

Abstract
Gold-catalyzed intramolecular oxidation of terminal alkynes with an arenesulfinyl group as the tethered oxidant is a reaction in gold chemistry of high impact, as it introduced to the field the highly-valued concept of gold carbene generation via alkyne oxidation. The proposed intermediacy of α-oxo gold carbenes in these reactions, however, has never been substantiated. Detailed experimental studies suggest that the involvement of such reactive intermediates in the formation of dihydrobenzothiepinones is highly unlikely. Instead, a [3,3]-sigmatropic rearrangement of the initial cyclization intermediate offers a reaction path that can readily explain the high reaction efficiency and the lack of sulfonium formation. With internal alkyne substrates, however, the generation of a gold carbene species becomes competitive with the [3,3]-sigmatropic rearrangement. This reactive intermediate, nevertheless, does not proceed to afford the Friedel-Crafts type cyclization product. Extensive Density Functional Theory studies support the mechanistic conclusion that the cyclized product is formed via an intramolecular [3,3]-sigmatropic rearrangement instead of the previously proposed Friedel-Crafts type cyclization. With the new mechanistic insight, the product scope of this versatile formation of mid-sized sulfur-containing cycloalkenones has been expanded readily to various dihydrobenzothiocinones, a tetrahydrobenzocyclononenone, and even those without the entanglement of a fused benzene ring. Besides gold, Hg(OTf)2 can be an effective catalyst, thereby offering a cheap alternative for this intramolecular redox reaction.
Introduction
Homogeneous gold catalysis1 is a rather recent phenomenon but has generated tremendous interest and excitement in the synthetic community. Its explosive growth owes much to various aspects of novel and unprecedented reactivities of gold complexes and to synergistic efforts from many research groups around the world.
One particularly rewarding, and still evolving, area in gold catalysis is the oxidation of alkynes using oxygen-delivering nucleophilic oxidants.2 These oxidants are composed of two parts: a negatively charged, thus nucleophilic, oxygen and a positively charged, thus potent, leaving group (nucleofuge). The gold catalysis follows the sequence outlined in Scheme 1: firstly, the nucleophilic oxygen from the oxidant would attack a gold-activated C-C triple bond in an anti addition; the alkenylgold intermediate thus formed, i.e., A, would then undergo γ-elimination, expelling the nucleofugal part of the oxidant and generating an α-oxo gold carbene intermediate, i.e., B. Since B could formally be formed through selective oxidation of the carbene center of an α-carbene gold carbene C, the overall transformation in Scheme 1 makes stable and benign alkynes in the presence of a gold complex equivalent to the imaginary species C, offering a formal access to the reactivities of this provocative structure and foretelling the synthetic potential of this strategy.
Scheme 1.
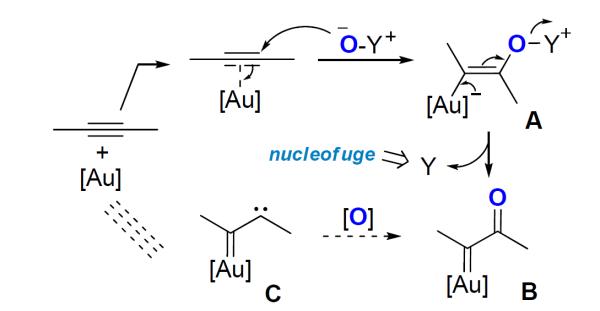
Gold-Catalyzed Oxidation of Alkynes Using Oxygen-Delivering Oxidants
Intramolecular Gold-Catalyzed Oxidation of Alkynes by Tethered Sulfoxides
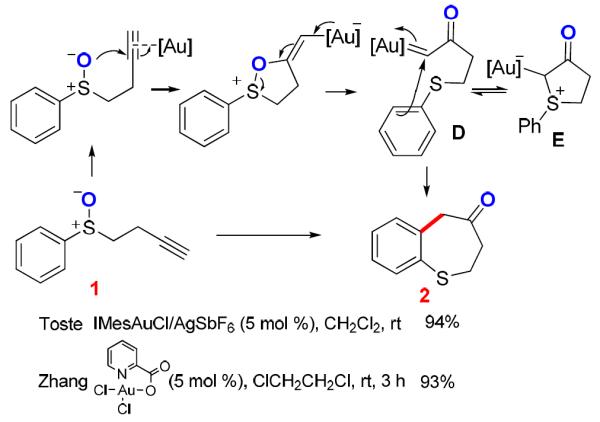
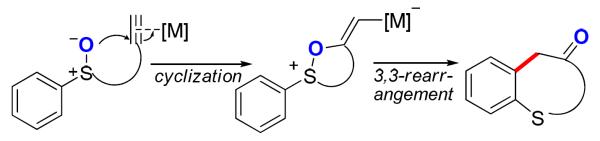
The application of this concept was first realized using tethered sulfoxides as the oxygen-delivering oxidant by Toste3 and us4 independently at around the same time, and dihydrobenzothiepinones (e.g., 2) were formed in typically excellent yields (e.g., Scheme 2). Along the same line as in Scheme 1, a common mechanism via an α-oxo gold carbene intermediate (e.g., D) was proposed.3,4 In addition, we hypothesized the sulfonium ion E as a resting state of the gold carbene, although it might instead act to trap the gold carbine irreversibly. Particularly noteworthy in these reactions are the mild nature of sulfoxides as oxidants and the extremely high efficiencies in the formation of the seven-membered thiepinone ring. In an attempt to further explore its synthetic utility, we coupled this transformation with a subsequent pinacol rearrangement upon modifying the phenyl group to hinder the benzothiepinone formation (Scheme 3), and β-diketones were isolated in fair to good yields. The reaction results were consistent with the formation of a gold carbene intermediate F.
Scheme 2.
Scheme 3.
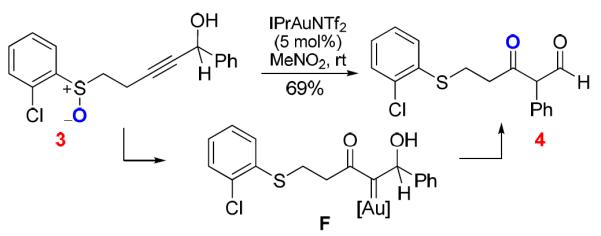
Sequential Oxidation/Pinacol-Type Rearrangement
Later, Davies5 employed a similar strategy to access such a gold carbene (Scheme 4), which is subsequently trapped by the tethered nascent sulfide to form a sulfur ylide G. This intermediate, similar in nature to our previously proposed resting state E, underwent a [2,3]-sigmatropic rearrangement to render the final sulfur heterocycle. The fact that this mechanistic rationale is seemingly the only viable one for this transformation strongly suggests that α-oxo gold carbenes of type B are accessible via the strategy outlined in Scheme 1 and, moreover, argues strongly that the sulfonium intermediate E should be formed in Scheme 2. This begs the question of why E, presumably much more stable than the gold carbene, does not impede the observed cyclization.
