

Abstract
Purpose
To describe immunohistopathological findings in autosomal dominant neovascular inflammatory vitreoretinopathy (ADNIV).
Methods
An enucleated eye specimen from a patient with Stage V ADNIV was examined using standard histopathological methods and lymphocyte markers.
Results
A c.731T>C CAPN5 mutation resulted in a p.Leu244Pro substitution in calpain-5. The eye showed exudative retinal detachment and neovascularization, intraocular fibrosis, and features of phthisis bulbi. Chronic inflammatory CD3-positive cell infiltrates were identified throughout the uvea, vitreous and retina, consistent with chronic uveitis.
Conclusion
Mutations in CAPN5 trigger autoimmune uveitis characterized by inflammatory T-cells and severe neovascularization.
Keywords: autosomal dominant neovascular inflammatory vitreoretinopathy, ADNIV, CAPN5, uveitis, CD3
Introduction
Mutations in the CAPN5 gene cause autosomal dominant neovascular inflammatory vitreoretinopathy (ADNIV), an inherited uveitis and vitreoretinal degeneration without systemic features.1–3 CAPN5 is the first nonsyndromic gene for autoimmune uveitis, and among the very few Mendelian autoimmune diseases where the gene is identified. CAPN5 encodes the protease calpain-5, a calcium-activated cysteine protease. Although the disease mechanisms are not understood, CAPN5 mutations represent a unique model for investigating autoimmunity.
In its early stages, ADNIV is characterized by inflammatory cells in the vitreous (Table 1).1–4 This stage is clinically similar to an idiopathic posterior uveitis. Chronic inflammation continues throughout the disease, which also features pigmentary retinal degeneration, loss of the electroretinogram b-wave, retinal and iris neovascularization, neovascular glaucoma, retinal detachment, and proliferative vitreoretinopathy.1–3 The retinal degeneration appears similar to retinitis pigmentosa, and the neovascularization is similar to proliferative diabetic retinopathy. After several decades, eyes become phthisical and blind (Table 1). We previously reported the immunohistopathological features of two eyes with a c.728G>T, p.Arg243 Leu mutation in CAPN5.5 We found CD3 cell infiltrates, suggesting a predominantly T-cell process. In this study, we examined an eye with end-stage ADNIV from a family with a different mutation in CAPN5.
Table 1.
Stages of ADNIV (autosomal dominant neovascular inflammatory vitreoretinopathy) disease and associated clinical findings
| Stage | Anterior segment | Posterior segment | Electroretinogram |
|---|---|---|---|
| Stage I | Rare cells | Rare cells | Reduced b-wave |
| Stage II | Mild cells Mild posterior subcapsular cataract |
Moderate cells Optic disc edema Cystoid macular edema Peripheral pigmentation of the retina |
Flat b-wave, normal a-wave |
| Stage III | Moderate cells Anterior subcapsular cataract Moderate posterior subcapsular cataract Iris synechiae |
Moderate cells Vitreous traction at posterior pole Epiretinal membrane Tractional retinal detachment Posterior vitreous bands Peripheral fibrosis Peripheral neovascularization Neovascularization of the disc Pigmentary retinopathy Cystoid macular edema |
Flat b-wave, reduced a-wave |
| Stage IV | Iris bombe, seclusio pupil Peripheral anterior synechiae Angle closure glaucoma Neovascular glaucoma |
Moderate cells Vitreous hemorrhage Posterior tractional retinal detachment Anterior tractional retinal detachment |
Non-recordable electroretinogram |
| Stage V | Phthisis | Combined tractional and rhegmatogenous retinal detachment Phthisis |
Methods
The patient clinical history was reviewed. An enucleated eye was received in 10% formalin (Sigma-Aldrich, St Louis, MO, USA). The eye was opened by pupil-optic nerve section and lightly decalcified. Histological staining with hemotoxylin and eosin and Masson’s Trichrome stain was performed according to the manufacturer’s protocol (Sigma-Aldrich, St Louis, MO, USA). Eye tissue sections were immunostained using a Ventana® Discovery Automated IHC System (Ventana Medical Systems, Oro Valley, AZ, USA). The following antibodies were used: anti-CD3; anti-glial fibrillary acidic protein (GFAP); anti-CD20 followed by horseradish peroxidase conjugated secondary antibodies and DAB+ detection, according to the manufacturer’s instructions (Dako, Carpinteria, CA, USA). Immunostained slides were counterstained with hematoxylin to highlight the nuclei.
