coding RNAs can interact with these chromatin remodelers to target them to specific loci, leading alternatively to gene activation or silencing.
Figure 3. Multiple levels of structural organization regulate gene expression in an interphase nucleus.

(Top) An actively transcribed (green) and repressed (red) segment of DNA occur on distinct regions of the same chromosome, directly regulated by chromatin remodelers and noncoding RNA. (Bottom Left) The higher-order chromatin structure surrounding these segments endows another level of regulation, with the cumulative effect of local modifications of nearby segments and neighboring chromosomes combining to create distinct repressive and activating environments. (Bottom Right) The position of the segments in relation to the global features of the nucleus imparts yet another level of regulation. All three levels regulate each other interdependently.
Cardiovascular disease is a scourge on the developed world, where it is the leading cause of morbidity and mortality. Despite the development of new therapeutic approaches,(2) heart failure is a particularly debilitating form of cardiovascular disease, with a ~50% death rate 5 years after diagnosis.(3) The last two decades have seen remarkable progress in our understanding of the molecular events that mediate heart failure, including its antecedent, cardiac hypertrophy. The role of transcriptional circuits has been extensively studied in cardiac development and disease; in contrast, the role of endogenous chromatin structure and its remodeling to facilitate transcriptional responses (thereby underpinning cell type-specific proteomes, and phenotypes) in the cardiovascular system is a recently emerging field. Our purpose herein is to succinctly summarize the current understanding of chromatin biology in the cardiovascular system, with a particular emphasis on the heart. We also aim to highlight concepts and techniques (Figure 2) from non-cardiovascular systems that reveal new avenues of investigation to understand cardiovascular biology and disease.
Figure 2. Methodologies and applications.
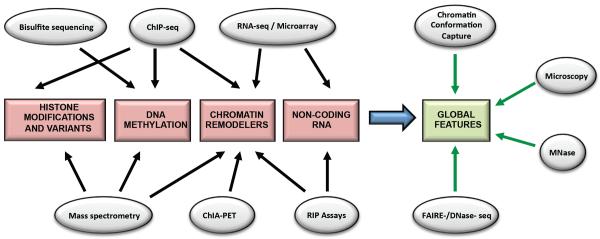
Various techniques currently utilized, alone or in combination, to analyze chromatin structure and regulation.
Histone Modifications and Histone Variants
Although histones have been known for some time to be regulated by PTM, recent advances in proteomic mass spectrometry over the past decade have advanced our understanding of the extent of this regulation, and dramatically increased the rote number of modifications that have been identified (>100 for some cell types).(4) An advantage of mass spectrometry is the ability to study combinations of PTMs on a single peptide or even intact protein.(5) One example of this approach comes from analysis of histone H3.2 PTMs in HeLa cells where differential PTM resulted in 150 different forms, or “protein species”, of H3.2.(6) In addition to combinatorial PTM identification, intact proteins can also be useful to study splice variants, and histone variants with high sequence similarity, where there are few unique tryptic peptides.
Functionally characterizing these modifications, on the other hand, remains a low throughput endeavor, and thus there are a handful of modifications for which specific transcriptional phenotypes have been ascribed and subsequently validated in multiple cell types. Commonly studied silencing marks, which by definition are associated with DNA regions with lower expression, include H3K9me3 (constitutive heterochromatin)(7) and H3K27me3 (facultative heterochromatin),(8) where constitutive heterochromatin is considered more permanently silenced. Commonly studied euchromatin marks include H3K4me3, which marks promoters and transcriptional elongation,(9) H3K4me1, which marks enhancers,(10) H3K27ac, which marks active enhancers,(11) and H3K36me3, which marks actively transcribed gene bodies.(12) In general, histone acetylation makes the DNA more euchromatic.(13) Less well-studied PTMs include phosphorylation, ubiquitination and SUMOylation, although all these modifications have been shown to exist on chromatin.
Nucleosomes can also be modified by the histone variants they contain. Histone variants have diverse roles, including association with transcriptional elongation in active gene bodies, kinetochore formation at the centromere, RNA Pol II recruitment at promoters, and chromatin remodeler recruitment after DNA breaks, as just some examples.(14) Finally, linker histone H1 is another class of histone, with its own panel of variants that associate peripherally with nucleosomes, organizing them into higher-order structure.(15)
Chromatin immunoprecipitation followed by next generation DNA sequencing (ChIP-seq) has become a widely used tool for studying the distribution of specific histone PTMs and variants (as well as other chromatin-bound proteins) across the genome. Integration of data sets from multiple ChIP-seq experiments enables localization of PTMs which may cooperate together to form a histone code,(16) or which may, even in the absence of existing on the same nucleosome, define chromatin domains.(17, 18)
Perhaps the most mature field of chromatin research in the heart is the regulation of histone modifications (Figure 1), acetylation in particular, which is governed by HDACs (histone deacetylases) and HATs (histone acetyltransferases).(19) Despite important insights from many cardiac studies on HDAC and HAT isoforms—primarily executed using pharmacologic and/or genetic gain/loss of function of individual isoforms or protein families—a critical unresolved question is which residues on the histone tails are modulated during disease, and how do the HDACs/HATs (and other modifying enzymes, such as histone methyltransferases) target the correct nucleosomes to bind and modify in their target genes. It is likely that a combination of histone variant expression, histone PTMs, DNA sequence and non-nucleosomal chromatin binding proteins together specify a disease-specific genomic structure and transcriptional response in cardiomyocytes (thus explaining how global inhibition of HDACs and HATs can have apparently specific effects), but a combination of proteomics to dissect HDAC/HAT complexes in the heart and genomics to map the localization of these molecules would be needed to test this conjecture.
One salient study in which ChIP-seq revealed a global principle of gene regulation in the heart has come in the area of development. During the process of differentiating human embryonic stem cells (hESCs) into cardiomyocytes, H3K27me3 decreases, while H3K36me3 and H3K4me3 increase at the promoters of genes that regulate cardiac differentiation.(20) This differs from cardiac structural proteins that also gain H3K36me3 and H3K4me3 during differentiation but lack inhibitory H3K27me3 even in less differentiated states (and even when these genes are silenced).(20) This suggests that repressive marks are not necessary to block transcription, but rather, are employed as an extra precaution (in addition to the lack of activating marks) to keep genes that promote differentiation tightly silenced in precursor lineages, since aberrant expression of a proteins whose expression directly induces the expression of other proteins (e.g. a developmental transcription factor) would be more detrimental than aberrant expression of a single structural protein.(20) A similar study performing ChIP-seq and RNA-seq during the differentiation of mouse ESCs into cardiomyocytes found that multiple genes showing a similar expression pattern during differentiation could be subdivided by chromatin marks to separate out genes coding for different gene ontology analysis-defined pathways.(21) They then used the presence of H3K4me1 alone, or H3K4me1 and H3K27ac together, to identify poised or active cardiac enhancers, respectively.(21) Interestingly, many poised enhancers in earlier lineages never became active in the cardiac linages, which the authors attribute to the plasticity of the precursor cells (i.e., enhancers poised for activation in other lineages).(21) Furthermore, the group of active enhancers was highly cell type-specific.(21) H3K27ac levels at active enhancers help explain the convergence of chromatin structural features and transcription factors to coordinate large-scale gene expression profiles. This study found that loci with high levels of H3K27ac surrounding a small dip in H3K27ac signal represented active enhancers which were open to transcription factor (TF) binding (hence the dip).(21) Taken together, these studies show how the chromatin patterns at promoters and enhancers progress during development to modulate the multiple large-scale gene expression patterns being driven by the—simultaneous, developmentally speaking—changing abundances of distinct transcription factors.
