

Abstract
The potential of the central nervous system (CNS) to regenerate is regulated by a complex interaction of neuronal intrinsic and extrinsic factors that remain poorly understood. Significant research has been dedicated to identifying these factors to facilitate design of therapies that will treat the functional impairment associated with CNS injuries. Over the last decade, the development of in vivo laser severing of single axons in C. elegans has established an invaluable model for the genetic identification of novel regeneration factors. In a recent study we report the unexpected identification of the core apoptotic proteins CED-4/Apaf-1 and the executioner caspase CED-3 as important factors that promote early events in regeneration in C. elegans. Other upstream regulators of apoptosis do not influence regeneration, indicating the existence of a novel mechanism for activation of CED-4 and CED-3 in neuronal repair. CED-4 and CED-3 function downstream of injury-induced calcium transients and appear to act through the conserved DLK-1 pathway to promote regeneration. We propose a working model for calcium-dependent localized activation of CED-4 and CED-3 caspase and discuss questions raised including mechanisms for spatially regulating activated CED-3 and the possible substrates that it might cleave to initiate regeneration.
Keywords: neuron regeneration, apoptosis, CED-3 caspase, CED-4/Apaf-1, DLK-1, calcium, C. elegans, laser axotomy
Introduction
Brain trauma, spinal cord injury and other insults to the vertebrate central nervous system (CNS) can result in permanent functional impairment due to the inability of CNS neurons to regenerate. This regeneration deficiency results from both neuronal-intrinsic and environmental factors that inhibit functional regrowth.1 Research aimed toward identifying the factors that regulate regeneration have been performed primarily in in vitro neuronal cultures and in vertebrate models such as rodents. Although these vertebrate models have proved highly fruitful, systematic genetic approaches that investigate the biology of neuronal regeneration are not practical in these experimental paradigms. In recent years, in vivo neuronal regeneration assays in model organisms such as C. elegans have been developed to add a new dimension to the molecular understanding of the factors that modulate and mediate regeneration.
In 2004, Yanik, et. al. utilized a femtosecond laser to axotomize individual fluorescently labeled C. elegans GABAergic motorneurons in vivo and then measured regenerative outgrowth in the transparent animal.2 Since this groundbreaking work, several others have exploited the genetically tractable nature of C. elegans to study the molecular pathways that regulate regeneration at the single cell level,3-7 including the use of forward and reverse genetic approaches to identify new factors that influence the ability of a severed neuron to extend new processes.8 C. elegans already has contributed significantly to our understanding of regeneration with the identification of the MAPKKK DLK-1 as an important factor that promotes regeneration.4,7 As is often the case, the dlk-1 regeneration pathway discovered in C. elegans has been shown to be conserved in higher organisms.9,10 In our recent work,11 we unexpectedly discovered that the core apoptotic proteins, CED-4 (Apaf-1 counterpart) and CED-3 caspase, promote early events in neuronal regeneration. Our data reveal a novel mechanism for activation of CED-4 and CED-3 distinct from that utilized in apoptosis and suggest these apoptotic proteins act upstream of DLK-1 to promote regeneration. Interestingly, the elucidation of the C. elegans CED-4/CED-3/DLK-1 pathway linked previously unconnected data from the vertebrate literature to suggest a regeneration pathway that may be conserved in higher organisms.
The Apoptotic Proteins CED-4 and Executioner Caspase CED-3 Promote Efficient Regeneration
An initial interest in how C. elegans eliminates the dissociated neuronal fragment that is generated by laser axotomy prompted us to investigate cell death genes in the context of neuronal injury and regeneration. Unexpectedly, axotomies performed in mutants defective for apoptosis revealed that the C. elegans caspase CED-3, the core apoptotic cell death executioner protein, is not required to eliminate the dissociated fragment, but rather is important in early post-axotomy events that promote neuronal regeneration. Regenerative outgrowth measured 24 h after laser axotomy in both the ALM mechanosensory neuron and D-type GABAergic motor neurons indicated that ced-3 neurons have ~50% less regrowth than wild type. Importantly, the ced-3(n2433) allele, which harbors a point mutation that disrupts the caspase active site to eliminate caspase activity,12 shows a defect similar to that of a CED-3 deletion allele, indicating that CED-3 caspase activity is required for efficient regrowth. Furthermore, CED-3 appears to act cell autonomously in regeneration since low-level expression of a ced-3 transgene only in the axotomized mechanosensory neurons of the ced-3 mutant is able to partially rescue the regeneration defect.
While ced-3 is important for promoting efficient regeneration, it is not absolutely required. As noted above, some outgrowth still occurs in the ced-3 mutant and by 3 d post-axotomy, total outgrowth catches up with that of wild type. The ability of ced-3 neurons to eventually regenerate to wild type levels suggests that a parallel regeneration pathway5 may compensate for the lack of CED-3 over time (see below).