Scheme 4.
Tandem Gold-catalyzed Intramolecular Alkyne Oxidation and 2,3-Sigmatropic Rearrangement via the Intermediacy of a Sulfur Ylide
Many researchers including us have subsequently applied this intramolecular strategy to other oxygen-delivering oxidants including nitrone,6N-oxides,7 epoxide8 and nitro9 successfully, under the assumption10 that α-oxo gold carbenes were generated.
Intermolecular Gold-Catalyzed Alkyne Oxidation Using Sulfoxides as Oxidants
With all these successes with intramolecular oxidations, intermolecular alkyne oxidation using sulfoxides were studied by Ujaque and Asensio.11 However, in their elegant work the experimental results do not support the formation of an α-oxo gold carbene intermediate of type B. Instead, a [3,3]-sigmatropic rearrangement is invoked to explain the reaction outcomes, which is further corroborated by computational studies (Scheme 5). This study prompted us to contemplate an alternative 3,3-rerrangement mechanism for the original intramolecular chemistry (see Scheme 2).3,4
Scheme 5.
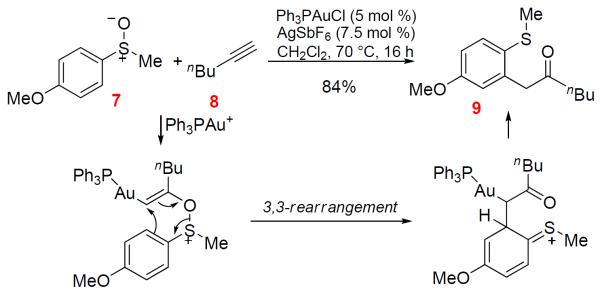
Gold-Catalyzed Alkyne Oxidation by an External Sulfoxide
Later, Liu1k reported a gold-catalyzed intermolecular oxidative ring expansion using Ph2SO as the external oxidant. In their proposed mechanism (Scheme 6), no α-oxo gold carbene is involved, and the reaction proceeds via a concerted ring expansion and ejection of nucleofugal Ph2S.
Scheme 6.
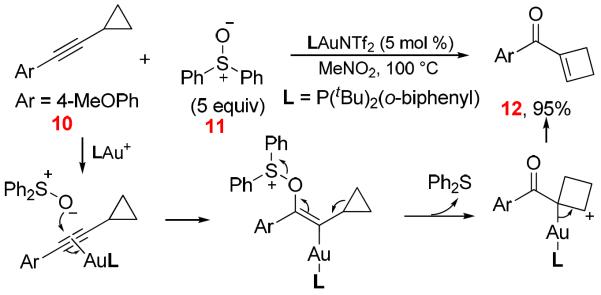
Gold-Catalyzed Alkyne Oxidation by an External Sulfoxide1k
All those studies using sulfoxides as oxidants seemingly suggest that the reaction pathways depend simply on whether the oxidant is tethered or not, i.e., [3,3]-rearrangement or concerted elimination when intermolecular and gold carbene formation when intramolecular. This, however, need not be the case. Due to the tremendous synthetic potential of α-oxo gold carbenes, coupled with the surprisingly efficient formation of seven-membered thiepinone 2 and the absence of reaction inhibition due to the formation of sulfonium E, we embarked on mechanistic studies of the dihydrobenzothiepinone formation both experimentally and theoretically. Herein we disclose our findings and the extension of this chemistry to the formation of even larger S-heterocycles and to non-aromatic substrates.
Results and discussion
Mechanistic studies of the benzothiepinone formation
In our continued effort to implement the general strategy shown in Scheme 1, we have recently reported the first case of successful generation of α-oxo gold carbenes using an external oxidant.12 This strategy was later expanded by us to 1) an efficient synthesis of oxetan-3-ones from propargyl alcohols (Scheme 7),13 2) a regioselective synthesis of α,β-unsaturated ketones from internal alkynes,14 3) a rapid [2+2+1] approach to 2,5-disubstituted oxazoles,15 4) a stereoselective synthesis of azetidin-3-ones from propargylic amides,16 and 5) a rapid access to chroman-3-ones.17 The suitable oxidants were not sulfoxides but pyridine N-oxides and quinoline N-oxides, and the formation of gold carbene intermediates, e.g., H, is strongly implicated in the formation of oxetan-3-ones (Scheme 7)13 and the azetidine-3-ones.16 To our delight, this strategy has also been adopted by other researchers in the development of versatile synthetic methods.18
Scheme 7.
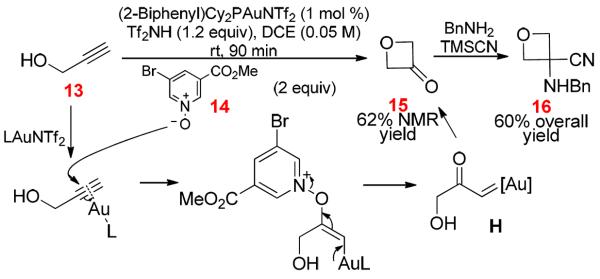
Generation of α-Oxo Gold Carbenes via Intermolecular Alkyne Oxidation and Its Application in Oxetan-3-One Formation
This easy access to α-oxo gold carbenes from alkynes using external pyridine/quinoline N-oxides provided us a valuable opportunity to probe the mechanism of the dihydrobenzothiepinone formation. Hence, a variety of oxidants and gold catalysts were examined with alkynyl sulfide 17 as the substrate, but most of the reactions were messy, and none gave the expected thiepinone 1. Instead, α-chloroketone 19 was formed in <10% yields in some cases (e.g., Scheme 8), which was the result of chloride abstraction5a,19 from the reaction solvent, dichloroethane. This is consistent with the intermediacy of the highly electrophilic α-oxo gold carbene D. In addition, the rearranged enone 20 was formed during isolation, which can be explained via a cyclization and elimination process. As a control experiment, no sulfoxide was observed after stirring a mixture of sulfide 17 and the oxidant, 8-isopropylquinoline N-oxides 18, in DCE for 20 h.
Scheme 8.