ChIP-seq for H3K4me3, H3K4me1 and H3K27ac was performed on adult mouse hearts and embryonic mouse hearts, day E14.5 and used, in conjunction with ChIP-seq for other chromatin proteins, to identify promoters and enhancers.(22) Typically, the enhancer is matched to the closest TSS or to the TSSs not separated from the enhancer by a CTCF binding event. However, this study compared the chromatin state at each enhancer to the presence of RNA Pol II at every promoter on the chromosome, to find the cis- and trans-regulated promoters whose activities are best correlated with that of the enhancer.(22) Rather than explain developmental or pathological processes specifically, this data offers insight into the structural (domains of interacting loci) and functional organization of the cardiac genome.
ChIP followed by microarray analysis of myocytes isolated from healthy or failing hearts of Dahl salt-sensitive rats shows that global distribution of H4K20me3, H3K27me2, H3ac, H4ac, H3K4me2 and H3K9me2 are significantly correlated between healthy and failing hearts.(23) However, H3K4me3 (as opposed to H3K4me2) and H3K9me3 (as opposed to H3K9me2) show a significant lack of correlation between healthy and disease states.(23) ChIP-seq for H3K4me3 and H3K9me3 were repeated on human heart samples from patients with valvular disease and preserved ejection fraction or patients with dilated cardiomyopathy and depressed ejection fraction.(23) Loci of 1kb with multiple reads mapped to it were deemed “high clusters” of H3K4me3 or H3K9me3, and were found to be enriched in cardiac specific genes.(23) In fact, even high clusters only found in one or the other of the two disease patient groups were part of pathways which overlapped between the two groups, even if the specific genes with the altered H3 PTMs did not.(23) This suggests that there is enrichment of H3K9me3 and H3K4me3 high-density regions at cell-specific genes, as opposed to, for example, predominately being found at developmentally silenced and housekeeping genes respectively, and as such, it is these PTMs that show the greatest change during disease.
Features of the “histone code”, if it exists, have been described to include writers, readers and erasers that respectively add, recognize or remove modifications from histones. There are two general approaches to studying the relationship amongst these types of chromatin modifiers: single gene-based approaches, which examine the behavior around well-characterized loci, drawing broad conclusions, and genome-wide or proteome-wide approaches, that are loci independent but that, for reasons of scale, lack extensive traditional analysis of individual target genes. One recent study(24) in the latter category determined the proteins associated with known trimethylation marks (five in total: H3K4me3, H3K36me3, H3K9me3, H3K27me3, and H4K20me3) as a discovery-based approach to map the proteomes of molecules ostensibly reading these histone PTMs.
Finally, histone variants, in addition to PTMs, play a role in heart disease. Quantitative proteomic analysis of proteins isolated from mouse cardiac nuclei by acid extraction found 54 histone protein variants in the heart, with a global decrease in the ratio of linker histone to core nucleosome in cardiac hypertrophy.(25) Additionally, histone H2A.Z was found to increase in a mouse model of hypertrophy, with shRNA experiments in isolated cells demonstrating its necessity in this process.(26) Proteins peripheral to the nucleosome and yet fundamentally involved in chromatin structure, including the H1 family (so-called linker histones(25)) and high mobility group, HMGA1(27) and HMGB2(25), specifically, have all been implicated in pathological cardiac growth. The role of other histone variants and non-nucleosomal chromatin structural proteins, and how they target specific nucleosomes in the heart, remains to be elucidated.
DNA Modifications
In mammals, DNA methylation occurs on cytosine residues with an adjacent guanine residue on the 3′ end, so called CpG sites, forming 5-methylcytosine.(28) The majority of CpG sites are methylated in vertebrates, however, there are regions of DNA, called CpG islands, which are enriched for CpG and are hypomethylated.(29) Analysis of multiple vertebrate genomes shows CpG islands enriched at the 5′ end of housekeeping genes, as well as some occurring at the 5′ or 3′ end of tissue-specific genes.(30) It was thought that disease-specific changes in methylation occurred at CpG islands and gene promoters, however a study on human colon cancer found that the majority of the changes were occurring in a new region, coined “CpG island shores,” which are regions within 2kb of CpG islands with less dense CpG nucleotides.(31)
Methyl CpG binding protein 2 (MeCP2) is a chromatin protein that can bind to 5-methylcytosine and facilitate gene compaction and silencing.(32) It is considered a general rule that cytosine methylation silences transcription, including repressing transposon expression.(33) However, there are nuances. Highly expressed genes tend to have hypomethylation in the promoter, but hypermethylation in the gene body. It is thus not surprising that the methylation patterns vary between cell types. Methylation array analysis of human heart, lung, and kidney found tissue-specific methylation patterns.(34)
DNA (cytosine-5-)-methyltransferase 1 (Dnmt1), acts on hemimethylated DNA to maintain methylation,(35) while Dnmt3A and Dnmt3B are responsible for de novo methylation,(36) with some exceptions.(37) DNA methylation can be maintained between cell divisions, making it truly epigenetic.(38) However, DNA demethylation can also occur during cell division (passively), and it has recently become appreciated that demethylation can also be an active process involving hydroxymethylation; hydroxymethyl-cytosine is enriched at enhancers and active promoters and exons in ESCs and may poise chromatin for transcription.(39) Additionally, 5-methylcytosine is enriched at nucleosomes and in exons.(40)
The most widely applied method for genome-wide analysis of DNA methylation is bisulfite sequencing, in which treatment of DNA with bisulfite converts unmethylated cytosines to uracil.(41) Following comparison with a reference genome, the mismatched bases are inferred to be unmethylated cytosines. Other methods that include targeted approaches using designer probes or methylation sensitive restriction enzyme digestion have advantages of increased coverage of select regions of the genome and of better specificity in terms of single base resolution and less bias for size of sequenced fragments, respectively. A final sequencing-based method is a variation of ChIP-seq, where antibody to methylated cytosine is used, so called methylated DNA immunoprecipitation (MeDIP).(42) MeDIP analysis is biased toward loci with high methylation,(42) and since the base methylation status is not read, single nucleotide resolution is not obtained.
There is an increasing recognition that specific genes may be differentially methylated in heart and vascular disease and, with the advent of the aforementioned genome-wide technologies, the extent of this regulation is becoming more evident. Some recent examples include a study on ventricular septal defects, the most common form of congenital heart disease, which found hypermethylation in the NOX5 gene, which plays a role in septum development.(43) Another study analyzed human left ventricular samples by MeDIP-chip (MeDIP followed by analysis on a microarray chip containing CpG island and promoter sequences) followed by bisulfite treatment and PCR for confirmation.(44) This study found altered methylation in disease corresponding to altered expression at 3 genes involved in angiogenesis.(44) Differential methylation can also affect intergenic regions. One study found human heart samples from end-stage cardiomyopathic patients were significantly hypomethylated in satellite repeat regions and these regions were up-regulated 27-fold in expression over control hearts.(45) In a separate study, pregnant mice exposed to the endocrine disrupting compound diethylstilbestrol birthed offspring which exhibited altered contractility when exercised, in part due to increased expression of Dnmt3a and increased methylation in the promoter of calsequestrin 2.(46) A study of selenium deficiency, associated with diastolic dysfunction and myocardial fibrosis in mice, found that low doses of selenium supplementation were capable of altering DNA methylation due to changes in the methionine-homocysteine cycle.(47) This suggests that methylation in the heart can be quite dynamic and potentially sensitive to a range of stimuli.