Given the hint that the most profound ced-3 defects might be in early regeneration, we focused on high-resolution analysis of neuronal regrowth during the first 5 h post-axotomy. Indeed, unlike wild type outgrowth, which initiates within ~45 min via the production of highly dynamic transient filopodia-like extensions at the cut site, we found that the initial regenerative outgrowth observed in severed ced-3 neurons often appear as short, wide, persistent bleb-like sprouts. Both time to initial outgrowths and numbers of filipodia extensions are reduced in ced-3 mutants. These data suggest CED-3 caspase contributes to the very early events in regeneration, specifically post-injury filopodia extension/dynamics.
Like many invertebrate systems, C. elegans have the ability to actively repair and reconnect the severed proximal and distal axonal fragments of a damaged neuron.11,13,14 Using a GFP fluorescence transfer protocol, we determined that ced-3 neurons are also defective in their ability to reconnect following axotomy. We found fewer ced-3 neurons track back to the dissociated distal fragment when compared with wild type (not surprising considering less regenerative outgrowth occurs in ced-3 neurons). However, of the neurons that do regrow back to the vicinity of the dissociated distal fragment, significantly fewer ced-3 neurons actually reconnect as compared with wild type. The “failure to reconnect” phenotype is diminished over longer timescales. Nonetheless, a deficiency in CED-3 caspase also impairs reparative timing of axotomized neurons.
The apoptotic cell death pathway was originally characterized in C. elegans and is conserved in higher organisms.15 Many of the apoptotic regulators involved in developmental apoptosis, germline apoptosis and radiation-induced apoptosis have been identified and well studied. An obvious question is whether the mechanism for activation of the CED-3 executioner caspase in regeneration is similar to that utilized in apoptosis. We tested genes involved in regulating developmental (egl-1, ced-9, ced-4 and ced-8), germline (lin-35) and radiation-induced apoptosis (ced-13) for roles in regeneration. Interestingly, only CED-4/Apaf-1, the direct upstream regulator of CED-3 in apoptosis, is required for regeneration. ced-4 mutants have a regeneration defect similar to ced-3 and the double mutant ced-4; ced-3 is impacted to the same degree as either single mutant, suggesting action in the same pathway. In addition, expression of a minimally toxic ced-3 transgene in the ced-4 mutant background is able to partially rescue the ced-4 defect. These data suggest ced-3 acts downstream of ced-4 to promote regeneration. Furthermore, since the other apoptotic regulators do not act in regeneration, a novel regulatory mechanism for activation of CED-4 and CED-3 in regeneration must be operative.
Neuronal injuries induce large intracellular calcium transients that have an important role in the initial neuronal response to injury and subsequent recovery.13,16-20 Influenced by this classic literature, we tested whether calcium might influence the CED-4/CED-3 regeneration pathway. Using the genetically-encoded calcium reporter cameleon, we (and others)13 measured large increases in intracellular calcium in wild type animals immediately consequent to laser axotomy in vivo. Mutations in the ER calcium-binding chaperone calreticulin (crt-1) significantly decrease axotomy-induced calcium transients, indicating that injury-induced calcium signals are partially dependent on CRT-1 and ER calcium supplies. Moreover, crt-1 mutants display initial outgrowth and 24 h regeneration defects similar to ced-3. These data further substantiate the critical role of calcium signaling in initiation of regeneration and indicate CRT-1 is required for C. elegans to mount a proper neuronal calcium response to injury.
How does calcium signaling interface with the CED-3/CED-4 pathway? We found that ced-3 and ced-4 mutants have basically normal calcium fluxes in response to axotomy, and thus mutations in ced-3 and ced-4 do not change the calcium signal itself. The ced-3; crt-1 and ced-4; crt-1 double mutants, however, show similar regeneration defects to each single mutant, indicating action in the same pathway. Expression of the ced-3 transgene partially rescues the crt-1 phenotype, suggesting CED-3 action downstream of CRT-1/calcium. Taken together, the data are consistent with a model in which injury-induced calcium signals, which are partially dependent on CRT-1, are needed for CED-4-dependent activation of CED-3 caspase activity that promotes efficient regeneration initiation.