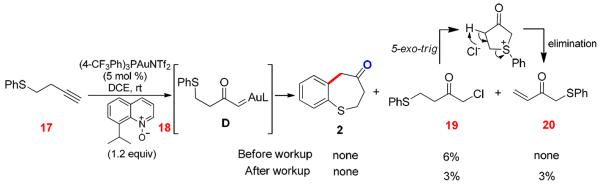
Access to Gold Carbene D via Intermolecular Oxidation
Pérez, Nolan and Echavarren recently reported in several interesting studies20 that gold catalysts could decompose α-diazo esters to access reactivities of gold carbenes such as C-H insertion and cyclopropanation. However, one experiment in our recent work10 led to our tentative conclusion that α-oxo gold carbenes might not be generated upon gold catalyzed decomposition of α-diazo ketones due to competing facile Wolff rearrangement. To exhaust all possible approaches to generating D, we nevertheless prepared α-diazo ketone 21 and treated it with Ph3PAuNTf221 (5 mol %). No thiepinone 2 was detected, and neither were the ketone products 19 and 20. Instead, the carboxylic acid 22 was formed via apparent sequential Wolff rearrangement22 and hydration in a 71% isolated yield (Scheme 9). This result is consistent with our previous observation10 and likewise suggests that the gold carbenoid I instead of the gold carbene D is most likely the reactive intermediate generated. It is worthwhile to point out that the mechanism of the Wolff rearrangement catalyzed by metal complexes including preferred silver salts has not been clearly elucidated.
Scheme 9.
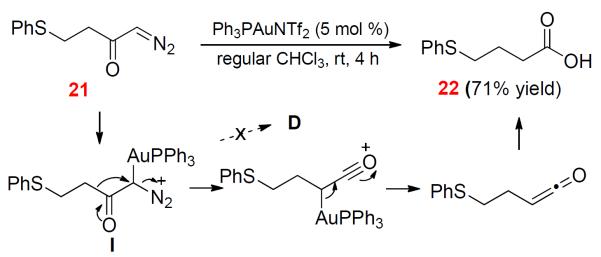
Attempt to Generate Gold Carbene D via Dediazotization
These results raise serious doubts about the nature of the gold intermediate en route to the thiepinone 2. If one of the reaction intermediates were indeed the gold carbene D, it might then be expected to abstract chloride, as in Scheme 8, and/or be trapped as the sulfonium species E. An alternative mechanism, in line with the intermolecular studies,11 would be a [3,3]-rearrangement for the key C-C bond forming step (Scheme 10). This would also readily explain the facile formation of seven-membered thiepinones and the absence of catalyst trapping by E.
Scheme 10.

An Alternative Mechanism for Benzothiepinone Formation.
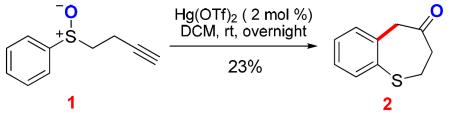
The revised mechanism suggests that other metals could catalyze this cycloisomerization as long as they could promote the initial 5-exo-dig cyclization. Indeed, Hg(OTf)2 promotes this reaction, albeit in a much lower efficiency (Eq. 1).
 |
(1) |
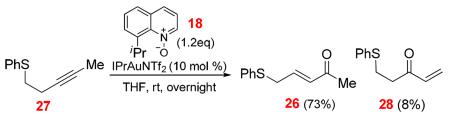
In the case of the sulfoxide 23 containing an internal alkyne (Scheme 11), Hg(OTf)2 catalyzed the cycloisomerization with a much higher efficiency. Besides the formation of the methylated thiepinone 24, the formation of the thiochroman 25 could also be explained by a [3,3]-rearrangement, which is preceded by a favored 6-endo-dig cyclization. When Ph3PAuNTf2 was used as the catalyst, a small amount of the enone 26 was detected, indicative of competing formation of carbene intermediate J. The thiochroman 25 could be also formed from J via a Friedel-Crafts type cyclization; however, this alternative was not supported by an intermolecular study. As shown in Eq. 2, following the intermolecular oxidation protocol14 we previously developed, the enone 26 was formed as the major regioisomer in a good yield; notably, neither 24 nor 25 was detected. As the gold carbene J was most likely the formed intermediate en route to 26, this result suggests that the apparent carbene 1,2-C-H insertion handily outcompetes cyclization, which in turn corroborates that 25 were formed via a [3,3]-rearrangement.
 |
(2) |
Scheme 11.
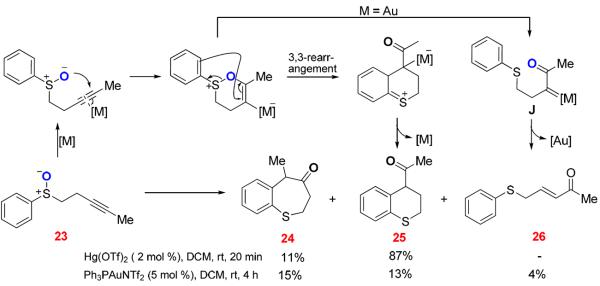
Gold-Catalyzed Oxidation of Internal Alkyne by a Tethered Sulfoxide
Formation of Benzene-Fused Larger S-Hetereocycles
With a better understanding of the reaction mechanism, we reasoned that benzene-fused larger S-heterocycles could be readily accessible via metal-catalyzed cycloisomerizations providing that the initial sulfoxide cyclization could be realized (Scheme 12).
Scheme 12.
Synthesis of Benzene-Fused Larger S-Heterocycles
In our early work,23 the cycloisomerization of sulfoxide 30a was examined only with dichloro-(2-picolinato)gold(III) (Table 1, entry 1) under the assumption that the formation of thiocine ring via gold carbene cyclization would be too challenging. Indeed, the reaction yield was modest. However, a quick screening of gold catalysts revealed that the common gold catalyst, Ph3PAuNTf2,21 was highly effective, and dihydrobenzothiocinone 30a was formed in 97% yield (entry 2), consistent with the [3,3]-rearrangement mechanism. Other gold catalysts were capable of catalyzing this reaction as well, albeit with lower efficiencies (entries 3–4). Hg(OTf)2 again was effective, although a competing hydration was observed, and the reaction was much faster than in the cases of gold catalysis (entry 7).
Table 1.
Catalyst Screening for the Formation of 30a 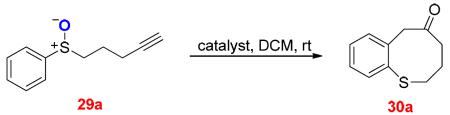
| entry | catalyst | time | yielda |
|---|---|---|---|
| 1 | dichloro(2-picolinato)gold(III) | 12 h | 52%b |
| 2 | Ph3PAuNTf2 | 4 h | 97% |
| 3 | IPrAuNTf2 | 4 h | 74%c |
| 4 | (4-CF3Ph)3PAuNTf2 | 4 h | 82% |
| 5 | Hg(OTf)2 | 40 min | 75%c |
NMR yield using diethyl phthalate as reference.
Data from our previous paper4, the reaction was run at 50 °C.
20% 29a left.
20% methyl ketone was formed due to alkyne hydration.