Results
A 52-year-old male had suffered bilateral chronic uveitis consistent with ADNIV since age 28. His grandmother and mother were affected, along with four sisters and one brother (Figure 1). This family carried a c.731T>C, p.Leu244Pro mutation in the CAPN5 gene.1 Over the course of his disease, he underwent multiple surgeries in the right eye, including cataract with lens implantation, vitrectomies for retina membranes and vitreous hemorrhages, and an Ahmed tube for neovascular and steroid response glaucoma. He eventually developed stage V ADNIV characterized by phthisis and no light perception.1 The eye became painful and was enucleated.
Figure 1.
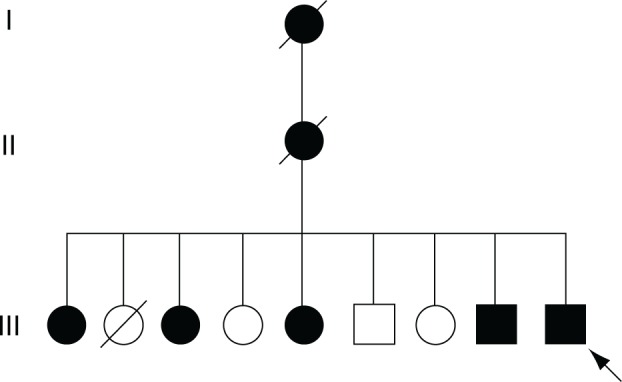
Pedigree of the enucleated eye.
Notes: A c.731T>C mutation in CAPN5 was inherited in a dominant fashion in a family with ADNIV (autosomal dominant neovascular inflammatory vitreoretinopathy). In the most recent generation, there were five affected, and four unaffected siblings. The enucleated eye was from a male sibling (arrow).
Gross examination of the eye revealed a shrunken, distorted phthisical globe (Figure 2A). The anterior to posterior dimension measured 17.8 mm, the superior to inferior dimension 22.0 mm, and the medial to lateral dimension 21.5 mm. A segment of optic nerve was unremarkable. A clear silicone Ahmed valve was found between the sclera and a tan-brown fibrous tissue. The cornea was depressed, brown, and opaque with a medial to lateral dimension of 12.0 mm and superior to inferior dimension of 10.0 mm. Pupil-optic nerve sectioning of the specimen revealed an anterior chamber filled with hemorrhage, and no distinct lens tissue was appreciated. The posterior chamber showed a vitreous filled with red-brown material, a detached retina, and subretinal hemorrhage. The ciliary body was detached. There was normally pigmented choroid. The optic nerve was atrophic, and there was replacement of axons by collagenous bands (Figure 2B).
Figure 2.
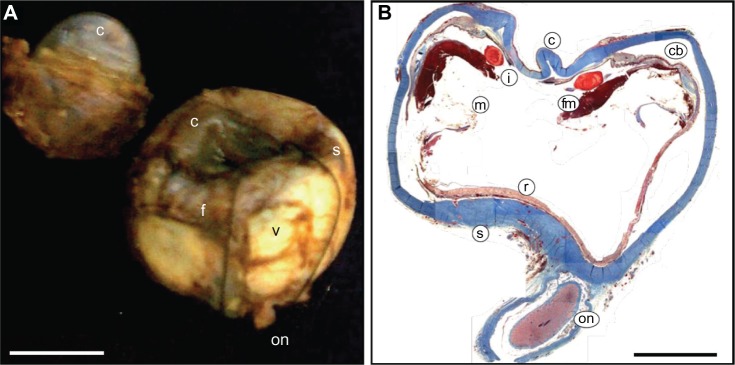
Stage V ADNIV (autosomal dominant neovascular inflammatory vitreoretinopathy).
