

Abstract
We performed an integrated genomic, transcriptomic and proteomic characterization of 373 endometrial carcinomas using array- and sequencing-based technologies. Uterine serous tumours and ∼25% of high-grade endometrioid tumours had extensive copy number alterations, few DNA methylation changes, low oestrogen receptor/progesterone receptor levels, and frequent TP53 mutations. Most endometrioid tumours had few copy number alterations or TP53 mutations, but frequent mutations in PTEN, CTNNB1, PIK3CA, ARID1A and KRAS and novel mutations in the SWI/SNF chromatin remodelling complex gene ARID5B. A subset of endometrioid tumours that we identified had a markedly increased transversion mutation frequency and newly identified hotspot mutations in POLE. Our results classified endometrial cancers into four categories: POLE ultramutated, microsatellite instability hypermutated, copy-number low, and copy-number high. Uterine serous carcinomas share genomic features with ovarian serous and basal-like breast carcinomas. We demonstrated that the genomic features of endometrial carcinomas permit a reclassification that may affect post-surgical adjuvant treatment for women with aggressive tumours.
Supplementary information
The online version of this article (doi:10.1038/nature12113) contains supplementary material, which is available to authorized users.
Subject terms: Cancer genomics, Endometrial cancer
An integrative genomic analysis of several hundred endometrial carcinomas shows that a minority of tumour samples carry copy number alterations or TP53 mutations and many contain key cancer-related gene mutations, such as those involved in canonical pathways and chromatin remodelling; a reclassification of endometrial tumours into four distinct types is proposed, which may have an effect on patient treatment regimes.
Supplementary information
The online version of this article (doi:10.1038/nature12113) contains supplementary material, which is available to authorized users.
Reclassification of uterine carcinomas
This paper from The Cancer Genome Atlas Research Network presents an in-depth genome-wide analysis of endometrial (uterine) carcinomas from more than 350 patients. Based on a series of genomic features including newly identified hotspot mutations in the DNA polymerase gene POLE, and novel mutations in the ARID5B DNA-binding protein, the authors propose a reclassification of endometrial tumours into four distinct types. This might have clinical relevance for post-surgical adjuvant treatment of women with aggressive tumours.
Supplementary information
The online version of this article (doi:10.1038/nature12113) contains supplementary material, which is available to authorized users.
Main
Endometrial cancer arises from the lining of the uterus. It is the fourth most common malignancy among women in the United States, with an estimated 49,500 new cases and 8,200 deaths in 2013 (ref. 1). Most patients present with low-grade, early-stage disease. The majority of patients with more aggressive, high-grade tumours who have disease spread beyond the uterus will progress within 1 year (refs 2, 3). Endometrial cancers have been broadly classified into two groups4. Type I endometrioid tumours are linked to oestrogen excess, obesity, hormone-receptor positivity, and favourable prognosis compared with type II, primarily serous, tumours that are more common in older, non-obese women and have a worse outcome. Early-stage endometrioid cancers are often treated with adjuvant radiotherapy, whereas serous tumours are treated with chemotherapy, similar to advanced-stage cancers of either histological subtype. Therefore, proper subtype classification is crucial for selecting appropriate adjuvant therapy.
Several previous reports suggest that PTEN mutations occur early in the neoplastic process of type I tumours and co-exist frequently with other mutations in the phosphatidylinositol-3-OH kinase (PI(3)K)/AKT pathway5,6. Other commonly mutated genes in type I tumours include FGFR2, ARID1A, CTNNB1, PIK3CA, PIK3R1 and KRAS7,8,9. Microsatellite instability (MSI) is found in approximately one-third of type I tumours, but is infrequent in type II tumours10. TP53, PIK3CA and PPP2R1A mutations are frequent in type II tumours11,12. Most of these studies have been limited to DNA sequencing only with samples of heterogeneous histological subtypes and tumour grades. We present a comprehensive, multiplatform analysis of 373 endometrial carcinomas including low-grade endometrioid, high-grade endometrioid, and serous carcinomas. This integrated analysis provides key molecular insights into tumour classification, which may have a direct effect on treatment recommendations for patients, and provides opportunities for genome-guided clinical trials and drug development.
Results
Tumour samples and corresponding germline DNA were collected from 373 patients, including 307 endometrioid and 66 serous (53) or mixed histology (13) cases. Local Institutional Review Boards approved all tissue acquisition. The clinical and pathological characteristics of the samples generally reflect a cross-section of individuals with recurrent endometrial cancer2,3 (Supplementary Table 1.1). The median follow-up of the cohort was 32 months (range, 1–195 months); 21% of the patients have recurred, and 11% have died. Comprehensive molecular analyses were performed at independent centres using six genomic or proteomic platforms (Supplementary Table 1.2). MSI testing performed on all samples using seven repeat loci (Supplementary Table 1.3) found MSI in 40% of endometrioid tumours and 2% of serous tumours.
Somatic copy number alterations
Somatic copy number alterations (SCNAs) were assessed in 363 endometrial carcinomas. Unsupervised hierarchical clustering grouped the tumours into four clusters (Fig. 1a). The first three copy-number clusters were composed almost exclusively (97%) of endometrioid tumours without significant differences in tumour grades. Cluster 1 tumours were nearly devoid of broad SCNAs, averaging less than 0.5% genome alteration, with no significant recurrent events. Cluster 1 tumours also had significantly increased non-synonymous mutation rates compared to all others (median 7.2 × 10−6 versus 1.7 × 10−6 mutations per megabase (Mb), P < 0.001). Copy-number clusters 2 and 3 consisted mainly of endometrioid tumours, distinguished by more frequent 1q amplification in cluster 3 than cluster 2 (100% of cluster 3 tumours versus 33% of cluster 2 tumours) and worse progression-free survival (P = 0.003, log-rank versus clusters 1 and 2; Fig. 1b).
Figure 1. SCNAs in endometrial carcinomas.