The DLK MAPKKK is critical for regeneration across species.4,7,9,10 In rodent models, DLK is important for retrograde transport of the injury signals p-STAT3 and p-cJun to promote axon extension.10 In C. elegans, the DLK-1 p38-like MAPK pathway is important for growth cone formation in regenerating D-type motor neurons.4,7 In addition, the JNK MAPK pathway, including the MAPK KGB-1, acts in parallel to the DLK-1 pathway in C. elegans to promote regeneration.5 We were interested in determining whether the apoptotic proteins CED-4 and CED-3 might act in either of these pathways. We found dlk-1 mutants have a regeneration defect in axotomized ALM neurons similar to ced-3 and ced-4 and the dlk-1; ced-3 and dlk-1; ced-4 double mutants show impairment similar to each of the single mutants. Transgenic ced-3 expression in dlk-1 mutants is unable to rescue the dlk-1 defect, suggesting that ced-3 may act upstream of dlk-1 to promote early events in regeneration. In contrast, the kgb-1 regeneration defect appeared similar to ced-3 but the kgb-1 ced-3 double mutant was significantly more impaired in regeneration than either single mutant, suggesting action in parallel pathways. Together, these studies define two parallel pathways, one involving ced-4, ced-3 and dlk-1, and the other involving kgb-1, that act in axon regeneration in severed mechanosensory neurons. The parallel kgb-1 regeneration pathway could partially compensate for the loss of ced-3, explaining why ced-3 defects are often mitigated by 3 d post-axotomy.
Working Model for Localized Activation of CED-4 and CED-3 to Promote Regeneration
Developmental apoptosis in C. elegans is initiated by expression of the BH3 domain protein EGL-1 in cells destined to die.21,22 EGL-1 binds to the Bcl-2-like protein CED-9, which is normally localized to the mitochondrial membrane in a complex with the pro-apoptotic protein CED-4.23 The interaction between EGL-1 and CED-9 causes a conformational change in CED-9 that induces CED-4 to dissociate.23 CED-4 is then free to homo-oligomerize and bind to the procaspase CED-3 through its caspase activation and recognition domain (CARD), which facilitates CED-3 autoactivation.24,25 Notably, however, we found that EGL-1 and CED-9 do not act in axon regeneration. How then are CED-4 and CED-3 activated to promote early events of regeneration? We noted that Apaf-1, the mammalian homolog of CED-4, binds calcium26 and CED-4 is predicted to contain two calcium-binding EF-hand domains.27 The importance of calcium signaling in regeneration, the rapid increase in intracellular calcium induced by axotomy, and our observation that CRT-1/calcium signaling acts upstream of CED-4 and CED-3, leads us to propose a working model for calcium-dependent CED-4/CED-3 activation during regeneration (Fig. 1). Axotomy-induced calcium transients, which are partially dependent on CRT-1, could be “sensed” by CED-4 through its calcium binding EF-hand domains, leading to oligomerization of CED-4. CED-4 could then bind procaspase CED-3, promoting localized autoactivation of CED-3 caspase activity, which in turn stimulates regeneration initiation through the DLK-1 regeneration pathway.
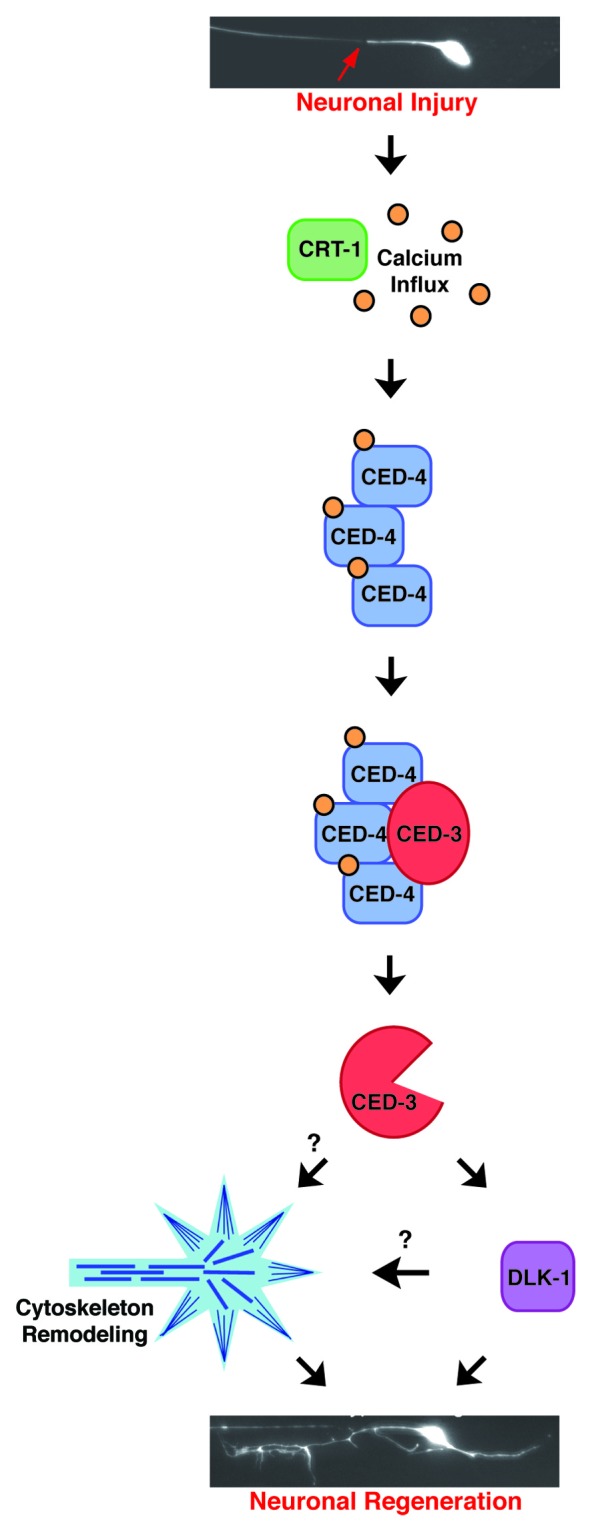
Figure 1. Working model for localized activation of CED-4 and CED-3 to promote regeneration. Neuronal injuries induce calcium transients that are partially dependent on ER calcium stores and CRT-1, possibly via calcium induced calcium release. CED-4 binds the free intracellular calcium through its calcium-binding EF-hand domains, which induces CED-4 oligomerization and recruitment of procaspase CED-3 for local activation of its caspase activity. CED-3 then promotes neuronal repair through the conserved DLK-1 regeneration pathway and potentially through regulation of cytoskeleton dynamics.38,50
Speculation on Localization, Regulation and Function of Activated CED-3 Caspase in Regeneration
Several questions still surround CED-3 caspase and its role in promoting axon regeneration. These include: (1) Where is CED-3 activated in an injured neuron? (2) How is activated CED-3 tightly regulated so it does not induce apoptosis? and (3) What are the substrates of CED-3 caspase activity that promote regeneration? Observations from our work and others can provide the bases for working hypotheses regarding the answers to these questions.