MeDIP followed by sequencing analysis of healthy and diseased human left ventricular samples showed significant differences in global methylation of CpG islands of gene promoters (cardiomyopathy samples hypomethylated versus control) and gene bodies (cardiomyopathy samples hypermethylated versus control), but not in 3′UTRs, enhancers, or intergenic regions.(48) The occurrence in disease of increased hypomethylation in promoters, increased hypermethylation in gene bodies, and lack of alteration in promoters of down-regulated genes together suggest that there is a global change in methylation during disease which helps up-regulate a subset of genes. If this overall mechanism is more universal, and how the specific pattern of methylation varies depending on the type of cardiovascular disease, remains unknown.
DNA methylation is also an important part of cellular reprogramming and differentiation. Multiple studies have evaluated the effectiveness of using 5-azacytidine (a nucleoside analogue which inhibits DNA methylation) on driving mesenchymal stem cells toward a cardiomyogenic lineage,(49) including a proteomic study identifying the cytoskeletal and metabolic proteins whose expression is altered after treatment with the compound.(50) Additionally, MeDIP analysis coupled with CpG island arrays on mouse brain, heart, liver and testis tissue taken at multiple developmental time periods (E15 to adult) shows that the majority of methylation differences are due to tissue specificity.(51) Thus, methylation is an important part of endowing cell specificity during differentiation and a deeper understanding of this regulation can be used to manipulate cellular reprogramming.
Noncoding RNA
Advances in genomic technologies have enabled investigation of the complete transcriptome of a cell. These analyses have reinforced the concept that RNA is not only an intermediary molecule between DNA and proteins, instead executing various tasks related to gene expression, macromolecular complex formation, mRNA translation and chromatin structure (Figure 1). In fact, around 98% of all transcriptional output in humans is non-protein-coding RNA,(52) with microRNAs being particularly well-studied in the molecular basis of cardiac development and disease(53, 54) including as potential biomarkers.(55) Of the two major techniques used to analyze the regulatory role of noncoding RNAs (ncRNA), the main advantages of RNA-seq over RNA microarrays are that the former does not require the target genomic sequence on the array and produces lower background levels.
Several recent investigations using RNA-seq in mouse models of heart disease have shown a widespread role for other forms of RNA processing, including splicing(56, 57), although the extent to which RNAs function to directly affect chromatin structure and gene expression in cardiovascular disease, is unknown.
High-throughput techniques have shown evidence for production of large amounts of long non-coding RNAs from the human genome.(58) These lncRNAs are molecules bigger than 200 bp and lack protein-coding potential. lncRNAs have been show to have many interesting actions(59) including gene silencing, enhancer activity, alternative splicing and as scaffolds for complexes of proteins bound to the genome (Figure 1). Two very recent studies have demonstrated the existence of tissue-specific lcRNAs involved in cardiac function, including the mesoderm-specific Fendrr, loss of which is embryonic lethal due to cardiac hypoplasia and functional defects,(60) and Braveheart, required for commitment of embryonic stem cells to the cardiac lineage.(61) The extent to which these or other ncRNAs participate in normal cardiac function in adults, including their roles in disease, remain to be determined.
Chromatin Remodelers
ATP-dependent chromatin remodelers are a class of chromatin proteins important for dynamic reorganization and maintenance of chromatin structure (Figure 1). There are 5 main families of chromatin remodeling complexes: ISWI and SWI/SNF (which are the best studied families), INO80, NURD/Mi-2/CHD, and SWRI.(62) These complexes can slide/space(63) or eject nucleosomes(64) to alter access to the DNA by transcription factors. Additionally, they can remove the H2A-H2B dimer(65) and exchange dimers to incorporate histone variants(66) as revealed by single molecule methods.(67) This set of nucleosome manipulations enables chromatin remodelers to participate in a variety of biological processes.(62) For example, the SWI/SNF family is involved in RNA polymerase regulation,(68) as well as alternative splicing(69) and transcriptional elongation.(70) The ISWI family also regulates elongation.(71) The NURD/Mi-2/CHD family can facilitate gene silencing, with Mi-2 able to associate simultaneously with HDACs and methylated DNA binding proteins, linking DNA methylation to histone deacetylation and transcriptional repression.(72) Outside of affecting transcription, the SWRI proteins are important for DNA repair,(73) and the ISWI family(74) and the SWI/SNF family(75) are important for cohesion between sister chromatids during DNA replication.
The role of chromatin remodelers in the heart has been reviewed elsewhere,(76) as has been the role of the SWI/SNF complex in cardiac progenitor cells.(77) Brg1, one of two ATPase subunits of the SWI/SNF family (BAF family in vertebrates), is critical for trabeculation, as well as the formation of the right ventricle, outflow tract, and septa during development, in part through its regulation of Bmp10 and Adamts1 expression as shown by mouse knockout studies.(78) Baf180 and Baf60C knockdown are both embryonic lethal resulting in underdeveloped ventricles, with Baf60C serving as a heart-specific Baf subunit that helps target the complex to cardiac enhancers.(79, 80) In disease, Brg1 is up-regulated following pressure overload and acts with HDACs and poly (ADP ribose) polymerase (PARPs) to coordinate the change in myosin heavy chain expression.(81) Inducible knockout of Brg1 before pressure overload attenuates both hypertrophy and fibrosis. Thus, like in development, the Baf complex can respond to environmental cues to regulate gene expression and cardiac morphology.
Members of the other chromatin remodeling complexes have also been analyzed, including by genetic manipulation in animal models. In zebrafish, a gain of function mutation in reptin, an ATPase in the INO80 family, results in cardiomyocyte hyperplasia during development after formation of the heart tube, while knockdown of Pontin, another ATPase in the complex, also results in hyperplasia.(82) Like with Brg1, Chd7, a member of the CHD family, has also been shown to interact with Tbx transcription factors, with heterozygous loss of function mutant mice having malformed aortic arches.(83) Taken together, these studies support the observations in the SWI/SNF (Baf) complex for different protein constituents to be able to tailor the function of the complex, for the complexes to modulate transcription factor activity, and for the complexes to participate in large-scale global transcriptional reprogramming.
As with the histone-modifying enzymes, it is important to better understand how these remodeling complexes are targeted to specific nucleosomes in different cell types, developmental stages, and disease states, with some evidence already existing for how histone PTMs facilitate this process.(80, 84) It is remains to be determined how the exchangeable protein components of these complexes can modulate their function and localization. Because many of the studies on molecular actions of ATP-dependent remodelers are performed on reconstituted chromatin in vitro, it remains unknown what effect the presence of other chromatin binding proteins and modifications have on the actions of these remodelers when they target the genome in vivo.
Global Structural Features of Genomes
What the genome looks like in vivo is unknown. All of the foregoing chromatin features (Figure 2) combine to package the genome into the nucleus in a non-random, dynamic structure (Figure 3); approaches to measure this structure directly are a key frontier in chromatin biology. Microscopy studies have provided insight into the regulation of nuclear size and morphology in cardiovascular disease,(85, 86) providing the rationale to investigate in a locus-specific, yet genome-wide manner, the properties of DNA packing in heart and vascular cells. The development of different Chromosome Conformation Capture (3C) variants during the last decade and the combination of these techniques with the new high-throughput sequencing methods has provided a global view of how chromatin architecture is involved in gene regulation by allowing the formation of active or repressive “microenvironments”.(87) These techniques share the advantage of much higher resolution, to the single base level,(88) in contrast with microscopy. All of the 3C variants have a common workflow of cell fixation, DNA digestion (using restriction enzymes) and DNA re-ligation (under diluted conditions to promote intramolecular ligations between cross-linked fragments) followed by sequencing of the resultant DNA library to examine the frequency of interactions between different loci.