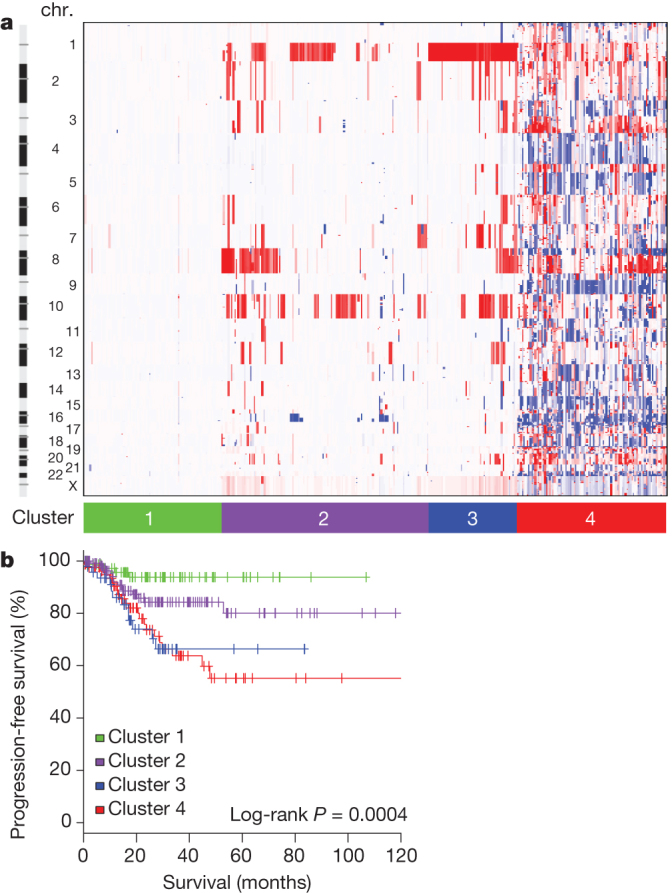
a, Tumours were hierarchically clustered into four groups based on SCNAs. The heat map shows SCNAs in each tumour (horizontal axis) plotted by chromosomal location (vertical axis). Chr., chromosome. b, Kaplan–Meier curves of progression-free survival for each copy-number cluster.
Most of the serous (50 out of 53; 94%) and mixed histology (8 out of 13; 62%) tumours clustered with 36 (12%) of the 289 endometrioid tumours, including 24% of grade 3 and 5% of grade 1 or 2, into copy-number cluster 4; a single group characterized by a very high degree of SCNAs (Supplementary Fig. 2.1; focal SCNAs with false discovery rate (FDR) < 0.15, and Supplementary Data 2.1). Cluster 4 tumours were characterized by significantly recurrent previously reported focal amplifications of the oncogenes MYC (8q24.12), ERBB2 (17q12) and CCNE1 (19q12)13, and by SCNAs previously unreported in endometrial cancers including those containing FGFR3 (4p16.3) and SOX17 (8q11.23). Cluster 4 tumours also had frequent TP53 mutations (90%), little MSI (6%), and fewer PTEN mutations (11%) than other endometrioid tumours (84%). Overall, these findings suggest that a subset of endometrial tumours contain distinct patterns of SCNAs and mutations that do not correlate with traditional tumour histology or grade.
As expected, tumours in the ‘serous-like’ cluster (cluster 4) had significantly worse progression-free survival than tumours in the endometrioid cluster groups (P = 0.003, log-rank, Fig. 1b). Potential therapeutically relevant SCNAs included the cluster 2 15q26.2 focal amplification, which contained IGF1R; and cluster 4 amplifications of ERBB2, FGFR1 and FGFR3, and LRP1B deletion, which was recently associated with resistance to liposomal doxorubicin in serous ovarian cancer14.
Exome sequence analysis
We sequenced the exomes of 248 tumour/normal pairs. On the basis of a combination of somatic nucleotide substitutions, MSI and SCNAs, the endometrial tumours were classified into four groups (Fig. 2a, b): (1) an ultramutated group with unusually high mutation rates (232 × 10−6 mutations per Mb) and a unique nucleotide change spectrum; (2) a hypermutated group (18 × 10−6 mutations per Mb) of MSI tumours, most with MLH1 promoter methylation; (3) a group with lower mutation frequency (2.9 × 10−6 mutations per Mb) and most of the microsatellite stable (MSS) endometrioid cancers; and (4) a group that consists primarily of serous-like cancers with extensive SCNA (copy-number cluster 4) and a low mutation rate (2.3 × 10−6 mutations per Mb). The ultramutated group consisted of 17 (7%) tumours exemplified by an increased C→A transversion frequency, all with mutations in the exonuclease domain of POLE, and an improved progression-free survival (Fig. 2a, c). POLE is a catalytic subunit of DNA polymerase epsilon involved in nuclear DNA replication and repair. We identified hotspot mutations in POLE at Pro286Arg and Val411Leu present in 13 (76%) of the 17 ultramutated samples. Significantly mutated genes (SMGs) identified at low FDRs (Q) in this subset included PTEN (94%, Q = 0), PIK3R1 (65%, Q = 8.3 × 10−7), PIK3CA (71%, Q = 9.1 × 10−5), FBXW7 (82%, Q = 1.4 × 10−4), KRAS (53%, Q = 9.2 × 10−4) and POLE (100%, Q = 4.2 × 10−3). Mutation rates in POLE mutant endometrial and previously reported ultramutated colorectal tumours exceeded those found in any other lineage including lung cancer and melanoma15,16,17. Germline susceptibility variants have been reported in POLE (Leu424Val) and POLD1 (Ser478Asn), but were not found in our endometrial normal exome-seq reads18.
Figure 2. Mutation spectra across endometrial carcinomas.

a, Mutation frequencies (vertical axis, top panel) plotted for each tumour (horizontal axis). Nucleotide substitutions are shown in the middle panel, with a high frequency of C-to-A transversions in the samples with POLE exonuclease mutations. CN, copy number. b, Tumours were stratified into the four groups by (1) nucleotide substitution frequencies and patterns, (2) MSI status, and (3) copy-number cluster. SNV, single nucleotide variant. c, POLE-mutant tumours have significantly better progression-free survival, whereas copy-number high tumours have the poorest outcome. d, Recurrently mutated genes are different between the four subgroups. Shown are the mutation frequencies of all genes that were significantly mutated in at least one of the four subgroups (MUSiC, asterisk denotes FDR < 0.05).
The MSI endometrioid tumours had a mutation frequency approximately tenfold greater than MSS endometrioid tumours, few SCNAs, frameshift deletions in RPL22, frequent non-synonymous KRAS mutations, and few mutations in FBXW7, CTNNB1, PPP2R1A and TP53. The MSS, copy-number low, endometrioid tumours had an unusually high frequency of CTNNB1 mutations (52%); the only gene with a higher mutation frequency than the MSI samples. The copy-number high group contained all of the remaining serous cases and one-quarter of the grade 3 endometrioid cases. Most of these tumours had TP53 mutations and a high frequency of FBXW7 (22%, Q = 0) and PPP2R1A (22%, Q = 1.7 × 10−16) mutations, previously reported as common in uterine serous but not endometrioid carcinomas. Thus, a subset of high-grade endometrioid tumours had similar SCNAs and mutation spectra as uterine serous carcinomas, suggesting that these patients might benefit from treatment approaches that parallel those for serous tumours.