CED-3 caspase is required to promote efficient regeneration in response to axotomy. However, CED-3 activation must be strictly regulated so as not to induce apoptotic cell death. Thus, it is likely that CED-3 is activated only locally at the site of injury. Cellular calcium transients are often locally modulated to control subcellular signaling. In our working model, the large increase in intracellular calcium that occurs at the site of neuronal injury and is restricted to that region13 could activate CED-4 and procaspase CED-3 already present, but latent, in the axon. Such a mechanism would be consistent with genetic data that favor CRT-1 action upstream of CED-3. Localized activation by calcium signals could also explain peculiar axotomy-induced responses we have observed in the disconnected distal axon fragment (independent of the nucleus). Interestingly, after axotomy, the disconnected distal axon initially extends filopodia-like exploratory processes similar to the proximal axon segment. Extension of these exploratory processes from the distal stump is deficient in severed ced-3 neurons. CED-3 caspase, or at the very least ced-3 mRNA, thus appears already present in the axon, waiting to be activated.
Whether procaspase CED-3 is present within the axon solely for purposes of regeneration and repair is uncertain. However, it should be appreciated that caspases have been increasingly found to have non-apoptotic roles in neurons, including activities in dendritic pruning, axon guidance and synaptogenesis.28 Furthermore, evidence for rapid localized activation of caspases has been published. In a model of learning and memory in zebra finch, pre-existing caspase-3 is rapidly and locally activated in the dendritic spines of the auditory forebrain in response to a memory-forming stimulus and is thought to influence synaptic remodeling.29 Localized activation of the fly caspase Dronc in dendrites during larval development is important for dendritic pruning in sensory neurons.30 These precedents support the feasibility of rapid and localized activation of pre-existing CED-3 in regenerating C. elegans neurons.
Avoiding apoptosis consequent to CED-3 activation might be more complicated than limiting the region of activity. Several native mechanisms for preventing autoactivation of procaspase CED-3 have been previously described. For example, an alternative splice form of CED-4, CED-4L, contains an insertion in the nucleotide-binding domain that inhibits its interaction with the CED-3 prodomain, limiting autoactivation of CED-331 (whether the CED-4S or CED-4L isoforms are functional in regeneration remains to be addressed). Autoactivation of CED-3 also can be inhibited by caspase-related CSP-3, which is catalytically inactive but binds the large subunit of the procaspase CED-3 to prevent activation.32 Similarly, catalytically inactive CSP-2 also can inhibit autoactivation of CED-3 in the gonad.33 Thus endogenous inhibitors, known and yet to be discovered, may contribute to limited autoactivation of CED-3 throughout the axon. Again, insight might be drawn from the localized activation of the D. melanogaster caspase Dronc, important in dendritic pruning of sensory neurons during metamorphosis. The E3 ubiquitin ligase DIAP1 targets Dronc for ubiquitination and degradation via the proteasome and may be responsible for restricting Dronc activation to specific dendrites.34,35 A similar mechanism for spatially restricting activated CED-3 could be active in regenerating neurons, but to our knowledge an E3 ubiquitin ligase that targets activated CED-3 for proteasomal degradation has yet to be identified. Interestingly, precedent exists for E3 ubiquitin ligase activity in regeneration. The RPM-1/Phr-1 E3 ligase is implicated in regeneration by targeting DLK-1/DLK for proteasomal degradation, which affects microtubule stability and growth cone morphology.7,36 Significant evidence also indicates the presence of the ubiquitin proteasome system in axons and its activity is required for regeneration.37 These observations suggest it is plausible that activated CED-3 might be spatially restricted within a regenerating neuron via ubiquitination and proteasomal degradation of the caspase, hypotheses remaining to be tested.