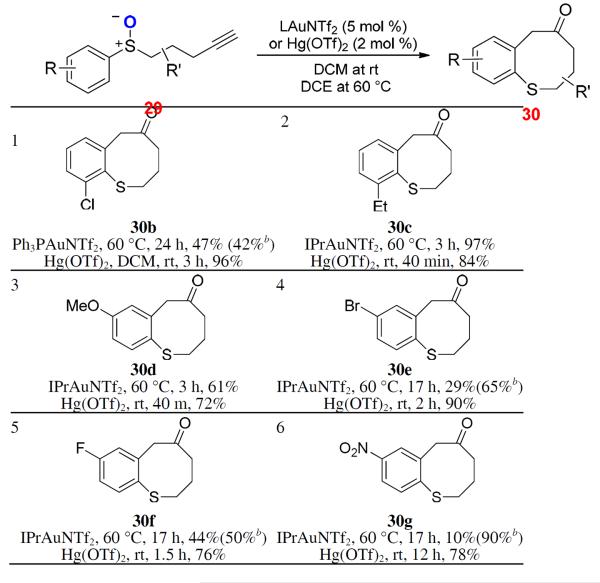
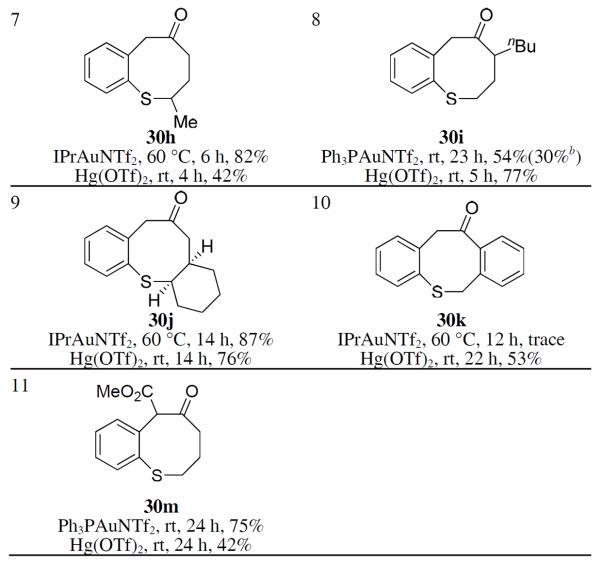
To probe the reaction scope, the benzene ring of 29a was substituted with groups of varying electronic natures at either the ortho or the para position. All the cases studied (Table 2, entries 1–6) worked smoothly and efficiently with Hg(OTf)2 as the catalyst. IPrAuNTf2 was found to be the best gold catalyst in most of the cases, but was generally less efficient than Hg(OTf)2. Moreover, Au catalysis was much slower than that by Hg(OTf)2 in these cases. Aliphatic substitutions at the pentyne chain were allowed (entries 7–9). Interestingly, the positions of their attachment strongly influenced the reaction outcomes for both Au and Hg but with opposite trends: with a 5-methyl group, IPrAuNTf2 was the better catalyst; with a n-butyl group at the 3 position, Hg(OTf)2 was better. cis-Fusion of the pentyne chain with a cyclohexane ring at the 4,5-positions caused no problem for the chemistry (entry 9), but a 3,4-fusion with a benzene ring only permitted the Hg catalysis in a moderate yield (entry 10). The sulfoxide 29m containing an electron-deficient C-C triple bond was allowed (entry 11), and in this case IPrAuNTf2 was better.
Table 2.
Scope studies of benzothiocinone formationa
|
|
[29] = 0.05 M. Isolated yield reported.
Unreacted substrate.
When sulfoxide 31 containing an ethyl group at the alkyne terminus was treated with Ph3PAuNTf2, both the cyclized product 32 and the enone 33 derived from an initial 6-exo-dig cyclization were formed (Eq. 3); in addition, the enone 34 formed from an initial 7-endo-dig cyclization was detected as trace material. Somewhat to our surprise, Hg(OTf)2 promoted the formation of 32 rapidly and efficiently, with only a trace amount of 33 formed. The Hg counterpart24 of α-oxo gold carbene B may be difficult to form. These results point to divergent mechanisms, similar to those shown in Scheme 11, where 32 is formed via a [3,3]-rearrangement of the initial 6-exo-dig cyclization intermediate, and 33 is formed via an α-oxo gold carbene intermediate. Indeed, no cyclized product was observed when the sulfide 34 was subjected to intermolecular oxidation by 8-methylquinoline N-oxide, confirming that the thiocinone 32 was not formed via a gold carbene intermediate (Eq. 4). Notably, when IPrAuNTf2 was used as the catalyst, 33 was formed at the expense of 32, which nevertheless is consistent with the fact that IPr is a much better σ-donor than Ph3P and hence can facilitate the formation of electrophilic gold carbene intermediates, thereby favoring the enone formation.
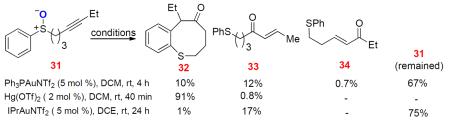 |
(3) |
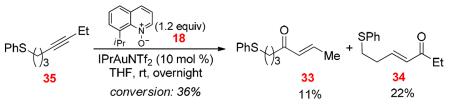 |
(4) |
Even nine-membered S-heterocycles could be readily prepared via this cycloisomerization approach (Eq. 5). Hg(OTf)2 turned out to be the catalyst of choice, and various gold complexes did not promote this reaction.
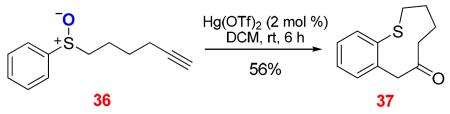 |
(5) |
With the aromatic ring merely provides a C-C double bond in the [3,3]-rearrangement, we reason that a simple C-C double bond would play a similar, if not a better, role. Indeed, when enone sulfoxide 38 was treated with IPrAuNTf2, cyclohexenone-fused dihydrothiocinone 39 was formed in 75% yield. A lower yield was observed with Hg(OTf)2 as the catalyst (Eq. 6). In addition, the enone could be reduced into the allylic alcohol 40. To our delight, it underwent a similar cyclization and [3,3]-rearrangement process albeit with decreased efficiency in the case of either a Au catalyst or Hg(OTf)2, affording the product 41 with the double bond migrated (Eq. 7)
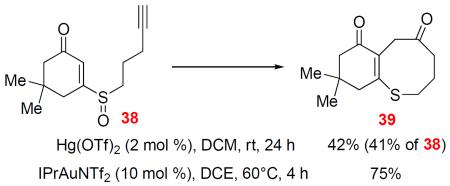 |
(6) |
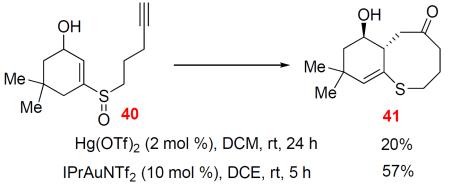 |
(7) |
Computational studies
To provide theoretical support for the [3,3]-sigmatropic rearrangement mechanism, density functional theory (DFT)25 studies have been performed with GAUSSIAN09 program26 using the PBE1PBE27 method. For C, H, O, N, P and S, the 6–31+G** basis set was used, and, for Au and Hg, the SDD basis set with an Effective Core Potential (ECP)28 was used. Geometry optimization was performed in dichloromethane using the SMD29 method. Harmonic vibration frequency calculations were carried out and the optimized structures are all shown to be either minima (with no imaginary frequency) or transition states (with one imaginary frequency). Reactions with three different substrates were calculated. Firstly, the reaction shown in Eq. 3 was explored by using the simplified H3P as the metal ligand. The likely competing occurrence of the gold carbene pathway and the [3,3]-sigmatropic rearrangement pathway in this reaction makes it an excellent case for theoretical studies. Then, the sulfoxide 29a, which differs from 31 by having a terminal alkyne, was examined using the experimentally employed catalyst IPrAu+. Finally, the reaction of the homopropargyl sulfoxide 1 (Scheme 2), reported by both Toste3 and us4 in the pioneer studies of gold carbene chemistry, was studied. All the calculations support the experimental conclusion that the cyclized products are formed via the [3,3]-sigmatropic rearrangement pathway, where no gold carbene intermediate is involved.