There were 48 genes with differential mutation frequencies across the four groups (Fig. 2d and Supplementary Data 3.1). ARID5B, a member of the same AT-rich interaction domain (ARID) family as ARID1A, was more frequently mutated in MSI (23.1%) than in either MSS endometrioid (5.6%) or high SCNA serous tumours (0%), a novel finding for endometrial cancer. Frameshifting RPL22 indels near a homopolymer at Lys 15 were almost exclusively found in the MSI group (36.9%). The TP53 mutation frequency (>90%) in serous tumours differentiated them from the endometrioid subtypes (11.4%). However, many (10 out of 20; 50%) endometrioid tumours with a non-silent TP53 mutation also had non-silent mutations in PTEN, compared to only 1 out of 39 (2.6%) serous tumours with non-silent TP53 mutations. Although TP53 mutations are not restricted to serous tumours, the co-existing PTEN mutations in the endometrioid cases suggest a distinct tumorigenic mechanism.
Comparisons of 66 SMGs between traditional histological subtypes are provided (Supplementary Methods 3), and SMGs across other subcohorts can be found in Supplementary Data 3.2. The spectrum of PIK3CA and PTEN mutations in endometrial cancer also differed from other solid tumours (Supplementary Methods 3). Integrated analysis may be useful for identifying histologically misclassified cases. For example, a single serous case was identified without a TP53 mutation or extensive SCNAs and with a KRAS mutation and high mutation rate. After re-review of the histological section, the case was deemed consistent with a grade 3 endometrioid tumour, demonstrating how molecular analysis could reclassify tumour histology and potentially affect treatment decisions.
Multiplatform subtype classifications
All of the endometrial tumours were examined for messenger RNA expression (n = 333), protein expression (n = 293), microRNA expression (n = 367), and DNA methylation (n = 373) (Supplementary Methods 4–7). Unsupervised k-means clustering of mRNA expression from RNA sequencing identified three robust clusters termed ‘mitotic’, ‘hormonal’ and ‘immunoreactive’ (Supplementary Fig. 4.1) that were significantly correlated with the four integrated clusters; POLE, MSI, copy-number low and copy-number high (P < 0.0001). Supervised analysis identified signature genes of the POLE cluster (n = 17) mostly involved in cellular metabolism (Fig. 3a). Among the few signature genes in the MSI cluster was decreased MLH1 mRNA expression, probably due to its promoter methylation. Increased progesterone receptor (PGR) expression was noted in the copy-number low cluster, suggesting responsiveness to hormonal therapy. The copy-number high cluster, which included most of the serous and serous-like endometrioid tumours, exhibited the greatest transcriptional activity exemplified by increased cell cycle deregulation (for example, CCNE1, PIK3CA, MYC and CDKN2A) and TP53 mutation (Supplementary Figs 4.2 and 4.3). This is consistent with reports that increased CDKN2A can distinguish serous from endometrioid carcinomas19. Approximately 85% of cases in the copy-number high cluster shared membership with the ‘mitotic’ mRNA subtype.
Figure 3. Gene expression across integrated subtypes in endometrial carcinomas.
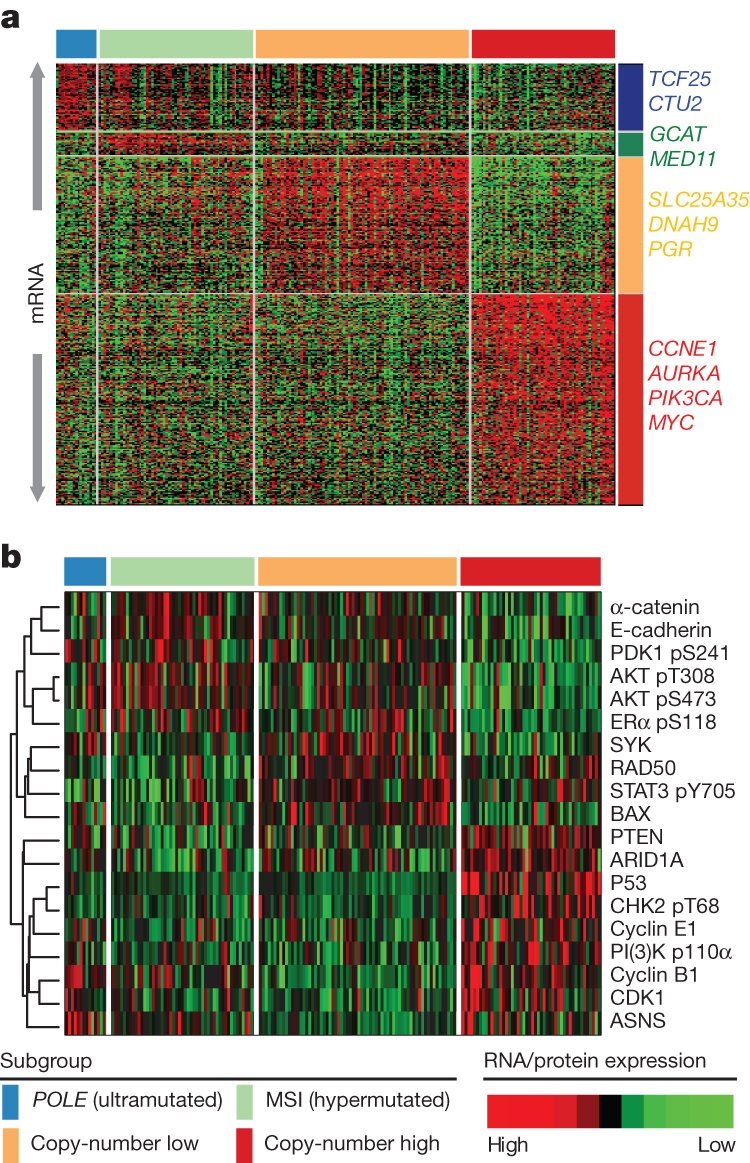
a, Supervised analysis of ∼1,500 genes significantly associated with integrated subtypes. b, Heat map of protein expression clusters, supervised by integrated subtypes. Samples are in columns; genes or proteins are in rows.
Supervised clustering of the reverse phase protein array (RPPA) expression data was consistent with loss of function for many of the mutated genes (Fig. 3b). TP53 was frequently mutated in the copy-number high group (P = 2.5 × 10−27) and its protein expression was also increased, suggesting that these mutations are associated with increased expression. By contrast, PTEN (P = 2.8 × 10−19) and ARID1A (P = 1.2 × 10−6) had high mutation rates in the remaining groups, but their expression was decreased, suggesting inactivating mutations in both genes. The copy-number high group also had decreased levels of phospho-AKT, consistent with downregulation of the AKT pathway. The copy-number low group had raised RAD50 expression, which is associated with DNA repair, explaining some of the differences between the copy-number high and low groups. The POLE group had high expression of ASNS and CCNB1, whereas the MSI tumours had both high phospho-AKT and low PTEN expression.