It also will be critical to determine the substrates of CED-3 caspase activity that act to promote regeneration. Cytoskeletal proteins are attractive possibilities for CED-3 regeneration substrates since after axonal injury the local cytoskeleton must be repaired and reorganized for growth cone formation and axon extension. In vitro studies have identified several C. elegans cytoskeletal proteins, including actin, tubulin and myosin chains, as substrates for CED-3 caspase.38 Interestingly, a calcium-dependent protease, calpain, regulates cytoskeletal dynamics as well as membrane sealing and possibly axoplasmic vesicle-plasma membrane fusion during growth cone formation and axon extension39-43; therefore, there is precedent for calcium-activated proteases regulating cytoskeleton dynamics in early events of regeneration. Moreover, these findings suggest that neuronal calpains of C. elegans should be tested for their roles in regeneration.
Our data suggests CED-3 acts at least in part through the DLK-1 pathway to promote regeneration; and thus another consideration for candidate substrates is that CED-3 might target regulators of the DLK-1 pathway. Two negative regulators of the DLK-1 pathway include the RPM-1 E3 ubiquitin ligase that can target DLK-1 for degradation by the proteasome5,44,45 and the phosphatase VHP-1 that can dephosphorylate p38 MAPK (PMK-3) to inhibit its activity and regeneration.5,46 Proteolysis of either by CED-3 could lead to enhanced regeneration through the DLK-1 pathway.
A Conserved Regeneration Pathway
The CED-3 regeneration pathway involving axotomy-induced calcium signals, calreticulin, CED-4/Apaf-1, CED-3 caspase and DLK-1 may be a conserved regeneration pathway that operates from invertebrates to mammals. Injury-induced calcium signals are observed in C. elegans and rodent culture models and are implicated in cytoskeleton reorganization and growth cone formation.13,16-20 Calreticulin expression is dramatically induced in injury-conditioned rat dorsal root ganglion (DRG) neurons.47 Caspase activation is implicated in growth cone formation and promoting chemotropic responses in vertebrate culture models.48,49 Finally, the dual leucine zipper kinase (DLK) is a MAPKKK that activates p38 MAPK and is critical for neuronal regeneration. In C. elegans, DLK-1 is required for growth cone formation consequent to axotomy.4,7 In DRG cultures harvested from DLK gene-trap mice, neurons do not regrow as well as wild type neurons.9 In addition, in DLK KO mice peripheral motor and sensory neurons do not display enhanced regeneration in vivo following a conditioning lesion due to loss of the DLK-promoted retrograde transport of p-STAT3 and p-cJun, which act as injury signals to the cell body to potentiate the regeneration response.10 Members of this proposed regeneration pathway are implicated in neuronal regeneration from nematodes to mammals but never have been linked together in the regeneration literature. Our genetic studies of regeneration link these previously unconnected observations and suggest an order to their activities in a regeneration pathway that may be conserved in higher organisms.