Although this is the early days for application of these techniques to the cardiovascular system in a genome-wide manner, some very interesting studies have emerged in the area of heart failure. Previous studies had demonstrated that the transcription factor Nkx2-5 mediates formation of a chromatin hub in a regulatory element upstream of the atrial natriuretic factor (ANF) gene,(89) a known indicator of hypertrophy. Using a 3C approach to map this chromatin hub, it was subsequently revealed how this hub ensures ANF structural availability and expression during heart development, but is not necessary in the setting of pressure-overload hypertrophy and failure, when ANF is reactivated. New enhancers can be identified by analyzing the gene expression microarray databases generated from previous publications(90) but also by using 3C technology, although this approach has not been applied to the cardiovascular system to our knowledge. ChIP-seq has been applied for this endpoint in the heart,(91)—for the transcriptional co-activator normally present in enhancers, p300, and five cardiac transcription factors essential for initiating or maintaining cardiac gene expression: GATA4, NKX2-5, TBX5, SRF and MEF2A. Genetic variation in several of these transcription factors has recently been reported in humans with congenital heart disease,(92) highlighting the potential translational impact of studies to understand their regulation at the genomic level.
A recent study examining global dynamics of chromatin function involved ChIP-seq-based determination of changes RNA POL II binding and H3K9-ac occupancy across the genome following pressure overload in mice.(93) Four patterns of change were revealed 4 days after stress, determined by RNA POL II behavior but also showing characteristic features for H3K9ac: promoter clearance of POL II (i.e. a paused polymerase begins transcribing), de novo recruitment of POL II across the promoter and gene body, a combination of the previous two, and decreased POL II binding. These modes of RNA POL II behavior were used by the authors to distinguish between transcripts regulated at the transcriptional or post-transcriptional level, although these studies also hold the potential to discriminate functionally different genomic regions based on their occupancy by RNA POL II and/or H3K9ac, regardless of the known annotation of the DNA (e.g. as coding or noncoding).
Several other approaches for examining chromatin functionality and/or nucleosome positioning (which itself can influence gene expression and genomic packaging(94)) have been developed and applied in a genome-wide manner in recent years. These include Formaldehyde-Assisted Isolation of Regulatory Elements, followed by sequencing (FAIRE-seq), which is used to identify regulatory elements of actively transcribed genes.(95) DNase-seq takes advantage of DNase I endonuclease to cut at DNase I sensitive sites, usually accessible DNA not protected by packaging, followed by sequencing, to detect regulatory elements and linker DNA.(96) Unlike DNase I digestion, micrococcal nuclease (MNase) digestion is less restrained (not limited by accessibility) and cuts nucleosome-depleted linker DNA.(97) DNA adenine methyltransferases identification (DamID) is another widely used method, in which a protein of interest is tagged with E. coli adenine methyltransferase (not normally expressed or present in eukaryotic cells),(98) to understand DNA-protein interactions. Any regions of the genome that interact with the tagged protein also are methylated, enriched by pull down of antibody recognizing methylated sites or enzymatic digestion (which eliminates potential variability due to antibody affinity), and then mapped and analyzed.(99) DamID has been used to map HP1 to heterochromatin associated with transposons.(100) This technique has also been used to assess DNA regions that associate with the nuclear lamina and mechanism of laminar recruitment of chromatin. One such study has identified a GAGA sequence motif, common to DNA regions associated with lamina, as a potential sequence-dependent mechanism for recruitment of DNA to the nuclear membrane in mouse fibroblasts.(101) Together, these genome-wide studies of nucleosome positioning are revealing the principles for endogenous chromatin structure from the standpoint of DNA sequence specification.(94, 102)
Future Perspectives
The rapid advance of the study of chromatin (Figure 2), with the advent of next generation sequencing technologies, proteomics and computational biology, have driven many investigators to propose models of chromatin states(17, 18) which, in our opinion,(103) should include direct measurements of structural features that can be correlated with function (often transcription, although there are other functions(104) the genome serves in the cell) of super-genic regions of the genome. Such efforts to codify the behavior of chromatin and to understand intermediate structural elements—between the scale of the nucleosome and the entire nucleus—are a critical step to understanding how the same genome encodes multiple cell types.
In the cardiovascular realm, a major challenge is to determine what aspects of the biology of chromatin understood from simpler organisms are relevant in the human heart and vasculature (Table 1; Table references:(25, 48, 105–108)). Key aspects of chromatin biology discussed in this review that remain unknown include: What is the role of DNA methylation in higher organisms and is this modification truly stable in differentiated cells?(109) How do the numerous histone variants, not present in lower organisms but known to be expressed in mammals,(25) contribute to genome specification amongst different cell types? What is the logic that governs interaction between chromatin remodelers and transcription factors, and how is this temporally regulated in disease to activate genome-wide changes in transcription? What are the unique structural adaptations that the multi-nucleated, largely heterochromatic, adult cardiomyocyte carries out to package its genome? We believe that investigating new areas of chromatin biology in cardiovascular system will be most effective if executed in two parallel approaches that differ in conception: we should try to “confirm” observations from lower organisms and non-cardiovascular cells in the heart and vasculature; however, a uniquely fruitful approach, we contend, is to explore anew in an unbiased manner (not eschewing what is known in lower organisms) the behavior of chromatin in various stages of cardiovascular development, normal physiology and disease. The latter approach hypothesizes that multicellular eukaryotes have evolved different, albeit not necessarily more complex or more effective, mechanisms to solve the problem of genome packaging and regulation.
Table 1.
Current state of understanding of various chromatin structural regulators in the cardiovascular system.