Unsupervised clustering of DNA methylation data generated from Illumina Infinium DNA methylation arrays revealed four unique subtypes (MC1–4) that support the four integrative clusters. A heavily methylated subtype (MC1) reminiscent of the CpG island methylator phenotype (CIMP) described in colon cancers and glioblastomas20,21,22 was associated with the MSI subtype and attributable to promoter hypermethylation of MLH1. A serous-like cluster (MC3) with minimal DNA methylation changes was composed primarily of serous tumours and some endometrioid tumours (Supplementary Fig. 7.1) and contained most of the copy-number high tumours.
Integrative clustering using the iCluster framework returned two major clusters split primarily on serous and endometrioid histology highlighting TP53 mutations, lack of PTEN mutation and encompassing almost exclusively copy-number high tumours23 (Supplementary Fig. 8.1). We developed a new clustering algorithm, called SuperCluster, to derive overall subtypes based on sample cluster memberships across all data types (Supplementary Fig. 9.1). SuperCluster identified four clusters that generally confirmed the contributions of individual platforms to the overall integrated clusters. No major batch effects were identified for any platform (Supplementary Methods 10).
Structural aberrations
To identify somatic chromosomal aberrations, we performed low-pass, paired-end, whole-genome sequencing on 106 tumours with matched normals. We found recurrent translocations involving genes in several pathways including WNT, EGFR–RAS–MAPK, PI(3)K, protein kinase A, retinoblastoma and apoptosis. The most frequent translocations (5 out of 106) involved a member of the BCL family (BCL2, BCL7A, BCL9 and BCL2L11). Four of these were confirmed by identification of the translocation junction point and two were also confirmed by high-throughput RNA sequencing (RNA-Seq). In all cases the translocations result in in-frame fusions and are predicted to result in activation or increased expression of the BCL family members (Supplementary Fig. 3.2). Translocations involving members of the BCL family leading to reduced apoptosis have been described in other tumour types24 and our results suggest that similar mechanisms may be operative here.
Pathway alterations
Multiple platform data were integrated to identify recurrently altered pathways in the four endometrial cancer integrated subgroups. Because of the high background mutation rate and small sample size, we excluded the POLE subgroup from this analysis. Considering all recurrently mutated, homozygously deleted, and amplified genes, we used MEMo25 to identify gene networks with mutually exclusive alteration patterns in each subgroup. The most significant module was found in the copy-number low group and contained CTNNB1, KRAS and SOX17 (Fig. 4a). The very strong mutual exclusivity between mutations in these three genes suggests that alternative mechanisms activate WNT signalling in endometrioid endometrial cancer. Activating KRAS mutations have been shown to increase the stability of β-catenin via glycogen synthase kinase 3β (GSK-3β), leading to an alternative mechanism of β-catenin activation other than adenomatous polyposis coli degradation26. SOX17, which mediates proteasomal degradation of β-catenin27,28, is mutated exclusively in the copy-number low group (8%) at recurrent positions (Ala96Gly and Ser403Ile) not previously described. Other genes with mutually exclusive alteration patterns in this module were FBXW7, FGFR2 and ERBB2 (ref. 29). ERBB2 was focally amplified with protein overexpression in 25% of the serous or serous-like tumours, suggesting a potential role for human epidermal growth factor receptor 2 (HER2)-targeted inhibitors. A small clinical trial of trastuzumab found no activity in endometrial carcinoma, but accrued few HER2 fluorescence in situ hybridization (FISH)-amplified serous carcinomas30.
Figure 4. Pathway alterations in endometrial carcinomas.

a, The RTK/RAS/β-catenin pathway is altered through several mechanisms that exhibit mutually exclusive patterns. Alteration frequencies are expressed as a percentage of all cases. The right panel shows patterns of occurrence. b, The PI(3)K pathway has mutually exclusive PIK3CA and PIK3R1 alterations that frequently co-occur with PTEN alterations in the MSI and copy-number low subgroups. c, Heat map display of top 1,000 varying pathway features within PARADIGM consensus clusters. Samples were arranged in order of their consensus cluster membership. The genomic subtype for each sample is displayed below the consensus clusters.
PIK3CA and PIK3R1 mutations were frequent and showed a strong tendency for mutual exclusivity in all subgroups, but unlike other tumour types, they co-occurred with PTEN mutations in the MSI and copy-number low subgroups as previously reported5,9 (Fig. 4b). The copy-number high subgroup showed mutual exclusivity between alterations of all three genes. Overall, 93% of endometrioid tumours had mutations that suggested potential for targeted therapy with PI(3)K/AKT pathway inhibitors.
Consensus clustering of copy number, mRNA expression and pathway interaction data for 324 samples yielded five PARADIGM clusters with distinct pathway activation patterns31 (Fig. 4c and Supplementary Methods 11). PARADIGM cluster 1 had the lowest level of MYC pathway activation and highest level of WNT pathway activation, consistent with its composition of copy-number low cases having frequent CTNNB1 mutations. PARADIGM cluster 3 was composed predominantly of the copy-number high cases, with relatively high MYC/MAX signalling but low oestrogen receptor/FOXA1 signalling and p53 activity. Only TP53 truncation and not missense mutations were implicated as loss-of-function mutations, suggesting different classes of p53 mutations may have distinct signalling consequences. PARADIGM cluster 5 was enriched for hormone receptor expression.
Comparison to ovarian and breast cancers
The clinical and pathologic features of uterine serous carcinoma and high-grade serous ovarian carcinoma (HGSOC) are quite similar. HGSOC shares many similar molecular features with basal-like breast carcinoma32. Focal SCNA patterns were similar between these three tumour subtypes and unsupervised clustering identified relatedness (Fig. 5a and Supplementary Fig. 12.1). Supervised analysis of transcriptome data sets showed high correlation between tumour subtypes (Supplementary Fig. 12.2). The MC3 DNA methylation subtype with minimal DNA methylation changes was also similar to basal-like breast and HGSOCs (Supplementary Fig. 12.3). A high frequency of TP53 mutations is shared across these tumour subtypes (uterine serous, 91%; HGSOC, 96%; basal-like breast, 84%)33,34, as is the very low frequency of PTEN mutations (uterine serous, 2%; HGSOC, 1%; basal-like breast, 1%). Differences included a higher frequency of FBXW7, PPP2R1A and PIK3CA mutations in uterine serous compared to basal-like breast and HGSOCs (Fig. 5b). We showed that uterine serous carcinomas share many molecular features with both HGSOCs and basal-like breast carcinomas, despite more frequent mutations, suggesting new opportunities for overlapping treatment paradigms.