Caspases have been a major research focus for their roles in apoptosis. Recent discoveries have begun to highlight caspases for their functions in other physiological processes such as dendritic pruning, axon guidance, synaptogenesis and possibly growth cone formation. Our work has cemented the role of a caspase in the promotion of efficient neuronal regeneration and placed its activity in a potentially conserved regeneration pathway. Future work will provide insights into how caspases are locally activated and spatially regulated to cleave substrates that promote regeneration. In the new age of regenerative medicine, lessons learned from C. elegans single neuron axotomies might hold clues to promoting regrowth in human injury.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/worm/article/22285
References
- 1.Sun F, He Z. Neuronal intrinsic barriers for axon regeneration in the adult CNS. Curr Opin Neurobiol. 2010;20:510–8. doi: 10.1016/j.conb.2010.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yanik MF, Cinar H, Cinar HN, Chisholm AD, Jin Y, Ben-Yakar A. Neurosurgery: functional regeneration after laser axotomy. Nature. 2004;432:822. doi: 10.1038/432822a. [DOI] [PubMed] [Google Scholar]
- 3.Gabel CV, Antoine F, Chuang CF, Samuel AD, Chang C. Distinct cellular and molecular mechanisms mediate initial axon development and adult-stage axon regeneration in C. elegans. Development. 2008;135:1129–36. doi: 10.1242/dev.013995. [DOI] [PubMed] [Google Scholar]
- 4.Hammarlund M, Nix P, Hauth L, Jorgensen EM, Bastiani M. Axon regeneration requires a conserved MAP kinase pathway. Science. 2009;323:802–6. doi: 10.1126/science.1165527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nix P, Hisamoto N, Matsumoto K, Bastiani M. Axon regeneration requires coordinate activation of p38 and JNK MAPK pathways. Proc Natl Acad Sci U S A. 2011;108:10738–43. doi: 10.1073/pnas.1104830108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wu Z, Ghosh-Roy A, Yanik MF, Zhang JZ, Jin Y, Chisholm AD. Caenorhabditis elegans neuronal regeneration is influenced by life stage, ephrin signaling, and synaptic branching. Proc Natl Acad Sci U S A. 2007;104:15132–7. doi: 10.1073/pnas.0707001104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yan D, Wu Z, Chisholm AD, Jin Y. The DLK-1 kinase promotes mRNA stability and local translation in C. elegans synapses and axon regeneration. Cell. 2009;138:1005–18. doi: 10.1016/j.cell.2009.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen L, Wang Z, Ghosh-Roy A, Hubert T, Yan D, O’Rourke S, et al. Axon regeneration pathways identified by systematic genetic screening in C. elegans. Neuron. 2011;71:1043–57. doi: 10.1016/j.neuron.2011.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Itoh A, Horiuchi M, Bannerman P, Pleasure D, Itoh T. Impaired regenerative response of primary sensory neurons in ZPK/DLK gene-trap mice. Biochem Biophys Res Commun. 2009;383:258–62. doi: 10.1016/j.bbrc.2009.04.009. [DOI] [PubMed] [Google Scholar]
- 10.Shin JE, Cho Y, Beirowski B, Milbrandt J, Cavalli V, DiAntonio A. Dual leucine zipper kinase is required for retrograde injury signaling and axonal regeneration. Neuron. 2012;74:1015–22. doi: 10.1016/j.neuron.2012.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pinan-Lucarre B, Gabel CV, Reina CP, Hulme SE, Shevkoplyas SS, Slone RD, et al. The core apoptotic executioner proteins CED-3 and CED-4 promote initiation of neuronal regeneration in Caenorhabditis elegans. PLoS Biol. 2012;10:e1001331. doi: 10.1371/journal.pbio.1001331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xue D, Shaham S, Horvitz HR. The Caenorhabditis elegans cell-death protein CED-3 is a cysteine protease with substrate specificities similar to those of the human CPP32 protease. Genes Dev. 1996;10:1073–83. doi: 10.1101/gad.10.9.1073. [DOI] [PubMed] [Google Scholar]
- 13.Ghosh-Roy A, Wu Z, Goncharov A, Jin Y, Chisholm AD. Calcium and cyclic AMP promote axonal regeneration in Caenorhabditis elegans and require DLK-1 kinase. J Neurosci. 2010;30:3175–83. doi: 10.1523/JNEUROSCI.5464-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Neumann B, Nguyen KC, Hall DH, Ben-Yakar A, Hilliard MA. Axonal regeneration proceeds through specific axonal fusion in transected C. elegans neurons. Dev Dyn. 2011;240:1365–72. doi: 10.1002/dvdy.22606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Conradt B. Genetic control of programmed cell death during animal development. Annu Rev Genet. 2009;43:493–523. doi: 10.1146/annurev.genet.42.110807.091533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chu GK, Tator CH. Calcium influx is necessary for optimal regrowth of transected neurites of rat sympathetic ganglion neurons in vitro. Neuroscience. 2001;102:945–57. doi: 10.1016/S0306-4522(00)00514-5. [DOI] [PubMed] [Google Scholar]