| Studied in CV Development | Studied in CV Disease | Well-studied Players in CV System | Comment | |
|---|---|---|---|---|
| Histone Modifying Enzymes | Extensive | Extensive | HDAC: Class I,II HAT: p300, CBP HMT: PRC2,, MLL HDM: Jumonji domain-containing proteins |
Extensive KD/OE and pharmacological studies (105) Limited understanding for genomic distribution of specific modifications and target loci |
| Histone Variants | Limited | Moderate | H2A.z | Variants expressed in the heart identified (25) but only a few understood mechanistically (26) |
| Histone Post-translational Modifications | Moderate | Moderate | Acetylation, Methylation | Global positioning analyzed by ChIP-seq (20, 21, 23) Proteomic analyses of PTM abundance, PTM combinations, and PTM interactions with “reader” proteins are still necessary |
| Non-nucleosomal Structural Proteins | Limited | Moderate | HMGA, HMGB | Some structural proteins have been well-studied in other systems, but their functions have yet to be addressed in the heart (25, 27) |
| ATP-dependent Chromatin Remodelers | Moderate | Moderate | Brg1, BAF complex | Members of major families studied in the heart (79, 80), though disease studies in this field are still incomplete |
| DNA Methylation | Moderate | Moderate | meC, Dnmt | Some loci specific studies (106) Few global studies (48) |
| Noncoding RNA | Extensive | Extensive | miRNAs | Specific miRNAs studied extensively (53, 54) for transcriptional regulation, but the role of noncoding RNA for regulating chromatin structure is needed (60, 61, 107) |
| Nucleosome Positioning | Limited | Limited | - | Emerging field in chromatin biology not yet explored in the heart |
| Chromatin Conformation | Limited | Limited | Enhancers and chromatin loops | Studies in the heart of this type are recent and include measuring long-range interactions (89) and nuclear organization by microscopy (108) |
Acknowledgments
List of Grants: NIH HL105699, HL115238 & UL1TR000124, AHA 13PRE14430015
References
- 1.Richmond TJ, Davey CA. The structure of DNA in the nucleosome core. Nature. 2003;423(6936):145–50. doi: 10.1038/nature01595. [DOI] [PubMed] [Google Scholar]
- 2.Nair AP, Timoh T, Fuster V. Contemporary medical management of systolic heart failure. Circulation journal : official journal of the Japanese Circulation Society. 2012;76(2):268–77. doi: 10.1253/circj.cj-11-1424. [DOI] [PubMed] [Google Scholar]
- 3.Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Borden WB, et al. Heart Disease and Stroke Statistics--2013 Update: A Report From the American Heart Association. Circulation. 2013;127(1):e6–e245. doi: 10.1161/CIR.0b013e31828124ad. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Young NL, DiMaggio PA, Plazas-Mayorca MD, Baliban RC, Floudas CA, Garcia BA. High throughput characterization of combinatorial histone codes. Mol Cell Proteomics. 2009;8(10):2266–84. doi: 10.1074/mcp.M900238-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Siuti N, Kelleher NL. Decoding protein modifications using top-down mass spectrometry. Nat Methods. 2007;4(10):817–21. doi: 10.1038/nmeth1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Garcia BA, Pesavento JJ, Mizzen CA, Kelleher NL. Pervasive combinatorial modification of histone H3 in human cells. Nat Methods. 2007;4(6):487–9. doi: 10.1038/nmeth1052. [DOI] [PubMed] [Google Scholar]
- 7.Nakayama J, Rice JC, Strahl BD, Allis CD, Grewal SI. Role of histone H3 lysine 9 methylation in epigenetic control of heterochromatin assembly. Science. 2001;292(5514):110–3. doi: 10.1126/science.1060118. [DOI] [PubMed] [Google Scholar]
- 8.Cao R, Wang L, Wang H, Xia L, Erdjument-Bromage H, Tempst P, et al. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science. 2002;298(5595):1039–43. doi: 10.1126/science.1076997. [DOI] [PubMed] [Google Scholar]
- 9.Strahl BD, Ohba R, Cook RG, Allis CD. Methylation of histone H3 at lysine 4 is highly conserved and correlates with transcriptionally active nuclei in Tetrahymena. Proc Natl Acad Sci U S A. 1999;96(26):14967–72. doi: 10.1073/pnas.96.26.14967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Koch CM, Andrews RM, Flicek P, Dillon SC, Karaoz U, Clelland GK, et al. The landscape of histone modifications across 1% of the human genome in five human cell lines. Genome research. 2007;17(6):691–707. doi: 10.1101/gr.5704207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(50):21931–6. doi: 10.1073/pnas.1016071107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kolasinska-Zwierz P, Down T, Latorre I, Liu T, Liu XS, Ahringer J. Differential chromatin marking of introns and expressed exons by H3K36me3. Nature genetics. 2009;41(3):376–81. doi: 10.1038/ng.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Struhl K. Histone acetylation and transcriptional regulatory mechanisms. Genes & development. 1998;12(5):599–606. doi: 10.1101/gad.12.5.599. [DOI] [PubMed] [Google Scholar]
- 14.Talbert PB, Henikoff S. Histone variants--ancient wrap artists of the epigenome. Nature reviews Molecular cell biology. 2010;11(4):264–75. doi: 10.1038/nrm2861. [DOI] [PubMed] [Google Scholar]
- 15.Ramakrishnan V. Histone H1 and chromatin higher-order structure. Critical reviews in eukaryotic gene expression. 1997;7(3):215–30. doi: 10.1615/critreveukargeneexpr.v7.i3.20. [DOI] [PubMed] [Google Scholar]
- 16.Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403(6765):41–5. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- 17.Ernst J, Kheradpour P, Mikkelsen TS, Shoresh N, Ward LD, Epstein CB, et al. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature. 2011;473(7345):43–9. doi: 10.1038/nature09906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Filion GJ, van Bemmel JG, Braunschweig U, Talhout W, Kind J, Ward LD, et al. Systematic protein location mapping reveals five principal chromatin types in Drosophila cells. Cell. 2010;143(2):212–24. doi: 10.1016/j.cell.2010.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Haberland M, Montgomery RL, Olson EN. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat Rev Genet. 2009;10(1):32–42. doi: 10.1038/nrg2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Paige SL, Thomas S, Stoick-Cooper CL, Wang H, Maves L, Sandstrom R, et al. A temporal chromatin signature in human embryonic stem cells identifies regulators of cardiac development. Cell. 2012;151(1):221–32. doi: 10.1016/j.cell.2012.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wamstad JA, Alexander JM, Truty RM, Shrikumar A, Li F, Eilertson KE, et al. Dynamic and coordinated epigenetic regulation of developmental transitions in the cardiac lineage. Cell. 2012;151(1):206–20. doi: 10.1016/j.cell.2012.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shen Y, Yue F, McCleary DF, Ye Z, Edsall L, Kuan S, et al. A map of the cis-regulatory sequences in the mouse genome. Nature. 2012;488(7409):116–20. doi: 10.1038/nature11243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kaneda R, Takada S, Yamashita Y, Choi YL, Nonaka-Sarukawa M, Soda M, et al. Genome-wide histone methylation profile for heart failure. Genes Cells. 2009;14(1):69–77. doi: 10.1111/j.1365-2443.2008.01252.x. [DOI] [PubMed] [Google Scholar]