Figure 5. Genomic relationships between endometrial serous-like, ovarian serous, and basal-like breast carcinomas.
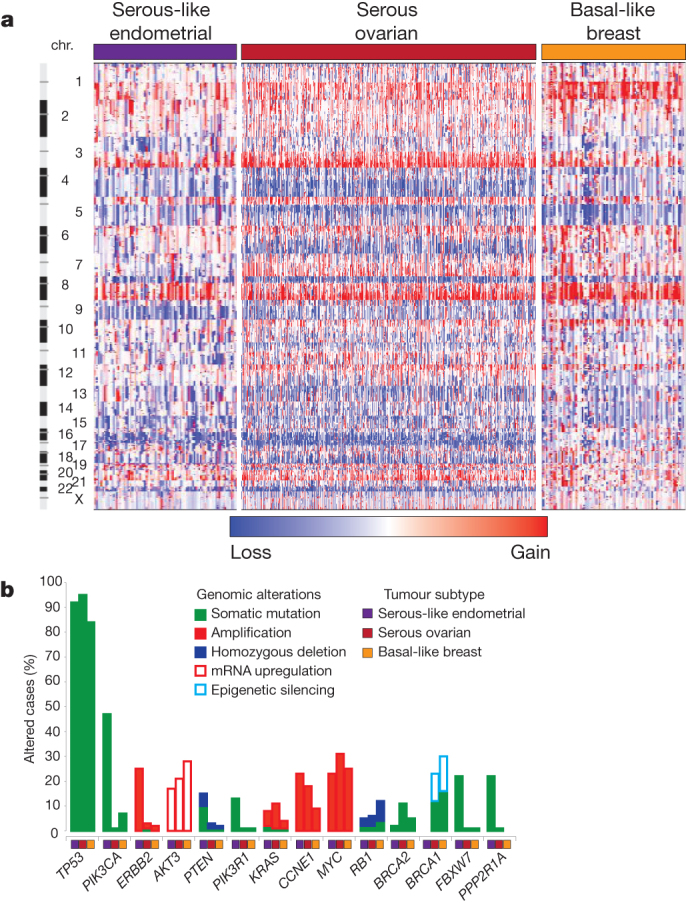
a, SCNAs for each tumour type. b, Frequency of genomic alterations present in at least 10% of one tumour type.
Discussion
This integrated genomic and proteomic analysis of 373 endometrial cancers provides insights into disease biology and diagnostic classification that could have immediate therapeutic application. Our analysis identified four new groups of tumours based on integrated genomic data, including a novel POLE subtype in ∼10% of endometrioid tumours. Ultrahigh somatic mutation frequency, MSS, and common, newly identified hotspot mutations in the exonuclease domain of POLE characterize this subtype. SCNAs add a layer of resolution, revealing that most endometrioid tumours have few SCNAs, most serous and serous-like tumours exhibit extensive SCNAs, and the extent of SCNA roughly correlates with progression-free survival.
Endometrial cancer has more frequent mutations in the PI(3)K/AKT pathway than any other tumour type studied by The Cancer Genome Atlas (TCGA) so far. Endometrioid endometrial carcinomas share many characteristics with colorectal carcinoma including a high frequency of MSI (40% and 11%, respectively), POLE mutations (7% and 3%, respectively) leading to ultrahigh mutation rates, and frequent activation of WNT/CTNNB1 signalling; yet endometrial carcinomas have novel exclusivity of KRAS and CTNNB1 mutations and a distinct mechanism of pathway activation. Uterine serous carcinomas share many similar characteristics with basal-like breast and HGSOCs; three tumour types with high-frequency non-silent TP53 mutations and extensive SCNA. However, the high frequency of PIK3CA, FBXW7, PPP2R1A and ARID1A mutations in uterine serous carcinomas are not found in basal-like breast and HGSOCs. The frequency of mutations in PIK3CA, FBXW7 and PPP2R1A was ∼30% higher than in a recently reported study of 76 uterine serous carcinomas11, but similar to another study12. Uterine serous carcinomas have ERBB2 amplification in 27% of tumours and PIK3CA mutations in 42%, which provide translational opportunities for targeted therapeutics.
Early stage type I endometrioid tumours are often treated with adjuvant radiotherapy, whereas similarly staged type II serous tumours are treated with chemotherapy. High-grade serous and endometrioid endometrial carcinomas are difficult to subtype correctly, and intra-observer concordance among speciality pathologists is low7,34,35,36. Our molecular characterization data demonstrate that ∼25% of tumours classified as high-grade endometrioid by pathologists have a molecular phenotype similar to uterine serous carcinomas, including frequent TP53 mutations and extensive SCNA. The compelling similarities between this subset of endometrioid tumours and uterine serous carcinomas suggest that genomic-based classification may lead to improved management of these patients. Clinicians should carefully consider treating copy-number-altered endometrioid patients with chemotherapy rather than adjuvant radiotherapy and formally test such hypotheses in prospective clinical trials. Furthermore, the marked molecular differences between endometrioid and serous-like tumours suggest that these tumours warrant separate clinical trials to develop the independent treatment paradigms that have improved outcomes in other tumour types, such as breast cancer.
Methods Summary
Biospecimens were obtained from 373 patients after Institutional Review Board-approved consents. DNA and RNA were co-isolated using a modified AllPrep kit (Qiagen). We used Affymetrix SNP 6.0 microarrays to detect SCNAs in 363 samples and GISTIC analysis to identify recurrent events37. The exomes of 248 tumours were sequenced to a read-depth of at least ×20. We performed low-pass whole-genome sequencing on 107 tumours to a mean depth of ×6. Consensus clustering was used to analyse mRNA, miRNA, RPPA and methylation data with methods previously described38,39,40. Integrated cross-platform analyses were performed using MEMo, iCluster and PARADIGM25,31.
Supplementary information
This file contains Supplementary Methods 1-12, which includes Supplementary Figures and Tables and additional references (see pages 1 and 2 for details). (PDF 14394 kb)
This zipped file contains Supplementary Data files 1.1, 2.1, 3.1, 3.2 and 5.1 (see Supplementary Information document for details). (ZIP 698 kb)
Acknowledgements
We wish to thank all patients and families who contributed to this study. We thank M. Sheth and L. Lund for administrative coordination of TCGA activities, G. Monemvasitis for editing the manuscript, and C. Gunter for critical reading of the manuscript. This work was supported by the following grants from the US National Institutes of Health: 5U24CA143799-04, 5U24CA143835-04, 5U24CA143840-04, 5U24CA143843-04, 5U24CA143845-04, 5U24CA143848-04, 5U24CA143858-04, 5U24CA143866-04, 5U24CA143867-04, 5U24CA143882-04, 5U24CA143883-04, 5U24CA144025-04, U54HG003067-11, U54HG003079-10 and U54HG003273-10.
