

Abstract
Existing computational methods for drug repositioning either rely only on the gene expression response of cell lines after treatment, or on drug-to-disease relationships, merging several information levels. However, the noisy nature of the gene expression and the scarcity of genomic data for many diseases are important limitations to such approaches. Here we focused on a drug-centered approach by predicting the therapeutic class of FDA-approved compounds, not considering data concerning the diseases. We propose a novel computational approach to predict drug repositioning based on state-of-the-art machine-learning algorithms. We have integrated multiple layers of information: i) on the distances of the drugs based on how similar are their chemical structures, ii) on how close are their targets within the protein-protein interaction network, and iii) on how correlated are the gene expression patterns after treatment. Our classifier reaches high accuracy levels (78%), allowing us to re-interpret the top misclassifications as re-classifications, after rigorous statistical evaluation. Efficient drug repurposing has the potential to significantly impact the whole field of drug development. The results presented here can significantly accelerate the translation into the clinics of known compounds for novel therapeutic uses.
Keywords: Drug repositioning, Connectivity map, CMap, ATC code, Mode of action, Machine learning, SVM, Integrative genomics, SMILES, Anthelmintics, Antineoplastic, Oxamniquine, Niclosamide
Background
Despite the enormous increase of financial investments in pharmaceutical R&D, the number of newly approved drugs has greatly diminished during the past decade [1]. Finding new uses for approved drugs has consequently become a major alternative strategy for the pharma industry. This practice, usually referred to as drug repositioning, is highly attractive because of its potential to speed up the process of drug development, hence reducing costs in addition to providing new treatments for unmet medical needs [2]. In this regard, compounds that have passed through phases II or III in the drug discovery pipeline but never made it to the market due to efficacy issues bear great potential for drug repositioning approaches. Successful drug repositioning requires that a known drug has a positive impact on a different disease, but its highest value resides in that its use for the novel indication surpasses the currently available therapeutic options for that condition. Experimental approaches to drug repositioning generally involve high-throughput assays where libraries of approved compounds are tested against biological targets of interest. The effects of a large number of Food and Drug Administration (FDA)-approved compounds on gene expression have been measured on several cultured human cell lines (the Connectivity Map, CMap) [3], and these information has been used to investigate similarities between drugs mechanisms of action [4]. Further, the CMap data has been systematically re-analyzed in search of differential expression patterns of the genes encoding the drug targets [5]. Computational approaches more specifically aimed at drug repositioning have been designed to find correlations between disease-associated and drug-associated expression signatures under the assumption that an effective drug should be able to counterbalance the perturbations caused by a disease. Remarkably, this kind of approach has already led to the identification and experimental validation of novel therapeutic indications for the antiepileptic topiramate in inflammatory bowel disease (IBD) [6]. Genetic risk effects associated with druggable genes in complex diseases have also been considered in order to attempt drug repositioning [7]. Finally, more comprehensive methods that take into account chemical, molecular and biological aspects of the drug-disease interactions have also been recently proposed [8]. Although the above studies have demonstrated that computational approaches to drug repositioning are feasible, there are still large margins for improvement. For instance, deriving drug repositioning from drug-disease interactions alone can be difficult due to the complexity, variability and sparsity of data currently available for the diseases, and to the intrinsic nature of publicly available gene expression data, which derive from patients already treated with other drugs in most of the cases. In order to overcome such limitations, in this study we have decided to establish a methodological approach focusing primarily on drug characteristics. Aiming at enhancing the predictive power of the available computational methods, we have developed a novel approach based on machine-learning classification algorithms, where mismatches between known and predicted drug classifications are purposely interpreted as potential alternative therapeutic indications. We have studied 410 drugs by integrating different layers of information based on their similarities, including their chemical structures, molecular targets and induced gene expression signatures.