The reaction pathways of the sulfoxide 31
As shown in Scheme 13, the complex of 31 and H3PAu+, R, may undergo 7-endo-dig (TS-7-endo-dig, ΔGosol = 12.9 kcal/mol) or 6-exo-dig (TS-6-exo-dig, ΔGosol = 12.1 kcal/mol) cyclization to afford the intermediates INT1 or INT2, respectively. The 6-exo-dig cyclization (Path-b) is slightly more favorable (with full models the energy difference increase to 1.3 kcal/mol),30 qualitatively consistent with the experimental observations that the products resulting from the 6-exo-dig cyclization (i.e., 32 and 33, Eq. 3) were dominant. Another reactant complex R', in which the gold coordinates with the sulfoxide oxygen, is 1.0 kcal/mol less stable than R, and its syn addition to the tethered alkyne (TS-6-exo-dig-syn) has an energy barrier of 19.4 kcal/mol, much higher than that of TS-6-exo-dig-anti.10
Scheme 13.

The calculated reaction pathways for substrate 31. (A) The 7-endo-dig (Path-a) and 6-exo-dig cyclization (Path-b) of the reactant complex R; (B) The gold carbene intermediate pathway (Path-b1) and the [3,3]-sigmatropic rearrangement pathway (Path-b2) of the alkenylgold intermediate INT2. The selected bond lengths are in angstroms, and the relative free energies in dichloromethane (ΔG°sol) are in kcal/mol. Calculated at the PBE1PBE/6-31+G**/SDD level.
There are two likely pathways for the alkenylgold intermediate INT2 to proceed. One is Path-b1, where it undergoes S-O bond breaking (TS-OS) to afford the α-oxo gold carbene intermediate, of which the singlet state is the ground state (INT3-Singlet).30 The C-Au bond length in INT2 is 2.059Å, which is decreased to 2.010Å in INT3-Singlet, indicating a significant interaction between the carbocation and the Au atom, which can be ascribed to the π-donation of the 5d electrons of the Au atom.31 The NBO32 charge on Au is increase from +0.220 in INT2 to +0.371 in INT3-Singlet.
As shown in Figure 2, interestingly, in INT3-Singlet, the carbonyl group (C2-O) is nearly perpendicular to the C3-C1-Au plane. The angle O-C2-C1 is 109.4°, smaller than the other two angles O-C2-C4 (126.7) and C1-C2-C4 (123.8), showing that the carbonyl oxygen atom become closer to C1. The calculated Wiberg bond index33 of O-C1 is 0.2112, indicating that to some extent the lone pair of the carbonyl oxygen atom fills the empty p orbital on C1, stabilizing the carbocation. These features are much more pronounced in INT3-H-Singlet: the angle O-C2-C1 decreases dramatically to 84.8°, and the O-C1 distance decreases to 1.803Å, and the calculated Wiberg bond index of O-C1 increases to 0.6154. Without the electron-donating ethyl group, the carbonyl oxygen atom has a very strong interaction with C1. The structure of the α-oxo gold carbene intermediate may be rationalized by the resonance structures shown in Figure 3. The interaction between the lone pair of the carbonyl oxygen atom and the empty p orbital of the carbocation is supported by the molecular orbital analysis. In the α-oxo gold carbene, in addition to the π-donation of the 5d electrons of the Au atom, the lone pair of the carbonyl oxygen atom is also involved in the stabilization of the carbocation.
Figure 2.
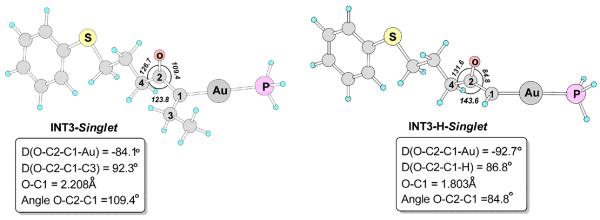
The optimized singlet states of α-oxo gold carbene structures with terminal ethyl (INT3-Singlet) and H group (INT3-H-Singlet). The selected bond lengths are in angstroms, angles are in degree. Calculated in dichloromethane at the PBE1PBE/6-31+G**/SDD level.
Figure 3.
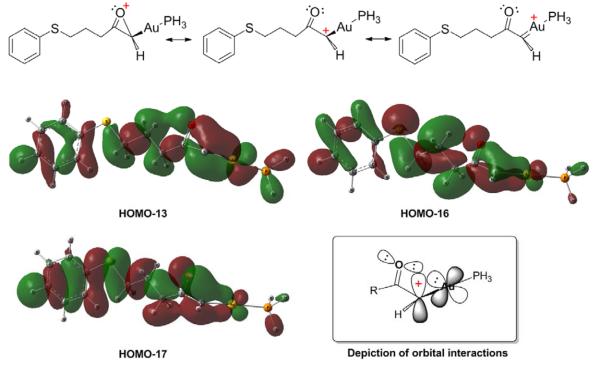
The selected molecular orbitals and the depiction of the orbital interactions in the singlet state α-oxo gold carbene INT3-H-Singlet.
For the gold carbene intermediate INT3-Singlet, the 1, 2-H shift (TS-HT) is very easy, with only a small energy barrier of 2.3 kcal/mol to form the enone product 33. In contrast, the energy barrier for the Friedel-Crafts type electrophilic aromatic substitution reaction via TS-CC is much higher (12.6 kcal/mol). The trapping of the gold carbene by the tethered sulfide to form of the sulfonium ion K has an intermediary energy barrier of 8.6 kcal/mol. These results indicate that the benzothiocinone 32 cannot be obtained from the gold carbene intermediates since the Friedel-Crafts pathway leading to its formation is much less favorable than the 1,2-H shift. The 1,2-H shift, hence, is the predominant route for the decomposition of the gold carbene intermediate, consistent with the experimental observation (Eq. 3). Even for substrates where the 1,2-H shift pathway is not available, the formation of sulfoniums of type E (see Scheme 2) could be substantially favored over the Friedel-Crafts type cyclization.