PowerPoint slides
Author Contributions
The TCGA Research Network contributed collectively to this study. Biospecimens were provided by the tissue source sites and processed by the biospecimen core resource. Data generation and analyses were performed by the genome sequencing centres, cancer genome characterization centres and genome data analysis centres. All data were released through the data coordinating centre. The National Cancer Institute and National Human Genome Research Institute project teams coordinated project activities. We also acknowledge the following TCGA investigators who made substantial contributions to the project: N.S. (manuscript coordinator); J. Gao (data coordinator); C.K. and L. Ding (DNA sequence analysis); W.Z. and Y.L. (mRNA sequence analysis); H.S. and P.W.L. (DNA methylation analysis); A.D.C. and I.P. (copy number analysis); S.L. and A. Hadjipanayis (translocations); N.S., N.W. G.C., C.C.B. and C.Y. (pathway analysis); Andy C. and A.G.R. (miRNA sequence analysis); R. Broaddus, P.J.G., G.B.M. and R.A.S. (pathology and clinical expertise); G.B.M., H.L. and R.A. (reverse phase protein arrays); P.J.G. and R.B. (disease experts); G.B.M. and R.K. (manuscript editing); D.A.L. and E.R.M. (project chairs).
Competing interests
The author declares no competing financial interests.
Footnotes
(Participants are arranged by area of contribution and then by institution.)
The primary and processed data used to generate the analyses presented here are deposited at the Data Coordinating Center (https://tcga-data.nci.nih.gov/tcga/tcgaDownload.jsp); all of the primary sequence files are deposited in CGHub (https://cghub.ucsc.edu/). Sample lists, data matrices and supporting data can be found at: (https://tcga-data.nci.nih.gov/docs/publications/ucec_2013/). The data can be explored via the cBio Cancer Genomics Portal (http://cbioportal.org). Reprints and permissions information is available at www.nature.com/reprints. The authors declare no competing financial interests. Readers are welcome to comment on the online version of the paper. Correspondence and requests for materials should be addressed to D.A.L. (levine2@mskcc.org).
Change history
6/12/2013
Nature 497, 67–73 (2013); doi:10.1038/nature12113 In the ‘Results’ section of this Article, the range in the sentence “The median follow-up of the cohort was 32 months (range, 1–19 months); 21% of the patients have recurred, and 11% have died.” should have been 1–195 months. This error has been corrected in the HTML and PDF versions of the paper.
Contributor Information
Douglas A. Levine, Email: levine2@mskcc.org
The Cancer Genome Atlas Research Network:
Gad Getz, Stacey B. Gabriel, Kristian Cibulskis, Eric Lander, Andrey Sivachenko, Carrie Sougnez, Mike Lawrence, Cyriac Kandoth, David Dooling, Robert Fulton, Lucinda Fulton, Joelle Kalicki-Veizer, Michael D. McLellan, Michelle O’Laughlin, Heather Schmidt, Richard K. Wilson, Kai Ye, Li Ding, Elaine R. Mardis, Adrian Ally, Miruna Balasundaram, Inanc Birol, Yaron S. N. Butterfield, Rebecca Carlsen, Candace Carter, Andy Chu, Eric Chuah, Hye-Jung E. Chun, Noreen Dhalla, Ranabir Guin, Carrie Hirst, Robert A. Holt, Steven J. M. Jones, Darlene Lee, Haiyan I. Li, Marco A. Marra, Michael Mayo, Richard A. Moore, Andrew J. Mungall, Patrick Plettner, Jacqueline E. Schein, Payal Sipahimalani, Angela Tam, Richard J. Varhol, A. Gordon Robertson, Andrew D. Cherniack, Itai Pashtan, Gordon Saksena, Robert C. Onofrio, Steven E. Schumacher, Barbara Tabak, Scott L. Carter, Bryan Hernandez, Jeff Gentry, Helga B. Salvesen, Kristin Ardlie, Gad Getz, Wendy Winckler, Rameen Beroukhim, Stacey B. Gabriel, Matthew Meyerson, Angela Hadjipanayis, Semin Lee, Harshad S. Mahadeshwar, Peter Park, Alexei Protopopov, Xiaojia Ren, Sahil Seth, Xingzhi Song, Jiabin Tang, Ruibin Xi, Lixing Yang, Dong Zeng, Raju Kucherlapati, Lynda Chin, Jianhua Zhang, J. Todd Auman, Saianand Balu, Tom Bodenheimer, Elizabeth Buda, D. Neil Hayes, Alan P. Hoyle, Stuart R. Jefferys, Corbin D. Jones, Shaowu Meng, Piotr A. Mieczkowski, Lisle E. Mose, Joel S. Parker, Charles M. Perou, Jeff Roach, Yan Shi, Janae V. Simons, Mathew G. Soloway, Donghui Tan, Michael D. Topal, Scot Waring, Junyuan Wu, Katherine A. Hoadley, Stephen B. Baylin, Moiz S. Bootwalla, Phillip H. Lai, Timothy J. Triche Jr, David J. Van Den Berg, Daniel J. Weisenberger, Peter W. Laird, Hui Shen, Lynda Chin, Jianhua Zhang, Gad Getz, Juok Cho, Daniel DiCara, Scott Frazer, David Heiman, Rui Jing, Pei Lin, Will Mallard, Petar Stojanov, Doug Voet, Hailei Zhang, Lihua Zou, Michael Noble, Mike Lawrence, Sheila M. Reynolds, Ilya Shmulevich, B. Arman Aksoy, Yevgeniy Antipin, Giovanni Ciriello, Gideon Dresdner, Jianjiong Gao, Benjamin Gross, Anders Jacobsen, Marc Ladanyi, Boris Reva, Chris Sander, Rileen Sinha, S. Onur Sumer, Barry S. Taylor, Ethan Cerami, Nils Weinhold, Nikolaus Schultz, Ronglai Shen, Stephen Benz, Ted Goldstein, David Haussler, Sam Ng, Christopher Szeto, Joshua Stuart, Christopher C. Benz, Christina Yau, Wei Zhang, Matti Annala, Bradley M. Broom, Tod D. Casasent, Zhenlin Ju, Han Liang, Guoyan Liu, Yiling Lu, Anna K. Unruh, Chris Wakefield, John N. Weinstein, Nianxiang Zhang, Yuexin Liu, Russell Broaddus, Rehan Akbani, Gordon B. Mills, Christopher Adams, Thomas Barr, Aaron D. Black, Jay Bowen, John Deardurff, Jessica Frick, Julie M. Gastier-Foster, Thomas Grossman, Hollie A. Harper, Melissa Hart-Kothari, Carmen Helsel, Aaron Hobensack, Harkness Kuck, Kelley Kneile, Kristen M. Leraas, Tara M. Lichtenberg, Cynthia McAllister, Robert E. Pyatt, Nilsa C. Ramirez, Teresa R. Tabler, Nathan Vanhoose, Peter White, Lisa Wise, Erik Zmuda, Nandita Barnabas, Charlenia Berry-Green, Victoria Blanc, Lori Boice, Michael Button, Adam Farkas, Alex Green, Jean MacKenzie, Dana