Results and discussion
Computational pipeline
We have integrated different techniques and data sources in order to build a classifier whose outcome is a therapeutic class for a given drug. The steps of our computational strategy are summarized in Figure 1. We have first re-analyzed the CMap gene expression data using state-of-the-art methods for probe annotation and normalization. The drug-drug similarities for the gene expression layer have been based on the ranks of the genes in each drug-induced expression profile and their associated p-values. Next, pairwise similarities for the molecular structures have been assessed by computing the distances between the corresponding binary fingerprints. Finally, target-based similarities have been obtained by taking into account known common targets and their distances across the global human protein-protein interaction network. Subsequently, we have combined the drug similarities into a single information layer used to train a multi-class SVM (Support Vector Machine) classifier [9]. Receiver Operating Characteristic (ROC) curves show that integrating information coming from different sources into a single kernel improves the performance of the corresponding classifier (Figure 2). This has also been confirmed by testing the performance of the classifiers built on a single information layer. Similarity (kernel) matrices have the advantage of being directly comparable even when computed from highly heterogeneous data. However, our kernels, designed to weigh information according to what is considered a priori to be relevant (molecular structure features in the fingerprints, differentially expressed genes in profiles, known protein-protein interactions), have shown poor results when used to train kernel classifiers. Conversely, proper projections of the data by using Classical Multidimensional Scaling (cMDS, or Principal Coordinate Analysis [10], see Section “Materials and Methods”) and subsequent computation of a classical kernel (Gaussian) has allowed higher classification rates, thus providing an effective way to transform a kernel directly built from biological data to another that is technically efficient. The therapeutic class of each drug has been extracted from the Anatomical Therapeutic Chemical classification (ATC, see Section “Materials and Methods”), as defined by the World Health Organization (WHO). By using the ATC second level (therapeutic subgroup) and the classes with at least 8 drugs, with the best kernel, we have applied random sub-sampling and re-training iterations both to improve classification performance and to reduce over-fitting, thus contributing to the final 78% accuracy, and producing reliable hints for repositioning. The final classification for each drug has been thus obtained by choosing the most frequently predicted ATC code. On the other hand, hints for repositioning have been obtained as the most frequent misclassifications. Thus, a classifier is built in order to obtain knowledge about known samples, as opposed to predict classes for new ones. Moreover, the inverted use of the classifier results, where correct classifications are used to assess the reliability of the misclassifications, puts automatic classifiers in a new perspective that could provide more interesting applications in the future.
Figure 1.
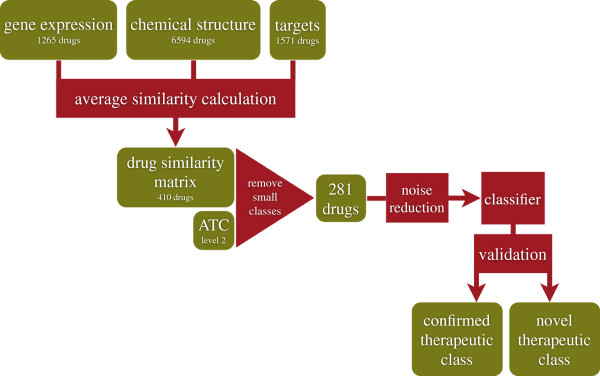
Flowchart of the analysis. Green boxes indicate data, red boxes indicate processes.
Figure 2.

Classification performance and data integration. Receiving Operator Curve for the three separated kernels (GEX: gene expression; CHEM: chemical structure; TAR: molecular targets) and the final joint kernel in respect with ATC classes.
Our computational strategy of drug repositioning crosses previous work at different levels. A recent study [11] has tackled our same task of predicting ATC codes by collecting chemical data for 3,883 drugs. Although our study is focused on a more specific level of ATC codes (level 2 as opposed to level 1) and includes a smaller database of drugs (a sizeable portion of drugs are not in CMap), our method provides higher classification performance (78% as compared to 73%). The predictive power of gene expression alone with respect to ATC codes has been also investigated in two related studies [4,5]. While focusing on drugs mechanisms of action, the first study has detected scarce correlation between similarities obtained through the gene expression profiles and those based on ATC codes. The second study has shown how this correlation could be improved by alternative data processing strategies. However, in both cases no attempt has been made at directly predicting ATC codes. More recently, an approach based on comparing multi-layered drug-drug and drug-disease similarities has been proposed to produce possible treatment predictions [8]. The results have been validated through Area Under the ROC Curve (AUC). From a methodological point of view, the classification accuracy of our approach cannot be directly compared with such score, which is meant to test edge predictions, as opposed to class predictions. From a methodological point of view, the main novelty of our work resides in the development of a data integration framework for efficiently predicting drugs ATC codes and in its use as a tool for drug repositioning.