- 17.Henley J, Poo MM. Guiding neuronal growth cones using Ca2+ signals. Trends Cell Biol. 2004;14:320–30. doi: 10.1016/j.tcb.2004.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kamber D, Erez H, Spira ME. Local calcium-dependent mechanisms determine whether a cut axonal end assembles a retarded endbulb or competent growth cone. Exp Neurol. 2009;219:112–25. doi: 10.1016/j.expneurol.2009.05.004. [DOI] [PubMed] [Google Scholar]
- 19.Mattson MP, LaFerla FM, Chan SL, Leissring MA, Shepel PN, Geiger JD. Calcium signaling in the ER: its role in neuronal plasticity and neurodegenerative disorders. Trends Neurosci. 2000;23:222–9. doi: 10.1016/S0166-2236(00)01548-4. [DOI] [PubMed] [Google Scholar]
- 20.Zheng JQ, Poo MM. Calcium signaling in neuronal motility. Annu Rev Cell Dev Biol. 2007;23:375–404. doi: 10.1146/annurev.cellbio.23.090506.123221. [DOI] [PubMed] [Google Scholar]
- 21.Conradt B, Horvitz HR. The TRA-1A sex determination protein of C. elegans regulates sexually dimorphic cell deaths by repressing the egl-1 cell death activator gene. Cell. 1999;98:317–27. doi: 10.1016/S0092-8674(00)81961-3. [DOI] [PubMed] [Google Scholar]
- 22.Thellmann M, Hatzold J, Conradt B. The Snail-like CES-1 protein of C. elegans can block the expression of the BH3-only cell-death activator gene egl-1 by antagonizing the function of bHLH proteins. Development. 2003;130:4057–71. doi: 10.1242/dev.00597. [DOI] [PubMed] [Google Scholar]
- 23.Yan N, Gu L, Kokel D, Chai J, Li W, Han A, et al. Structural, biochemical, and functional analyses of CED-9 recognition by the proapoptotic proteins EGL-1 and CED-4. Mol Cell. 2004;15:999–1006. doi: 10.1016/j.molcel.2004.08.022. [DOI] [PubMed] [Google Scholar]
- 24.Yang X, Chang HY, Baltimore D. Essential role of CED-4 oligomerization in CED-3 activation and apoptosis. Science. 1998;281:1355–7. doi: 10.1126/science.281.5381.1355. [DOI] [PubMed] [Google Scholar]
- 25.Seshagiri S, Miller LK. Caenorhabditis elegans CED-4 stimulates CED-3 processing and CED-3-induced apoptosis. Curr Biol. 1997;7:455–60. doi: 10.1016/S0960-9822(06)00216-8. [DOI] [PubMed] [Google Scholar]
- 26.Bao Q, Lu W, Rabinowitz JD, Shi Y. Calcium blocks formation of apoptosome by preventing nucleotide exchange in Apaf-1. Mol Cell. 2007;25:181–92. doi: 10.1016/j.molcel.2006.12.013. [DOI] [PubMed] [Google Scholar]
- 27.Yuan J, Horvitz HR. The Caenorhabditis elegans cell death gene ced-4 encodes a novel protein and is expressed during the period of extensive programmed cell death. Development. 1992;116:309–20. doi: 10.1242/dev.116.2.309. [DOI] [PubMed] [Google Scholar]
- 28.Hyman BT, Yuan J. Apoptotic and non-apoptotic roles of caspases in neuronal physiology and pathophysiology. Nat Rev Neurosci. 2012;13:395–406. doi: 10.1038/nrn3228. [DOI] [PubMed] [Google Scholar]
- 29.Huesmann GR, Clayton DF. Dynamic role of postsynaptic caspase-3 and BIRC4 in zebra finch song-response habituation. Neuron. 2006;52:1061–72. doi: 10.1016/j.neuron.2006.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Williams DW, Kondo S, Krzyzanowska A, Hiromi Y, Truman JW. Local caspase activity directs engulfment of dendrites during pruning. Nat Neurosci. 2006;9:1234–6. doi: 10.1038/nn1774. [DOI] [PubMed] [Google Scholar]
- 31.Shaham S, Horvitz HR. An alternatively spliced C. elegans ced-4 RNA encodes a novel cell death inhibitor. Cell. 1996;86:201–8. doi: 10.1016/S0092-8674(00)80092-6. [DOI] [PubMed] [Google Scholar]
- 32.Geng X, Shi Y, Nakagawa A, Yoshina S, Mitani S, Shi Y, et al. Inhibition of CED-3 zymogen activation and apoptosis in Caenorhabditis elegans by caspase homolog CSP-3. Nat Struct Mol Biol. 2008;15:1094–101. doi: 10.1038/nsmb.1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Geng X, Zhou QH, Kage-Nakadai E, Shi Y, Yan N, Mitani S, et al. Caenorhabditis elegans caspase homolog CSP-2 inhibits CED-3 autoactivation and apoptosis in germ cells. Cell Death Differ. 2009;16:1385–94. doi: 10.1038/cdd.2009.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kuo CT, Zhu S, Younger S, Jan LY, Jan YN. Identification of E2/E3 ubiquitinating enzymes and caspase activity regulating Drosophila sensory neuron dendrite pruning. Neuron. 2006;51:283–90. doi: 10.1016/j.neuron.2006.07.014. [DOI] [PubMed] [Google Scholar]