Nicholson, Steve E. Kalloger, C. Blake Gilks, Beth Y. Karlan, Jenny Lester, Sandra Orsulic, Mark Borowsky, Mark Cadungog, Christine Czerwinski, Lori Huelsenbeck-Dill, Mary Iacocca, Nicholas Petrelli, Brenda Rabeno, Gary Witkin, Elena Nemirovich-Danchenko, Olga Potapova, Daniil Rotin, Andrew Berchuck, Michael Birrer, Phillip DiSaia, Laura Monovich, Erin Curley, Johanna Gardner, David Mallery, Robert Penny, Sean C. Dowdy, Boris Winterhoff, Linda Dao, Bobbie Gostout, Alexandra Meuter, Attila Teoman, Fanny Dao, Narciso Olvera, Faina Bogomolniy, Karuna Garg, Robert A. Soslow, Douglas A. Levine, Mikhail Abramov, John M. S. Bartlett, Sugy Kodeeswaran, Jeremy Parfitt, Fedor Moiseenko, Blaise A. Clarke, Marc T. Goodman, Michael E. Carney, Rayna K. Matsuno, Jennifer Fisher, Mei Huang, W. Kimryn Rathmell, Leigh Thorne, Linda Van Le, Rajiv Dhir, Robert Edwards, Esther Elishaev, Kristin Zorn, Russell Broaddus, Paul J. Goodfellow, David Mutch, Nikolaus Schultz, Yuexin Liu, Rehan Akbani, Andrew D. Cherniack, Ethan Cerami, Nils Weinhold, Hui Shen, Katherine A. Hoadley, Ari B. Kahn, Daphne W. Bell, Pamela M. Pollock, Chen Wang, David A.Wheeler, Eve Shinbrot, Beth Y. Karlan, Andrew Berchuck, Sean C. Dowdy, Boris Winterhoff, Marc T. Goodman, A. Gordon Robertson, Rameen Beroukhim, Itai Pashtan, Helga B. Salvesen, Peter W. Laird, Michael Noble, Joshua Stuart, Li Ding, Cyriac Kandoth, C. Blake Gilks, Robert A. Soslow, Paul J. Goodfellow, David Mutch, Russell Broaddus, Wei Zhang, Gordon B. Mills, Raju Kucherlapati, Elaine R. Mardis, Douglas A. Levine, Brenda Ayala, Anna L. Chu, Mark A. Jensen, Prachi Kothiyal, Todd D. Pihl, Joan Pontius, David A. Pot, Eric E. Snyder, Deepak Srinivasan, Ari B. Kahn, Kenna R. Mills Shaw, Margi Sheth, Tanja Davidsen, Greg Eley Martin L. Ferguson, John A. Demchok, Liming Yang, Mark S. Guyer, Bradley A. Ozenberger, Heidi J. Sofia, Cyriac Kandoth, Nikolaus Schultz, Andrew D. Cherniack, Rehan Akbani, Yuexin Liu, Hui Shen, A. Gordon Robertson, Itai Pashtan, Ronglai Shen, Christopher C. Benz, Christina Yau, Peter W. Laird, Li Ding, Wei Zhang, Gordon B. Mills, Raju Kucherlapati, Elaine R. Mardis, and Douglas A. Levine
References
- 1.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J. Clin. 2013;63:11–30. doi: 10.3322/caac.21166. [DOI] [PubMed] [Google Scholar]
- 2.Fleming GF, et al. Phase III trial of doxorubicin plus cisplatin with or without paclitaxel plus filgrastim in advanced endometrial carcinoma: a Gynecologic Oncology Group Study. J. Clin. Oncol. 2004;22:2159–2166. doi: 10.1200/JCO.2004.07.184. [DOI] [PubMed] [Google Scholar]
- 3.Sutton G, et al. Whole abdominal radiotherapy in the adjuvant treatment of patients with stage III and IV endometrial cancer: a gynecologic oncology group study. Gynecol. Oncol. 2005;97:755–763. doi: 10.1016/j.ygyno.2005.03.011. [DOI] [PubMed] [Google Scholar]
- 4.Lax SF, Kurman RJ. A dualistic model for endometrial carcinogenesis based on immunohistochemical and molecular genetic analyses. Verh. Dtsch. Ges. Pathol. 1997;81:228–232. [PubMed] [Google Scholar]
- 5.Cheung LW, et al. High frequency of PIK3R1 and PIK3R2 mutations in endometrial cancer elucidates a novel mechanism for regulation of PTEN protein stability. Cancer Discov. 2011;1:170–185. doi: 10.1158/2159-8290.CD-11-0039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Levine RL, et al. PTEN mutations and microsatellite instability in complex atypical hyperplasia, a precursor lesion to uterine endometrioid carcinoma. Cancer Res. 1998;58:3254–3258. [PubMed] [Google Scholar]
- 7.McConechy MK, et al. Use of mutation profiles to refine the classification of endometrial carcinomas. J. Pathol. 2012;228:20–30. doi: 10.1002/path.4056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Byron SA, et al. FGFR2 point mutations in 466 endometrioid endometrial tumors: relationship with MSI, KRAS, PIK3CA, CTNNB1 mutations and clinicopathological features. PLoS ONE. 2012;7:e30801. doi: 10.1371/journal.pone.0030801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Urick ME, et al. PIK3R1 (p85α) is somatically mutated at high frequency in primary endometrial cancer. Cancer Res. 2011;71:4061–4067. doi: 10.1158/0008-5472.CAN-11-0549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zighelboim I, et al. Microsatellite instability and epigenetic inactivation of MLH1 and outcome of patients with endometrial carcinomas of the endometrioid type. J. Clin. Oncol. 2007;25:2042–2048. doi: 10.1200/JCO.2006.08.2107. [DOI] [PubMed] [Google Scholar]
- 11.Kuhn E, et al. Identification of molecular pathway aberrations in uterine serous carcinoma by genome-wide analyses. J. Natl. Cancer Inst. 2012;104:1503–1513. doi: 10.1093/jnci/djs345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Le Gallo M, et al. Exome sequencing of serous endometrial tumors identifies recurrent somatic mutations in chromatin-remodeling and ubiquitin ligase complex genes. Nature Genet. 2012;44:1310–1315. doi: 10.1038/ng.2455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Salvesen HB, et al. Integrated genomic profiling of endometrial carcinoma associates aggressive tumors with indicators of PI3 kinase activation. Proc. Natl Acad. Sci. USA. 2009;106:4834–4839. doi: 10.1073/pnas.0806514106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cowin PA, et al. LRP1B deletion in high-grade serous ovarian cancers is associated with acquired chemotherapy resistance to liposomal doxorubicin. Cancer Res. 2012;72:4060–4073. doi: 10.1158/0008-5472.CAN-12-0203. [DOI] [PubMed] [Google Scholar]
- 15.The Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature487, 330–337 (2012) [DOI] [PMC free article] [PubMed]