- 24.Vermeulen M, Eberl HC, Matarese F, Marks H, Denissov S, Butter F, et al. Quantitative interaction proteomics and genome-wide profiling of epigenetic histone marks and their readers. Cell. 2010;142(6):967–80. doi: 10.1016/j.cell.2010.08.020. [DOI] [PubMed] [Google Scholar]
- 25.Franklin S, Chen H, Mitchell-Jordan S, Ren S, Wang Y, Vondriska TM. Quantitative analysis of the chromatin proteome in disease reveals remodeling principles and identifies high mobility group protein B2 as a regulator of hypertrophic growth. Mol Cell Proteomics. 2012;11(6):M111–014258. doi: 10.1074/mcp.M111.014258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen IY, Lypowy J, Pain J, Sayed D, Grinberg S, Alcendor RR, et al. Histone H2A.z is essential for cardiac myocyte hypertrophy but opposed by silent information regulator 2alpha. The Journal of biological chemistry. 2006;281(28):19369–77. doi: 10.1074/jbc.M601443200. [DOI] [PubMed] [Google Scholar]
- 27.Fedele M, Fidanza V, Battista S, Pentimalli F, Klein-Szanto AJ, Visone R, et al. Haploinsufficiency of the Hmga1 gene causes cardiac hypertrophy and myelolymphoproliferative disorders in mice. Cancer Res. 2006;66(5):2536–43. doi: 10.1158/0008-5472.CAN-05-1889. [DOI] [PubMed] [Google Scholar]
- 28.Cooper DN, Taggart MH, Bird AP. Unmethylated domains in vertebrate DNA. Nucleic acids research. 1983;11(3):647–58. doi: 10.1093/nar/11.3.647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bird A, Taggart M, Frommer M, Miller OJ, Macleod D. A fraction of the mouse genome that is derived from islands of nonmethylated, CpG-rich DNA. Cell. 1985;40(1):91–9. doi: 10.1016/0092-8674(85)90312-5. [DOI] [PubMed] [Google Scholar]
- 30.Gardiner-Garden M, Frommer M. CpG islands in vertebrate genomes. Journal of molecular biology. 1987;196(2):261–82. doi: 10.1016/0022-2836(87)90689-9. [DOI] [PubMed] [Google Scholar]
- 31.Irizarry RA, Ladd-Acosta C, Wen B, Wu Z, Montano C, Onyango P, et al. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nature genetics. 2009;41(2):178–86. doi: 10.1038/ng.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nikitina T, Shi X, Ghosh RP, Horowitz-Scherer RA, Hansen JC, Woodcock CL. Multiple modes of interaction between the methylated DNA binding protein MeCP2 and chromatin. Molecular and cellular biology. 2007;27(3):864–77. doi: 10.1128/MCB.01593-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Newell-Price J, Clark AJ, King P. DNA methylation and silencing of gene expression. Trends Endocrinol Metab. 2000;11(4):142–8. doi: 10.1016/s1043-2760(00)00248-4. [DOI] [PubMed] [Google Scholar]
- 34.Xie L, Weichel B, Ohm JE, Zhang K. An integrative analysis of DNA methylation and RNA-Seq data for human heart, kidney and liver. BMC Syst Biol. 2011;5(Suppl 3):S4. doi: 10.1186/1752-0509-5-S3-S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yoder JA, Soman NS, Verdine GL, Bestor TH. DNA (cytosine-5)-methyltransferases in mouse cells and tissues. Studies with a mechanism-based probe. Journal of molecular biology. 1997;270(3):385–95. doi: 10.1006/jmbi.1997.1125. [DOI] [PubMed] [Google Scholar]
- 36.Okano M, Bell DW, Haber DA, Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999;99(3):247–57. doi: 10.1016/s0092-8674(00)81656-6. [DOI] [PubMed] [Google Scholar]
- 37.Bestor TH. The DNA methyltransferases of mammals. Human molecular genetics. 2000;9(16):2395–402. doi: 10.1093/hmg/9.16.2395. [DOI] [PubMed] [Google Scholar]
- 38.Johnson R, Richter N, Bogu GK, Bhinge A, Teng SW, Choo SH, et al. A genome-wide screen for genetic variants that modify the recruitment of REST to its target genes. PLoS genetics. 2012;8(4):e1002624. doi: 10.1371/journal.pgen.1002624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Irier HA, Jin P. Dynamics of DNA methylation in aging and Alzheimer's disease. DNA Cell Biol. 2012;31(Suppl 1):S42–8. doi: 10.1089/dna.2011.1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chodavarapu RK, Feng S, Bernatavichute YV, Chen PY, Stroud H, Yu Y, et al. Relationship between nucleosome positioning and DNA methylation. Nature. 2010;466(7304):388–92. doi: 10.1038/nature09147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cokus SJ, Feng S, Zhang X, Chen Z, Merriman B, Haudenschild CD, et al. Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature. 2008;452(7184):215–9. doi: 10.1038/nature06745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mohn F, Weber M, Schubeler D, Roloff TC. Methylated DNA immunoprecipitation (MeDIP) Methods in molecular biology. 2009;507:55–64. doi: 10.1007/978-1-59745-522-0_5. [DOI] [PubMed] [Google Scholar]
- 43.Zhu C, Yu ZB, Chen XH, Ji CB, Qian LM, Han SP. DNA hypermethylation of the NOX5 gene in fetal ventricular septal defect. Exp Ther Med. 2011;2(5):1011–5. doi: 10.3892/etm.2011.294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Movassagh M, Choy MK, Goddard M, Bennett MR, Down TA, Foo RS. Differential DNA methylation correlates with differential expression of angiogenic factors in human heart failure. PloS one. 2010;5(1):e8564. doi: 10.1371/journal.pone.0008564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Haider S, Cordeddu L, Robinson E, Movassagh M, Siggens L, Vujic A, et al. The landscape of DNA repeat elements in human heart failure. Genome biology. 2012;13(10):R90. doi: 10.1186/gb-2012-13-10-r90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Haddad R, Kasneci A, Mepham K, Sebag IA, Chalifour LE. Gestational exposure to diethylstilbestrol alters cardiac structure/function, protein expression and DNA methylation in adult male mice progeny. Toxicol Appl Pharmacol. 2013;266(1):27–37. doi: 10.1016/j.taap.2012.10.018. [DOI] [PubMed] [Google Scholar]
- 47.Metes-Kosik N, Luptak I, Dibello PM, Handy DE, Tang SS, Zhi H, et al. Both selenium deficiency and modest selenium supplementation lead to myocardial fibrosis in mice via effects on redox-methylation balance. Mol Nutr Food Res. 2012;56(12):1812–24. doi: 10.1002/mnfr.201200386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Movassagh M, Choy MK, Knowles DA, Cordeddu L, Haider S, Down T, et al. Distinct epigenomic features in end-stage failing human hearts. Circulation. 2011;124(22):2411–22. doi: 10.1161/CIRCULATIONAHA.111.040071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Antonitsis P, Ioannidou-Papagiannaki E, Kaidoglou A, Papakonstantinou C. In vitro cardiomyogenic differentiation of adult human bone marrow mesenchymal stem cells. The role of 5-azacytidine. Interact Cardiovasc Thorac Surg. 2007;6(5):593–7. doi: 10.1510/icvts.2007.157875. [DOI] [PubMed] [Google Scholar]
- 50.Ye NS, Chen J, Luo GA, Zhang RL, Zhao YF, Wang YM. Proteomic profiling of rat bone marrow mesenchymal stem cells induced by 5-azacytidine. Stem Cells Dev. 2006;15(5):665–76. doi: 10.1089/scd.2006.15.665. [DOI] [PubMed] [Google Scholar]