Alternatively, after a conformational conversion (TS-INT2-conf), INT2' may undergo a [3,3]-sigmatropic rearrangement (TS-33) to afford INT4, which will lead to the benzothiocinone product 32 after 1, 2-H shift.30 The lengths of the breaking O-S bond and the forming C-C bond are 2.023 Å and 3.261 Å, respectively, comparable to that of the former theoretical study (2.565 Å and 3.251 Å).11
The barriers of the competing product-forming steps from INT2 are close to each other for Path-b1 (TS-OS, ΔGosol = 6.7 kcal/mol) and Path-b2 (TS-33, ΔGosol = 7.0 kcal/mol), which is consistent with the formation of both 32 and 33 experimentally. With full models the energy diffence between TS-OS and TS-33 is 0.2 kcal/mol.30
When using Hg(OTf)2 as the catalyst, Hg cannot stabilize the carbocation like Au does due to a much weaker relativistic effect,34 thus the energy of the S-O bond breaking transition state will increase relative to that of the [3, 3] sigmatropic rearrangement. The calculations show that Hg-TS-33 is much more favorable than Hg-TS-OS,30 thus the cyclic product 32 was obtained predominantly.
As we were preparing this manuscript, DFT studies35 of the gold-catalyzed rearrangements of alkynyl aryl sulfoxides appeared. Notably, only the reaction pathways involving the generation of gold carbene intermediates and their subsequent Friedel-Crafts cyclizations, identical to the original proposed mechanisms by Toste3 and us,4 were calculated. The [3,3]-sigmatropic rearrangement pathway and the gold carbene pathways involving the 1,2-hydride migration and the formation of sulfonium species were not considered. Although the Friedel-Crafts cyclizations by gold carbene moieties have calculated energy barriers ranging from 6.0 kcal/mol to 22.8 kcal/mol, they are, as we have shown both experimentally and by calculation, not likely to be the processes leading to the cyclized product.
The reaction pathways of sulfoxide 29a
Based on the systematic theoretical studies with the sulfoxide 31, similar reaction pathways were also obtained with the sulfoxide 29a, which instead is a terminal alkyne. To reflect the fact that IPrAuNTf2 was also an effective catalyst (Table 1), the calculation used the full model of IPr as the metal ligand36 (Scheme 14 and Figure 4).
Scheme 14.

The calculated reaction pathways of the substrate 29a. The selected bond lengths are in angstroms, and the relative free energies in dichloromethane (ΔG°sol) are in kcal/mol. Calculated at the PBE1PBE/6-31+G**/SDD level.
Figure 4.

The main optimized structures on the reaction pathways shown in Scheme 14. The selected bond lengths are in angstroms, and the relative free energies in dichloromethane (ΔG°sol) are in kcal/mol. Calculated at the PBE1PBE/6-31+G**/SDD level.
As shown in Scheme 14, the 6-exo-dig cyclization (IPr-TS-6-exo-dig) is much favorable than 7-endo-dig cyclization (IPr-TS-7-endo-dig) by 5.8 kcal/mol, which is consistent with the experimental observations that only product resulting from the 6-exo-dig cyclization (i.e., 30a) was observed. In the competing product-forming steps from IPr-INT2, the barrier of [3,3]-sigmatropic rearrangement (IPr-TS-33, ΔGosol = 4.9 kcal/mol) is lower than that of O-S bond breaking (IPr-TS-OS, ΔGosol = 6.0 kcal/mol), which is consistent with the formation of 30a experimentally. The α-oxo gold carbene intermediate IPr-INT3-Singlet has similar structural features as INT3-H-Singlet (Figure 2).
The preference to the [3,3]-sigmatropic rearrangement pathway by the sulfoxide 29a, in relation to the case of 31, can be qualitatively rationalized: when the electron-donating ethyl group of 31 is changed to the hydrogen in 29a, the stabilization of the corresponding transition state TS-OS by the ethyl group is lost, whereas during the [3,3]-sigmatropic rearrangement the steric congestion caused by the ethyl group in TS-33 is relieved, and the electrophilicity of the carbon center ipso to AuL in IPr-TS-33 should increases. All the factors should synergistically promote the benzothiocinone formation relative to the gold carbene pathway.
The reaction pathways of sulfoxide 1
As discussed in the introduction, the gold-catalyzed transformation of 1 is a pioneer reaction in gold carbene chemistry. While the experimental studies suggest strongly that gold carbene intermediates are not involved in the formation of benzothiepinones, DFT calculations of the reaction would provide further support for the conclusion. As shown in Scheme 15 and Figure 5, the calculated reaction pathways using IMe as the metal ligand are similar to that of 29a. The 5-exo-dig cyclization (IMe-TS-5-exo-dig) is favorable than 6-endo-dig cyclization (IMe-TS-6-endo-dig) by 6.2 kcal/mol, and the barrier of [3,3]-sigmatropic rearrangement (IMe-TS-33, ΔGosol = 3.2 kcal/mol) is more favorable than that of O-S bond breaking (IMe-TS-OS, ΔGosol = 4.6 kcal/mol).
Scheme 15.

The calculated reaction pathways for substrate 1. The selected bond lengths are in angstroms, and the relative free energies in dichloromethane (ΔG°sol) are in kcal/mol. Calculated at the PBE1PBE/6-31+G**/SDD level.
Figure 5.

The main optimized structures on the reaction pathways shown in Scheme 11. The selected bond lengths are in angstroms, and the relative free energies in dichloromethane (ΔG°sol) are in kcal/mol. Calculated at the PBE1PBE/6-31+G**/SDD level.