- 16.Govindan R, et al. Genomic landscape of non-small cell lung cancer in smokers and never-smokers. Cell. 2012;150:1121–1134. doi: 10.1016/j.cell.2012.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pleasance ED, et al. A comprehensive catalogue of somatic mutations from a human cancer genome. Nature. 2010;463:191–196. doi: 10.1038/nature08658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Palles C, et al. Germline mutations affecting the proofreading domains of POLE and POLD1 predispose to colorectal adenomas and carcinomas. Nature Genet. 2013;45:136–144. doi: 10.1038/ng.2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bartosch C, et al. Endometrial carcinomas: a review emphasizing overlapping and distinctive morphological and immunohistochemical features. Adv. Anat. Pathol. 2011;18:415–437. doi: 10.1097/PAP.0b013e318234ab18. [DOI] [PubMed] [Google Scholar]
- 20.Toyota M, et al. CpG island methylator phenotype in colorectal cancer. Proc. Natl Acad. Sci. USA. 1999;96:8681–8686. doi: 10.1073/pnas.96.15.8681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hinoue T, et al. Genome-scale analysis of aberrant DNA methylation in colorectal cancer. Genome Res. 2012;22:271–282. doi: 10.1101/gr.117523.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Noushmehr H, et al. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell. 2010;17:510–522. doi: 10.1016/j.ccr.2010.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shen R, Olshen AB, Ladanyi M. Integrative clustering of multiple genomic data types using a joint latent variable model with application to breast and lung cancer subtype analysis. Bioinformatics. 2009;25:2906–2912. doi: 10.1093/bioinformatics/btp543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hockenbery D, Nunez G, Milliman C, Schreiber RD, Korsmeyer SJ. Bcl-2 is an inner mitochondrial membrane protein that blocks programmed cell death. Nature. 1990;348:334–336. doi: 10.1038/348334a0. [DOI] [PubMed] [Google Scholar]
- 25.Ciriello G, Cerami E, Sander C, Schultz N. Mutual exclusivity analysis identifies oncogenic network modules. Genome Res. 2012;22:398–406. doi: 10.1101/gr.125567.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li J, Mizukami Y, Zhang X, Jo WS, Chung DC. Oncogenic K-ras stimulates Wnt signaling in colon cancer through inhibition of GSK-3β. Gastroenterology. 2005;128:1907–1918. doi: 10.1053/j.gastro.2005.02.067. [DOI] [PubMed] [Google Scholar]
- 27.Zorn AM, et al. Regulation of Wnt signaling by Sox proteins: XSox17 α/β and XSox3 physically interact with β-catenin. Mol. Cell. 1999;4:487–498. doi: 10.1016/S1097-2765(00)80200-2. [DOI] [PubMed] [Google Scholar]
- 28.Sinner D, et al. Sox17 and Sox4 differentially regulate β-catenin/T-cell factor activity and proliferation of colon carcinoma cells. Mol. Cell. Biol. 2007;27:7802–7815. doi: 10.1128/MCB.02179-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pollock PM, et al. Frequent activating FGFR2 mutations in endometrial carcinomas parallel germline mutations associated with craniosynostosis and skeletal dysplasia syndromes. Oncogene. 2007;26:7158–7162. doi: 10.1038/sj.onc.1210529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fleming GF, et al. Phase II trial of trastuzumab in women with advanced or recurrent, HER2-positive endometrial carcinoma: a Gynecologic Oncology Group study. Gynecol. Oncol. 2010;116:15–20. doi: 10.1016/j.ygyno.2009.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vaske CJ, et al. Inference of patient-specific pathway activities from multi-dimensional cancer genomics data using PARADIGM. Bioinformatics. 2010;26:i237–i245. doi: 10.1093/bioinformatics/btq182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.The Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature490, 61–70 (2012) [DOI] [PMC free article] [PubMed]
- 33.The Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature474, 609–615 (2011) [DOI] [PMC free article] [PubMed]
- 34.Clarke BA, Gilks CB. Endometrial carcinoma: controversies in histopathological assessment of grade and tumour cell type. J. Clin. Pathol. 2010;63:410–415. doi: 10.1136/jcp.2009.071225. [DOI] [PubMed] [Google Scholar]
- 35.Yemelyanova A, et al. Utility of p16 expression for distinction of uterine serous carcinomas from endometrial endometrioid and endocervical adenocarcinomas: immunohistochemical analysis of 201 cases. Am. J. Surg. Pathol. 2009;33:1504–1514. doi: 10.1097/PAS.0b013e3181ac35f5. [DOI] [PubMed] [Google Scholar]
- 36.Gilks CB, Oliva E, Soslow RA. Poor inter-observer reproducibility in the diagnosis of high-grade endometrial carcinoma. Am. J. Surg. Pathol. 2012;91:248A. doi: 10.1097/PAS.0b013e31827f576a. [DOI] [PubMed] [Google Scholar]
- 37.Mermel CH, et al. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 2011;12:R41. doi: 10.1186/gb-2011-12-4-r41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gaujoux R, Seoighe C. A flexible R package for nonnegative matrix factorization. BMC Bioinformatics. 2010;11:367. doi: 10.1186/1471-2105-11-367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Houseman EA, et al. Model-based clustering of DNA methylation array data: a recursive-partitioning algorithm for high-dimensional data arising as a mixture of beta distributions. BMC Bioinformatics. 2008;9:365. doi: 10.1186/1471-2105-9-365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brunet JP, Tamayo P, Golub TR, Mesirov JP. Metagenes and molecular pattern discovery using matrix factorization. Proc. Natl Acad. Sci. USA. 2004;101:4164–4169. doi: 10.1073/pnas.0308531101. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
This file contains Supplementary Methods 1-12, which includes Supplementary Figures and Tables and additional references (see pages 1 and 2 for details). (PDF 14394 kb)
This zipped file contains Supplementary Data files 1.1, 2.1, 3.1, 3.2 and 5.1 (see Supplementary Information document for details). (ZIP 698 kb)