Notes: (A) The right eye is phthisical. The vitreous (v) is filled with hemorrhage and fibrosis (f). The cornea (c) is depressed and the sclera (s) is shrunken. The optic nerve (on) is atrophic. Scale bar = 1 cm. (B) Masson’s trichrome stain shows a square shaped, thickened sclera (s). The cornea (c) is distorted and the anterior chamber is collapsed. Fibrovascular membranes (fm) cause tractional detachment of the ciliary body (cb) and membranes (m) cause traction on the retina (r). The lens (l) is fragmented. The optic nerve (on) is atrophic. Scale bar = 0.5 cm.
Microscopic examination showed typical features of phthisis bulbi (Figure 2B). The cornea and sclera were thickened and formed a shrunken square outline. The intraocular contents were grossly disorganized. An anterior hyaloid fibrovascular membrane detached the ciliary body, which had underlying suprachoroidal effusions. The membrane extended over the iris and into a collapsed anterior chamber.
In the anterior segment, the corneal epithelium appeared normal, but Bowman’s membrane showed dystrophic calcium deposits consistent with calcific band keratopathy (Figure 3A). The stroma did not show evidence of neovascularization (Figure 3A). Descemet’s membrane was intact, but there was loss of endothelial cells. The lens capsule was intact, and there were severe cataractous changes (Figure 3B). There was hemorrhage in the anterior chamber. The iridocorneal angle was occluded by fibrous membranes, highlighted in a Masson’s trichrome stain (Figure 3C and D). The iris showed significant chronic inflammation consistent with chronic uveitis (Figure 3E and F).
Figure 3.

Anterior segment of the cornea.
Notes: (A) Cornea reveals folded Bowman’s membrane and stromal (st) disorganization without neovascularization. Epithelium (e) (H&E). (B) Lens (l) reveals cataractous changes including Morgagnian globules and lens fiber degeneration. Vitreous hemorrhage (vh) (H&E). (C and D) Iridocorneal angle (a) is occluded (arrow in C) by fibrous membrane (f) (C, H&E; D, MT) stromal (st); iris pigment epithelium (p). (E and F) Iris shows extensive inflammatory cell infiltrate (arrows in E, F) into iris stroma (s) (H&E). Descemet’s membrane (d), iris (i), iris stroma (s), iris pigment epithelium (p), membrane (m). Scale bar = 250 μm.
Abbreviations: H&E, hemotoxylin and eosin stain; MT, Masson’s trichrome.
In the posterior segment, the ciliary body was detached with chronic inflammatory cells in the supraciliary space. The vitreous was filled with hemorrhage, fibrous membranes, and showed a tractional retinal detachment (Figure 4). The retina showed extensive degeneration, and most of the nerve fiber layer, retinal ganglion cells, inner plexiform layer, outer plexiform layer, and photoreceptor cells were absent (Figure 5). Dense gliotic scars replaced the retina, which was confirmed by positive glial fibrillary acidic protein immunohistochemistry (Figure 5D). There was retinal neovascularization and subretinal hemorrhage. The retinal pigment epithelium showed significant cell loss. The vessels of the choriocapillaris were patent and without thrombi. The choroid was focally hyalinized and calcified, and there appeared to be inflammatory cell infiltrates that were overwhelmingly lymphocytic in nature (Figure 5A). Interestingly, other inflammatory cells, like neutrophils, eosinophils, and macrophages were rare.
Figure 4.

Vitreous hemorrhage and membrane.
Notes: (A) Vitreous hemorrhage (vh) and fibrovascular membranes (fm) cause tractional retinal detachment (r) (H&E). Vitreous membranes (m). (B) Tractional detachment and vitreous hemorrhage (MT). Vitreous hemorrhage (vh) and fibrovascular membranes (fm) cause tractional retinal detachment (r). (C and D) Vitreous membranes (m) and red blood cells (H&E). Scale bar = 250 μm.
Abbreviations: H&E, hemotoxylin and eosin stain; MT, Masson’s trichrome.
Figure 5.

Retina.
Notes: (A) Cell infiltrates (arrows) are present in the retina (r) and choroid (ch), and retinal pigment epithelium is atrophic (arrow head) (H&E). (B) An epiretinal membrane (erm) overlies the retina (MT). (C) Osseous metaplasia (om) replaces the retina and choroid is indicated (arrow). (D) Extensive gliosis of retina (r) is highlighted by a glial fibrillary acidic protein stain (brown); vitreous (v). Scale bar = 250 μm.