Conclusion
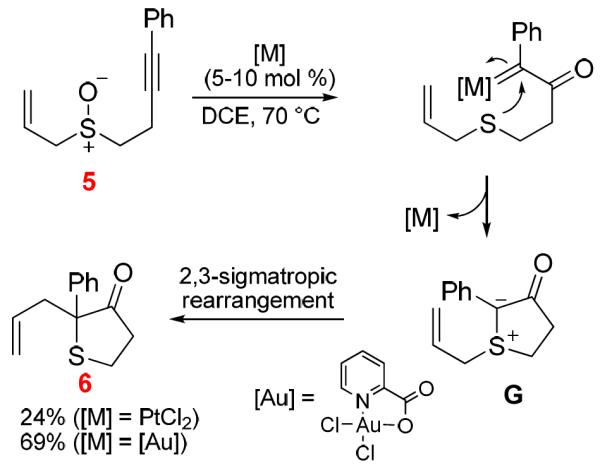
Through a combination of experiments and Density Functional Theory studies, we have addressed lingering questions on the mechanism of gold-catalyzed intramolecular oxidation of terminal alkynes with an arenesulfinyl group as the tethered oxidant. The previous proposed mechanism for the formation of benzothiepinone via Friedel-Crafts type cyclizations of α-oxo gold carbene intermediates is disproved experimentally by generating these reactive species via intermolecular oxidation, and by extensive DFT studies, which reveal a high energy barrier for such cyclizations relative to a 1,2-H shift or an intramolecular trapping by the tethered nascent sulfide. Instead, a [3,3]-sigmatropic rearrangement of the initial cyclization intermediate offers a reaction path that is consistent with both experimental results and the theoretical data. With internal alkyne substrates, however, the intermediacy of a gold carbene species becomes possible. This reactive intermediate, nevertheless, does not proceed to afford the Friedel-Crafts type cyclization product. Instead, a facile 1,2-H shift leads to exclusive formation of an enone product. The [3,3]-sigmatropic rearrangement can still compete with gold carbene formation, yielding mid-sized sulfur-containing cycloalkenone products. With this new mechanistic insight, the product scope of this versatile formation of mid-sized sulfur-containing cycloalkenones has been expanded to various dihydrobenzothiocinones and even a tetrahydrobenzocyclononenone. Besides benzenesulfinyl substrates, alkynes with a tethered alkenylsulfinyl group can likewise participate in the [3,3]-sigmatropic rearrangement, leading to mid-sized S-heterocycles without the entanglement of the fused benzene ring. This opens a new opportunity for accessing a diverse range of substituted mid-sized S-heterocycles. Besides gold, Hg(OTf)2 can be an effective catalyst, thereby offering a cheap alternative for this intramolecular redox reaction.
Supplementary Material
Figure 1.

The main optimized structures on the reaction pathways shown in Scheme 13. The selected bond lengths are in angstroms, and the relative free energies in dichloromethane (ΔG°sol) are in kcal/mol. Calculated at the PBE1PBE/6-31+G**/SDD level.
Acknowledgment
Biao Lu, Yingdong Luo and Liming Zhang thank NSF (CAREER CHE-0969157) and NIGMS (R01 GM084254) for generous financial support. Liming Zhang thanks PIRE-ECCI for supporting the collaboration between him and Yuxue Li. Yuxue Li thanks the National Natural Science Foundation of China (Grant No. 21172248, 21121062). Donald Aue acknowledges support from the Center for Scientific Computing at the CNSI and MRL (NSF MRSEC DMR-1121053 and NSF CNS-0960316) and the National Center for Supercomputing Applications, (NSF TG-CHE100123) utilizing the NCSA Gordon and Blacklight systems.
Footnotes
Supporting Information Available: Experimental procedures, compound characterization and spectra, all the structures in the calculated pathways, the relaxed potential energy surface (PES) scan and the calculated total energies and geometrical coordinates. This information is available free of charge via the internet at http://pubs.acs.org.
References
- (1).(a) Fürstner A, Davies PW. Angew. Chem., Int. Ed. 2007;46:3410–3449. doi: 10.1002/anie.200604335. [DOI] [PubMed] [Google Scholar]; (b) Hashmi ASK. Chem. Rev. 2007;107:3180–3211. doi: 10.1021/cr000436x. [DOI] [PubMed] [Google Scholar]; (c) Ma S, Yu S, Gu Z. Angew. Chem., Int. Ed. 2006;45:200–203. doi: 10.1002/anie.200502999. [DOI] [PubMed] [Google Scholar]; (d) Zhang L, Sun J, Kozmin SA. Adv. Synth. Catal. 2006;348:2271–2296. [Google Scholar]; (e) Arcadi A. Chem. Rev. 2008;108:3266–3325. doi: 10.1021/cr068435d. [DOI] [PubMed] [Google Scholar]; (f) Jiménez-Nuń̃ez E, Echavarren AM. Chem. Rev. 2008;108:3326–3350. doi: 10.1021/cr0684319. [DOI] [PubMed] [Google Scholar]; (g) Li Z, Brouwer C, He C. Chem. Rev. 2008;108:3239–3265. doi: 10.1021/cr068434l. [DOI] [PubMed] [Google Scholar]; (h) Patil NT, Yamamoto Y. Chem. Rev. 2008;108:3395–3442. doi: 10.1021/cr050041j. [DOI] [PubMed] [Google Scholar]; (i) Gorin DJ, Sherry BD, Toste FD. Chem. Rev. 2008;108:3351–3378. doi: 10.1021/cr068430g. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Abu Sohel SM, Liu R-S. Chem. Soc. Rev. 2009;38:2269–2281. doi: 10.1039/b807499m. [DOI] [PubMed] [Google Scholar]; (k) Wang S, Zhang G, Zhang L. Synlett. 2010:692–706. [Google Scholar]; (l) Corma A, Leyva-Perez A, Sabater MJ. Chem. Rev. 2011;111:1657–1712. doi: 10.1021/cr100414u. [DOI] [PubMed] [Google Scholar]; (m) Biannic B, Aponick A. Eur. J. Org. Chem. 2011;2011:6605–6617. [Google Scholar]
- (2).Xiao J, Li X. Angew. Chem., Int. Ed. 2011;50:7226–7236. doi: 10.1002/anie.201100148. [DOI] [PubMed] [Google Scholar]
- (3).Shapiro ND, Toste FD. J. Am. Chem. Soc. 2007;129:4160–4161. doi: 10.1021/ja070789e. [DOI] [PubMed] [Google Scholar]
- (4).Li G, Zhang L. Angew. Chem., Int. Ed. 2007;46:5156–5159. doi: 10.1002/anie.200701449. [DOI] [PubMed] [Google Scholar]
- (5).(a) Davies PW, Albrecht SJC. Angew. Chem., Int. Ed. 2009;48:8372–8375. doi: 10.1002/anie.200904309. [DOI] [PubMed] [Google Scholar]; (b) Davies PW. Pure Appl. Chem. 2010;82:1537–1544. [Google Scholar]