- 51.Liang P, Song F, Ghosh S, Morien E, Qin M, Mahmood S, et al. Genome-wide survey reveals dynamic widespread tissue-specific changes in DNA methylation during development. BMC genomics. 2011;12(1):231. doi: 10.1186/1471-2164-12-231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mattick JS. Non-coding RNAs: the architects of eukaryotic complexity. EMBO reports. 2001;2(11):986–91. doi: 10.1093/embo-reports/kve230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dorn GW., 2nd Decoding the cardiac message: the 2011 Thomas W. Smith Memorial Lecture. Circ Res. 2012;110(5):755–63. doi: 10.1161/CIRCRESAHA.111.256768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.van Rooij E, Olson EN. MicroRNA therapeutics for cardiovascular disease: opportunities and obstacles. Nature reviews Drug discovery. 2012;11(11):860–72. doi: 10.1038/nrd3864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kawashima T, Shioi T. MicroRNA, emerging role as a biomarker of heart failure. Circulation journal : official journal of the Japanese Circulation Society. 2011;75(2):268–9. doi: 10.1253/circj.cj-10-1254. [DOI] [PubMed] [Google Scholar]
- 56.Song HK, Hong SE, Kim T, Kim do H. Deep RNA sequencing reveals novel cardiac transcriptomic signatures for physiological and pathological hypertrophy. PLoS One. 2012;7(4):e35552. doi: 10.1371/journal.pone.0035552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lee JH, Gao C, Peng G, Greer C, Ren S, Wang Y, et al. Analysis of transcriptome complexity through RNA sequencing in normal and failing murine hearts. Circ Res. 2011;109(12):1332–41. doi: 10.1161/CIRCRESAHA.111.249433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cabili MN, Trapnell C, Goff L, Koziol M, Tazon-Vega B, Regev A, et al. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes & development. 2011;25(18):1915–27. doi: 10.1101/gad.17446611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lee JT. Epigenetic regulation by long noncoding RNAs. Science. 2012;338(6113):1435–9. doi: 10.1126/science.1231776. [DOI] [PubMed] [Google Scholar]
- 60.Grote P, Wittler L, Hendrix D, Koch F, Wahrisch S, Beisaw A, et al. The Tissue-Specific lncRNA Fendrr Is an Essential Regulator of Heart and Body Wall Development in the Mouse. Developmental cell. 2013;24(2):206–14. doi: 10.1016/j.devcel.2012.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Klattenhoff CA, Scheuermann JC, Surface LE, Bradley RK, Fields PA, Steinhauser ML, et al. Braveheart, a long noncoding RNA required for cardiovascular lineage commitment. Cell. 2013;152(3):570–83. doi: 10.1016/j.cell.2013.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Saha A, Wittmeyer J, Cairns BR. Chromatin remodelling: the industrial revolution of DNA around histones. Nature reviews Molecular cell biology. 2006;7(6):437–47. doi: 10.1038/nrm1945. [DOI] [PubMed] [Google Scholar]
- 63.Fazzio TG, Tsukiyama T. Chromatin remodeling in vivo: evidence for a nucleosome sliding mechanism. Mol Cell. 2003;12(5):1333–40. doi: 10.1016/s1097-2765(03)00436-2. [DOI] [PubMed] [Google Scholar]
- 64.Boeger H, Griesenbeck J, Strattan JS, Kornberg RD. Removal of promoter nucleosomes by disassembly rather than sliding in vivo. Mol Cell. 2004;14(5):667–73. doi: 10.1016/j.molcel.2004.05.013. [DOI] [PubMed] [Google Scholar]
- 65.Bruno M, Flaus A, Stockdale C, Rencurel C, Ferreira H, Owen-Hughes T. Histone H2A/H2B dimer exchange by ATP-dependent chromatin remodeling activities. Mol Cell. 2003;12(6):1599–606. doi: 10.1016/s1097-2765(03)00499-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mizuguchi G, Shen X, Landry J, Wu WH, Sen S, Wu C. ATP-driven exchange of histone H2AZ variant catalyzed by SWR1 chromatin remodeling complex. Science. 2004;303(5656):343–8. doi: 10.1126/science.1090701. [DOI] [PubMed] [Google Scholar]
- 67.Cairns BR. Chromatin remodeling: insights and intrigue from single-molecule studies. Nat Struct Mol Biol. 2007;14(11):989–96. doi: 10.1038/nsmb1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Armstrong JA, Papoulas O, Daubresse G, Sperling AS, Lis JT, Scott MP, et al. The Drosophila BRM complex facilitates global transcription by RNA polymerase II. Embo J. 2002;21(19):5245–54. doi: 10.1093/emboj/cdf517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Batsche E, Yaniv M, Muchardt C. The human SWI/SNF subunit Brm is a regulator of alternative splicing. Nat Struct Mol Biol. 2006;13(1):22–9. doi: 10.1038/nsmb1030. [DOI] [PubMed] [Google Scholar]
- 70.Corey LL, Weirich CS, Benjamin IJ, Kingston RE. Localized recruitment of a chromatin-remodeling activity by an activator in vivo drives transcriptional elongation. Genes Dev. 2003;17(11):1392–401. doi: 10.1101/gad.1071803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Morillon A, Karabetsou N, O'Sullivan J, Kent N, Proudfoot N, Mellor J. Isw1 chromatin remodeling ATPase coordinates transcription elongation and termination by RNA polymerase II. Cell. 2003;115(4):425–35. doi: 10.1016/s0092-8674(03)00880-8. [DOI] [PubMed] [Google Scholar]
- 72.Wade PA, Gegonne A, Jones PL, Ballestar E, Aubry F, Wolffe AP. Mi-2 complex couples DNA methylation to chromatin remodelling and histone deacetylation. Nat Genet. 1999;23(1):62–6. doi: 10.1038/12664. [DOI] [PubMed] [Google Scholar]
- 73.Kusch T, Florens L, Macdonald WH, Swanson SK, Glaser RL, Yates JR, 3rd, et al. Acetylation by Tip60 is required for selective histone variant exchange at DNA lesions. Science. 2004;306(5704):2084–7. doi: 10.1126/science.1103455. [DOI] [PubMed] [Google Scholar]
- 74.Hakimi MA, Bochar DA, Schmiesing JA, Dong Y, Barak OG, Speicher DW, et al. A chromatin remodelling complex that loads cohesin onto human chromosomes. Nature. 2002;418(6901):994–8. doi: 10.1038/nature01024. [DOI] [PubMed] [Google Scholar]
- 75.Huang J, Hsu JM, Laurent BC. The RSC nucleosome-remodeling complex is required for Cohesin's association with chromosome arms. Mol Cell. 2004;13(5):739–50. doi: 10.1016/s1097-2765(04)00103-0. [DOI] [PubMed] [Google Scholar]
- 76.Han P, Hang CT, Yang J, Chang CP. Chromatin remodeling in cardiovascular development and physiology. Circ Res. 2011;108(3):378–96. doi: 10.1161/CIRCRESAHA.110.224287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lei I, Gao X, Sham MH, Wang Z. SWI/SNF protein component BAF250a regulates cardiac progenitor cell differentiation by modulating chromatin accessibility during second heart field development. J Biol Chem. 2012;287(29):24255–62. doi: 10.1074/jbc.M112.365080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Stankunas K, Hang CT, Tsun ZY, Chen H, Lee NV, Wu JI, et al. Endocardial Brg1 represses ADAMTS1 to maintain the microenvironment for myocardial morphogenesis. Developmental cell. 2008;14(2):298–311. doi: 10.1016/j.devcel.2007.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lickert H, Takeuchi JK, Von Both I, Walls JR, McAuliffe F, Adamson SL, et al. Baf60c is essential for function of BAF chromatin remodelling complexes in heart development. Nature. 2004;432(7013):107–12. doi: 10.1038/nature03071. [DOI] [PubMed] [Google Scholar]
- 80.Wang Z, Zhai W, Richardson JA, Olson EN, Meneses JJ, Firpo MT, et al. Polybromo protein BAF180 functions in mammalian cardiac chamber maturation. Genes Dev. 2004;18(24):3106–16. doi: 10.1101/gad.1238104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hang CT, Yang J, Han P, Cheng HL, Shang C, Ashley E, et al. Chromatin regulation by Brg1 underlies heart muscle development and disease. Nature. 2010;466(7302):62–7. doi: 10.1038/nature09130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Rottbauer W, Saurin AJ, Lickert H, Shen X, Burns CG, Wo ZG, et al. Reptin and pontin antagonistically regulate heart growth in zebrafish embryos. Cell. 2002;111(5):661–72. doi: 10.1016/s0092-8674(02)01112-1. [DOI] [PubMed] [Google Scholar]