Abbreviations: H&E, hemotoxylin and eosin stain; MT, Masson’s trichrome.
Immunohistochemical lymphocyte markers were examined. CD3 cells (T-cell receptor and a specific molecular marker of T cells) were found throughout the uvea (Figure 6). Dense infiltrates were present in the iris and choroid. CD3 cells were also present in the supraciliary space, vitreous, and retina, similar to findings in other ADNIV eyes.5 In contrast to our previous ADNIV study, we also found some CD20 cells (B-cell marker) in the iris, but these were estimated to represent less than 15% of all positively stained lymphocytes (data not shown). Neither of these lymphocyte markers was found in control phthisical eyes.5
Figure 6.

Lymphocyte infiltration.
Notes: Cluster of Differentiation-3 (CD3) T-cell marker was identified in cells (brown color reaction, arrows) infiltrating the: (A) iris, (B) supraciliary space (ss), (C) retina (r), and (D) vitreous (v) and retinal blood cells (rbc). Scale bar = 250 μm.
Discussion
Study of CAPN5 mutations can provide critical insight into the mechanisms of ocular autoimmunity. These findings support the hypothesis that vitreous cells observed on clinical examination of ADNIV are largely due to T-cell infiltration. Regardless of the specific CAPN5 genotype, our study indicates the cellular immune response is similar. These inflammatory cells extend to the iris and anterior chamber. The inflammatory cells found in the supraciliary space, retina, and choroid, however, are not appreciated clinically. The finding of mixed T- and B-cell inflammation in the uvea of this eye is consistent with a chronic uveitis. In previously examined eyes, we did not find B-cells, and patient serum did not contain antiretinal antibodies.5 One difference is the patient genotype, but a more likely explanation is the stage at which the eyes were enucleated. Previous eyes without B-cells were obtained at autopsy, after the eyes had been phthisical for years, and the comparatively less severe inflammation suggested the uveitis had burnt out. Nevertheless, the relative fraction of T-cells was much higher than the fraction of B-cells. Since increased T-cells are relatively rare, compared to B-cells, the detection of large numbers of T-cells is much more significant.
Immunomodulation can be directed against T-cells, but past medical therapy for ADNIV has had limited efficacy.3 The eye in this case received repeated injections of triamcinolone acetonide (Kenalog®, Bristol-Myers Squibb, Princeton, NJ, USA) to control cystoid macular edema and intraocular inflammation, but developed steroid-induced glaucoma. We have reported on the use of surgical fluocinolone acetonide implants for the long-term sustained delivery of a local steroid.3 This device suppressed inflammatory cells and neovascularization, but was subject to failure if encapsulated by fibrotic membranes.6 Despite the control of inflammation, there was persistent retinal degeneration and vision loss. It is possible that earlier intervention with T-cell immunomodulators may be more successful.
Iris and retinal neovascularization causes hyphema and vitreous hemorrhages, and membrane formation contributes to tractional detachment at the posterior pole and vitreous base. Neovascular vessels, membranes, and recent hemorrhage were found in the examined eye. Although the clinical appearance of neovascularization is similar to proliferative diabetic retinopathy, significant capillary dropout or evidence of ischemia is absent (unpublished observations). This suggests that a non-ischemic signaling pathway activates neovascularization. In addition, the examined eye showed near complete fibrosis of the retina. This indicates the cell degenerative signals of mutant CAPN5 are resistant to steroids.
Calpains cause limited intracellular proteolysis, and target multiple intracellular signaling pathways.7,8 Since the disease is dominant, highly penetrant, and displays a severe phenotype, it seems likely that CAPN5 mutations lead gain of function1 where the proteolytic activity is increased or substrate specificity is altered.1 Excess calpain activity has been linked to several diseases, including multiple sclerosis, Alzheimer’s disease, diabetes, cancer, muscular dystrophy, and photoreceptor degeneration.9–12 Uncontrolled activation of inflammatory, neovascular, and fibrotic pathways would explain the various phenotypic stages of ADNIV. Identification of these intracellular signaling pathways may point to therapeutic targets to delay or reverse ADNIV and related eye diseases.3
Conclusion
Studies in mice demonstrated that CAPN5 knockout mice had no observable phenotype.13 Calpain-5 expression was found in embryonic C25 thymocytes and some CD3 cells, but there was no sign of autoimmunity.13 This supports the concept that the ADNIV disease alleles might lead to a gain of function. It emphasizes the importance of developing an animal model that expresses the CAPN5 mutations. We found calpain-5 expression in human photoreceptors,1 and we recently verified this in the mouse (Mahajan, unpublished data, 2013). It is plausible that the inflammatory trigger for ADNIV arises in the retina. Since CAPN5 is also expressed in T-cells, it will be important to distinguish the effects of ADNIV mutations in the retina from the adaptive immune system.