- 35.Rumpf S, Lee SB, Jan LY, Jan YN. Neuronal remodeling and apoptosis require VCP-dependent degradation of the apoptosis inhibitor DIAP1. Development. 2011;138:1153–60. doi: 10.1242/dev.062703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lewcock JW, Genoud N, Lettieri K, Pfaff SL. The ubiquitin ligase Phr1 regulates axon outgrowth through modulation of microtubule dynamics. Neuron. 2007;56:604–20. doi: 10.1016/j.neuron.2007.09.009. [DOI] [PubMed] [Google Scholar]
- 37.Gumy LF, Tan CL, Fawcett JW. The role of local protein synthesis and degradation in axon regeneration. Exp Neurol. 2010;223:28–37. doi: 10.1016/j.expneurol.2009.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Taylor RC, Brumatti G, Ito S, Hengartner MO, Derry WB, Martin SJ. Establishing a blueprint for CED-3-dependent killing through identification of multiple substrates for this protease. J Biol Chem. 2007;282:15011–21. doi: 10.1074/jbc.M611051200. [DOI] [PubMed] [Google Scholar]
- 39.Godell CM, Smyers ME, Eddleman CS, Ballinger ML, Fishman HM, Bittner GD. Calpain activity promotes the sealing of severed giant axons. Proc Natl Acad Sci U S A. 1997;94:4751–6. doi: 10.1073/pnas.94.9.4751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Howard MJ, David G, Barrett JN. Resealing of transected myelinated mammalian axons in vivo: evidence for involvement of calpain. Neuroscience. 1999;93:807–15. doi: 10.1016/S0306-4522(99)00195-5. [DOI] [PubMed] [Google Scholar]
- 41.Shi R, Asano T, Vining NC, Blight AR. Control of membrane sealing in injured mammalian spinal cord axons. J Neurophysiol. 2000;84:1763–9. doi: 10.1152/jn.2000.84.4.1763. [DOI] [PubMed] [Google Scholar]
- 42.Yoo S, Nguyen MP, Fukuda M, Bittner GD, Fishman HM. Plasmalemmal sealing of transected mammalian neurites is a gradual process mediated by Ca(2+)-regulated proteins. J Neurosci Res. 2003;74:541–51. doi: 10.1002/jnr.10771. [DOI] [PubMed] [Google Scholar]
- 43.Bennett V, Baines AJ. Spectrin and ankyrin-based pathways: metazoan inventions for integrating cells into tissues. Physiol Rev. 2001;81:1353–92. doi: 10.1152/physrev.2001.81.3.1353. [DOI] [PubMed] [Google Scholar]
- 44.Nakata K, Abrams B, Grill B, Goncharov A, Huang X, Chisholm AD, et al. Regulation of a DLK-1 and p38 MAP kinase pathway by the ubiquitin ligase RPM-1 is required for presynaptic development. Cell. 2005;120:407–20. doi: 10.1016/j.cell.2004.12.017. [DOI] [PubMed] [Google Scholar]
- 45.Collins CA, Wairkar YP, Johnson SL, DiAntonio A. Highwire restrains synaptic growth by attenuating a MAP kinase signal. Neuron. 2006;51:57–69. doi: 10.1016/j.neuron.2006.05.026. [DOI] [PubMed] [Google Scholar]
- 46.Mizuno T, Hisamoto N, Terada T, Kondo T, Adachi M, Nishida E, et al. The Caenorhabditis elegans MAPK phosphatase VHP-1 mediates a novel JNK-like signaling pathway in stress response. EMBO J. 2004;23:2226–34. doi: 10.1038/sj.emboj.7600226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Willis D, Li KW, Zheng JQ, Chang JH, Smit AB, Kelly T, et al. Differential transport and local translation of cytoskeletal, injury-response, and neurodegeneration protein mRNAs in axons. J Neurosci. 2005;25:778–91. doi: 10.1523/JNEUROSCI.4235-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Campbell DS, Holt CE. Apoptotic pathway and MAPKs differentially regulate chemotropic responses of retinal growth cones. Neuron. 2003;37:939–52. doi: 10.1016/S0896-6273(03)00158-2. [DOI] [PubMed] [Google Scholar]
- 49.Verma P, Chierzi S, Codd AM, Campbell DS, Meyer RL, Holt CE, et al. Axonal protein synthesis and degradation are necessary for efficient growth cone regeneration. J Neurosci. 2005;25:331–42. doi: 10.1523/JNEUROSCI.3073-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hirai S, Banba Y, Satake T, Ohno S. Axon formation in neocortical neurons depends on stage-specific regulation of microtubule stability by the dual leucine zipper kinase-c-Jun N-terminal kinase pathway. J Neurosci. 2011;31:6468–80. doi: 10.1523/JNEUROSCI.5038-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]