- 83.Hurd EA, Capers PL, Blauwkamp MN, Adams ME, Raphael Y, Poucher HK, et al. Loss of Chd7 function in gene-trapped reporter mice is embryonic lethal and associated with severe defects in multiple developing tissues. Mammalian genome : official journal of the International Mammalian Genome Society. 2007;18(2):94–104. doi: 10.1007/s00335-006-0107-6. [DOI] [PubMed] [Google Scholar]
- 84.Lange M, Kaynak B, Forster UB, Tonjes M, Fischer JJ, Grimm C, et al. Regulation of muscle development by DPF3, a novel histone acetylation and methylation reader of the BAF chromatin remodeling complex. Genes Dev. 2008;22(17):2370–84. doi: 10.1101/gad.471408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Koda M, Takemura G, Okada H, Kanoh M, Maruyama R, Esaki M, et al. Nuclear hypertrophy reflects increased biosynthetic activities in myocytes of human hypertrophic hearts. Circulation journal : official journal of the Japanese Circulation Society. 2006;70(6):710–8. doi: 10.1253/circj.70.710. [DOI] [PubMed] [Google Scholar]
- 86.Nikolova V, Leimena C, McMahon AC, Tan JC, Chandar S, Jogia D, et al. Defects in nuclear structure and function promote dilated cardiomyopathy in lamin A/C-deficient mice. J Clin Invest. 2004;113(3):357–69. doi: 10.1172/JCI19448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.van Steensel B, Dekker J. Genomics tools for unraveling chromosome architecture. Nature biotechnology. 2010;28(10):1089–95. doi: 10.1038/nbt.1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Simonis M, Kooren J, de Laat W. An evaluation of 3C-based methods to capture DNA interactions. Nature methods. 2007;4(11):895–901. doi: 10.1038/nmeth1114. [DOI] [PubMed] [Google Scholar]
- 89.Warren SA, Terada R, Briggs LE, Cole-Jeffrey CT, Chien WM, Seki T, et al. Differential role of Nkx2-5 in activation of the atrial natriuretic factor gene in the developing versus failing heart. Molecular and cellular biology. 2011;31(22):4633–45. doi: 10.1128/MCB.05940-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Narlikar L, Sakabe NJ, Blanski AA, Arimura FE, Westlund JM, Nobrega MA, et al. Genome-wide discovery of human heart enhancers. Genome research. 2010;20(3):381–92. doi: 10.1101/gr.098657.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.He A, Kong SW, Ma Q, Pu WT. Co-occupancy by multiple cardiac transcription factors identifies transcriptional enhancers active in heart. Proc Natl Acad Sci U S A. 2011;108(14):5632–7. doi: 10.1073/pnas.1016959108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kodo K, Nishizawa T, Furutani M, Arai S, Ishihara K, Oda M, et al. Genetic analysis of essential cardiac transcription factors in 256 patients with non-syndromic congenital heart defects. Circulation journal : official journal of the Japanese Circulation Society. 2012;76(7):1703–11. doi: 10.1253/circj.cj-11-1389. [DOI] [PubMed] [Google Scholar]
- 93.Sayed D, He M, Yang Z, Lin L, Abdellatif M. Transcriptional Regulation Patterns Revealed by High Resolution Chromatin Immunoprecipitation during Cardiac Hypertrophy. J Biol Chem. 2013;288(4):2546–58. doi: 10.1074/jbc.M112.429449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Segal E, Widom J. What controls nucleosome positions? Trends Genet. 2009;25(8):335–43. doi: 10.1016/j.tig.2009.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Giresi PG, Kim J, McDaniell RM, Iyer VR, Lieb JD. FAIRE (Formaldehyde-Assisted Isolation of Regulatory Elements) isolates active regulatory elements from human chromatin. Genome research. 2007;17(6):877–85. doi: 10.1101/gr.5533506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Boyle AP, Davis S, Shulha HP, Meltzer P, Margulies EH, Weng Z, et al. High-resolution mapping and characterization of open chromatin across the genome. Cell. 2008;132(2):311–22. doi: 10.1016/j.cell.2007.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Cui K, Zhao K. Genome-wide approaches to determining nucleosome occupancy in metazoans using MNase-Seq. Methods Mol Biol. 2012;833:413–9. doi: 10.1007/978-1-61779-477-3_24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.van Steensel B, Henikoff S. Identification of in vivo DNA targets of chromatin proteins using tethered dam methyltransferase. Nature biotechnology. 2000;18(4):424–8. doi: 10.1038/74487. [DOI] [PubMed] [Google Scholar]
- 99.Xiao R, Moore DD. DamIP: using mutant DNA adenine methyltransferase to study DNA-protein interactions in vivo. In: Ausubel Frederick M, et al., editors. Current protocols in molecular biology. Unit21. Chapter 21. 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.de Wit E, Greil F, van Steensel B. Genome-wide HP1 binding in Drosophila: developmental plasticity and genomic targeting signals. Genome research. 2005;15(9):1265–73. doi: 10.1101/gr.3198905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Zullo JM, Demarco IA, Pique-Regi R, Gaffney DJ, Epstein CB, Spooner CJ, et al. DNA sequence-dependent compartmentalization and silencing of chromatin at the nuclear lamina. Cell. 2012;149(7):1474–87. doi: 10.1016/j.cell.2012.04.035. [DOI] [PubMed] [Google Scholar]
- 102.Zhang Z, Wippo CJ, Wal M, Ward E, Korber P, Pugh BF. A packing mechanism for nucleosome organization reconstituted across a eukaryotic genome. Science. 2011;332(6032):977–80. doi: 10.1126/science.1200508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Chen H, Monte E, Parvatiyar MS, Rosa-Garrido M, Franklin S, Vondriska TM. Structural considerations for chromatin state models with transcription as a functional readout. FEBS letters. 2012;586(20):3548–54. doi: 10.1016/j.febslet.2012.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Solovei I, Kreysing M, Lanctot C, Kosem S, Peichl L, Cremer T, et al. Nuclear architecture of rod photoreceptor cells adapts to vision in mammalian evolution. Cell. 2009;137(2):356–68. doi: 10.1016/j.cell.2009.01.052. [DOI] [PubMed] [Google Scholar]
- 105.Ohtani K, Dimmeler S. Epigenetic regulation of cardiovascular differentiation. Cardiovasc Res. 2011;90(3):404–12. doi: 10.1093/cvr/cvr019. [DOI] [PubMed] [Google Scholar]
- 106.Udali S, Guarini P, Moruzzi S, Choi SW, Friso S. Cardiovascular epigenetics: From DNA methylation to microRNAs. Molecular aspects of medicine. 2012 doi: 10.1016/j.mam.2012.08.001. [DOI] [PubMed] [Google Scholar]
- 107.Schonrock N, Harvey RP, Mattick JS. Long noncoding RNAs in cardiac development and pathophysiology. Circ Res. 2012;111(10):1349–62. doi: 10.1161/CIRCRESAHA.112.268953. [DOI] [PubMed] [Google Scholar]
- 108.Mitchell-Jordan S, Chen H, Franklin S, Stefani E, Bentolila LA, Vondriska TM. Features of endogenous cardiomyocyte chromatin revealed by super-resolution STED microscopy. J Mol Cell Cardiol. 2012;53(4):552–8. doi: 10.1016/j.yjmcc.2012.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Guo JU, Ma DK, Mo H, Ball MP, Jang MH, Bonaguidi MA, et al. Neuronal activity modifies the DNA methylation landscape in the adult brain. Nature neuroscience. 2011;14(10):1345–51. doi: 10.1038/nn.2900. [DOI] [PMC free article] [PubMed] [Google Scholar]