- (6).(a) Yeom HS, Lee JE, Shin S. Angew. Chem., Int. Ed. 2008;47:7040–7043. doi: 10.1002/anie.200802802. [DOI] [PubMed] [Google Scholar]; (b) Yeom HS, Lee Y, Jeong J, So E, Hwang S, Lee JE, Lee SS, Shin S. Angew. Chem., Int. Ed. 2010;49:1611–1614. doi: 10.1002/anie.200906346. [DOI] [PubMed] [Google Scholar]
- (7).(a) Cui L, Peng Y, Zhang L. J. Am. Chem. Soc. 2009;131:8394–8395. doi: 10.1021/ja903531g. [DOI] [PubMed] [Google Scholar]; (b) Cui L, Zhang G, Peng Y, Zhang L. Org. Lett. 2009;11:1225–1228. doi: 10.1021/ol900027h. [DOI] [PubMed] [Google Scholar]; (c) Cui L, Ye L, Zhang L. Chem. Commun. 2010;46:3351–3353. doi: 10.1039/c001314e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).(a) Hashmi AS, Bührle M, Salathé R, Bats J. Adv. Synth. Catal. 2008;350:2059–2064. [Google Scholar]; (b) Li C-W, Lin G-Y, Liu R-S. Chem. Eur. J. 2010;16:5803–5811. doi: 10.1002/chem.201000009. [DOI] [PubMed] [Google Scholar]
- (9).Jadhav AM, Bhunia S, Liao H-Y, Liu R-S. J. Am. Chem. Soc. 2011;133:1769–1771. doi: 10.1021/ja110514s. [DOI] [PubMed] [Google Scholar]
- (10).Noey EL, Luo Y, Zhang L, Houk KN. J. Am. Chem. Soc. 2012;134:1078–1084. doi: 10.1021/ja208860x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Cuenca AB, Montserrai S, Hossain KM, Mancha G, Lledos A, Medio-Simon M, Ujaque G, Asensio G. Org. Lett. 2009;11:4906–4909. doi: 10.1021/ol9020578. [DOI] [PubMed] [Google Scholar]
- (12).Ye L, Cui L, Zhang G, Zhang L. J. Am. Chem. Soc. 2010;132:3258–3259. doi: 10.1021/ja100041e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Ye L, He W, Zhang L. J. Am. Chem. Soc. 2010;132:8550–8551. doi: 10.1021/ja1033952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Lu B, Li C, Zhang L. J. Am. Chem. Soc. 2010;132:14070–14072. doi: 10.1021/ja1072614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).He W, Li C, Zhang L. J. Am. Chem. Soc. 2011:8482–8485. doi: 10.1021/ja2029188. [DOI] [PubMed] [Google Scholar]
- (16).Ye L, He W, Zhang L. Angew. Chem., Int. Ed. 2011;50:3236–3239. doi: 10.1002/anie.201007624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Wang Y, Ji K, Lan S, Zhang L. Angew. Chem., Int. Ed. 2012:1915–1918. doi: 10.1002/anie.201107561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).(a) Bhunia S, Ghorpade S, Huple DB, Liu R-S. Angew. Chem., Int. Ed. 2012;51:2939–2942. doi: 10.1002/anie.201108027. [DOI] [PubMed] [Google Scholar]; (b) Vasu D, Hung H-H, Bhunia S, Gawade SA, Das A, Liu R-S. Angew. Chem., Int. Ed. 2011:6911–6914. doi: 10.1002/anie.201102581. [DOI] [PubMed] [Google Scholar]
- (19).(a) Pirrung MC, Zhang J, Lackey K, Sternbach DD, Brown F. J. Org. Chem. 1995;60:2112–2124. [Google Scholar]; (b) He W, Xie L, Xu Y, Xiang J, Zhang L. Org. Biomol. Chem. 2012;10:3168–3171. doi: 10.1039/c2ob25235j. [DOI] [PubMed] [Google Scholar]
- (20).(a) Fructos MR, Belderrain TR, de Fremont P, Scott NM, Nolan SP, Diaz-Requejo MM, Perez PJ. Angew. Chem. Int. Ed. 2005;44:5284–5288. doi: 10.1002/anie.200501056. [DOI] [PubMed] [Google Scholar]; (b) Fructos MR, de Frémont P, Nolan SP, Díaz-Requejo MM, Pérez PJ. Organometallics. 2006;25:2237–2241. [Google Scholar]; (c) Diaz-Requejo MM, Perez PJ. Chem. Rev. 2008;108:3379–3394. doi: 10.1021/cr078364y. [DOI] [PubMed] [Google Scholar]; (d) Prieto A, Fructos MR, Mar Díaz-Requejo M, Pérez PJ, Pérez-Galán P, Delpont N, Echavarren AM. Tetrahedron. 2009;65:1790–1793. [Google Scholar]
- (21).Mézailles N, Ricard L, Gagosz F. Org. Lett. 2005;7:4133–4136. doi: 10.1021/ol0515917. [DOI] [PubMed] [Google Scholar]
- (22).Kirmse W. Eur. J. Org. Chem. 2002;2002:2193–2256. [Google Scholar]
- (23).Li G, Zhang L. Angew. Chem., Int. Ed. 2007;46:5156–5159. doi: 10.1002/anie.200701449. [DOI] [PubMed] [Google Scholar]
- (24).Lambert JB, Mueller PH, Gaspar PP. J. Am. Chem. Soc. 1980;102:6615–16. [Google Scholar]
- (25).(a) Hohenberg P, Kohn W. Phys. Rev. 1964;136:B864–B871. [Google Scholar]; (b) Kohn W, Sham LJ. Phys. Rev. 1965;140:A1133–A1138. [Google Scholar]
- (26).Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery AJ, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas O, Foresman JB, Ortiz JV, J. C, Fox DJ. Gaussian 09, Revision A.02. Gaussian, Inc.; Wallingford, CT: 2009. [Google Scholar]
- (27).(a) Perdew JP, Burke K, Ernzerhof M. Phys. Rev. Lett. 1996;77:3865–3868. doi: 10.1103/PhysRevLett.77.3865. [DOI] [PubMed] [Google Scholar]; (b) Perdew JP, Burke K, Ernzerhof M. Phys. Rev. Lett. 1997;78:1396–1396. doi: 10.1103/PhysRevLett.77.3865. [DOI] [PubMed] [Google Scholar]
- (28).Fuentealba P, Preuss H, Stoll H, Von Szentpály L. Chem. Phys. Lett. 1982;89:418–422. [Google Scholar]
- (29).Marenich AV, Cramer CJ, Truhlar DG. J. Phys. Chem. 2009;113:6378–6396. doi: 10.1021/jp810292n. [DOI] [PubMed] [Google Scholar]
- (30).See supporting information for details.
- (31).Benitez D, Shapiro ND, Tkatchouk E, Wang Y, Goddard WA, III, Toste FD. Nat. Chem. 2009;1:482–486. doi: 10.1038/nchem.331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Reed AE, Weinstock RB, Weinhold F. J. Chem. Phys. 1985;83:735–746. [Google Scholar]
- (33).Wiberg KB. Tetrahedron. 1968;24:1083–1096. [Google Scholar]
- (34).(a) Pyykkö P. Angew. Chem. Int. Ed. 2004;43:4412–4456. doi: 10.1002/anie.200300624. [DOI] [PubMed] [Google Scholar]; (b) Pyykkö P. Inorg. Chim. Acta. 2005;358:4113–4130. [Google Scholar]
- (35).Fang R, Yang L. Organometallics. 2012:3043–3055. [Google Scholar]
- (36).Correa A, Marion N, Fensterbank L, Malacria M, Nolan SP, Cavallo L. Angew. Chem., Int. Ed. 2008;47:718–721. doi: 10.1002/anie.200703769. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.