Acknowledgments
The authors are supported by NIH Grant K08EY020530 and Research to Prevent Blindness (VBM) and NIH grant RO1EY020846 (JHL). The authors gratefully acknowledge MaryAnn Mahajan for microscopic photography.
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Mahajan VB, Skeie JM, Bassuk AG, et al. Calpain-5 mutations cause autoimmune uveitis, retinal neovascularization, and photoreceptor degeneration. PLoS Genet. 2012 Oct 4; doi: 10.1371/journal.pgen.1003001. Epub. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bennett SR, Folk JC, Kimura AE, Russell SR, Stone EM, Raphtis EM. Autosomal dominant neovascular inflammatory vitreoretinopathy. Ophthalmology. 1990;97:1125–1135. doi: 10.1016/s0161-6420(90)32447-8. [DOI] [PubMed] [Google Scholar]
- 3.Tlucek PS, Folk JC, Orien JA, Stone EM, Mahajan VB. Inhibition of neovascularization but not fibrosis with the fluocinolone acetonide implant in autosomal dominant neovascular inflammatory vitreoretinopathy. Arch Ophthalmol. 2012;130(11):1395–1401. doi: 10.1001/archophthalmol.2012.1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rowell HA, Bassuk AG, Mahajan VB. Monozygotic twins with CAPN5 autosomal dominant neovascular inflammatory vitreoretinopathy. Clin Ophthalmol. 2012;6:2037–2044. doi: 10.2147/OPTH.S40086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mahajan VB, Vallone JG, Lin JH, et al. T-cell infiltration in autosomal dominant neovascular inflammatory vitreoretinopathy. Mol Vis. 2010;16:1034–1040. [PMC free article] [PubMed] [Google Scholar]
- 6.Tlucek PS, Folk JC, Sobol WM, Mahajan VB. Surgical management of fibrotic encapsulation of the fluocinolone acetonide implant in CAPN5-associated proliferative vitreoretinopathy. Clin Ophthalmol. 2013;7:1093–1098. doi: 10.2147/OPTH.S43939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Goll DE, Thompson VF, Li H, Wei W, Cong J. The calpain system. Physiol Rev. 2003;83:731–801. doi: 10.1152/physrev.00029.2002. [DOI] [PubMed] [Google Scholar]
- 8.Sato K, Kawashima S. Calpain function in the modulation of signal transduction molecules. Biol Chem. 2001;382:743–751. doi: 10.1515/BC.2001.090. [DOI] [PubMed] [Google Scholar]
- 9.Zatz M, Starling A. Calpains and disease. N Engl J Med. 2005;352:2413–2423. doi: 10.1056/NEJMra043361. [DOI] [PubMed] [Google Scholar]
- 10.Huang Y, Wang KK. The calpain family and human disease. Trends Mol Med. 2001;7:355–362. doi: 10.1016/s1471-4914(01)02049-4. [DOI] [PubMed] [Google Scholar]
- 11.Richard I, Broux O, Allamand V, et al. Mutations in the proteolytic enzyme calpain 3 cause limb-girdle muscular dystrophy type 2A. Cell. 1995;81:27–40. doi: 10.1016/0092-8674(95)90368-2. [DOI] [PubMed] [Google Scholar]
- 12.Azuma M, Shearer TR. The role of calcium-activated protease calpain in experimental retinal pathology. Surv Ophthalmol. 2008;53:150–163. doi: 10.1016/j.survophthal.2007.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Franz T, Winckler L, Boehm T, Dear TN. Capn5 is expressed in a subset of T cells and is dispensable for development. Mol Cell Biol. 2004;24:1649–1654. doi: 10.1128/MCB.24.4.1649-1654.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]