Drug repositioning
Figure 3 shows the main trends of repositioning highlighting that, among our selected drug portofolio, the repositioning of antihelmintics to antineoplasic agents and of antineoplasic agents to antibacterials of systemic use were the most frequent drug reclassifications. Table 1 highlights the 12 top scoring drug repositionings identified by our model (presented in full in Additional file 1). We have found that our model correctly assigned the corresponding activity of the ophthalmologicals levobunolol and sulfacetamide to beta-blocking agents and antibacterials, respectively. In addition, it has accurately underlined drugs known structural similarities, such as the beta-adrenergic agonist dobutamine reclassified from cardiac therapy to beta blocking agents, and the antihelmintic ivermectin reclassified as an antibacterial. It is known that, despite being structurally similar to macrolide antibiotics and antifungal macrocyclic polyenes, irvemectin is actually devoid of antibacterial or antifungal activities [12]. Inspection of our results also indicates that our method has accurately predicted very plausible alternative therapeutic classes for known drugs. Antihistamines, known to have antipsychotic effects (chlorphenamine, thiethylperazine) or currently in use for their antipsychotic properties (hydroxyzine), were reclassified as psychoanaleptics/psycholeptics. Finally, the antiepileptic carbamazepine, known to have cardiovascular side effects, has been reclassified for cardiac therapy, and the diuretic spirolonactone, which has known anti-androgenic effects, has been repositioned in the class of sex hormones and modulators of the genital system. The repositioning of antihelmintics to antineoplasic agents is consistent with the fact that some antihelmintic drugs included in this study (benzimidazole antihelmintics) interfere with microtubule synthesis in the parasites and could have the potential to cause mitotic arrest in tumor cells. Anticancer properties related to microtubule disruption have already been reported for mebendazole and albendazole, which has inclusively been studied on Phase I clinical trials for patients with advanced cancer [13]. Mebendazole has been reported to show survival benefit in two preclinical models of glioblastoma multiforme, and to induce apoptosis of several cancer cell lines including melanoma, human adrenocortical carcinoma, and non-small cell lung cancer [14-18]. Praziquantel, which is chemically different from the benzimidazole antihelmintics, has been however best repositioned by our model as an antiepileptic, most likely due to its effects on calcium homeostasis. Of note, the antihelmintics niclosamide and oxamniquine have been also repositioned as anticancer agents. In agreement to our computational prediction, niclosamide has been recently shown multiple anticancer effects in tumors of the ovary and colon, and also in leukemia and myeloma [19-24]. Some of its molecular targets have been disclosed and include, among others, the Wnt/Frizzled 1 [25], the mammalian target of rapamycin complex 1 (mTOR) [26], and the signal transducer and activator of transcription 3 (STAT 3) [27] signaling pathways. Interestingly, oxamniquine exerts its antihelmintic effects by causing paralysis and contraction of the worms after interference with their DNA [28], a mechanism that could also account for its potential activity in cancer. Thus, not only our model has predicted the repositioning of several antihelmintics to anticancer agents in line with most recent literature, but has also suggested that a systematic investigation of this therapeutic class may disclose important information that could be of therapeutic use for anticancer treatment and/or drug discovery. The significance of the direct repositioning of antineoplasic drugs as systemic antibacterials is however more difficult to extrapolate because most of these drugs do not offer advantages to the antimicrobials in current use due to toxicity issues. Nonetheless, gefitinib, a more selective chemotherapeutic agent, presents the highest score for reposition as antibacterial. To the best of our knowledge, there is yet no prior evidence or related supportive information concerning this finding, which could pave the way for the development of a novel class of antibacterials.
Figure 3.

Repositioning overview. Direction of the arrows represent direction of repositioning from one ATC class to another. Thickness and opacity of the edges represent a score-weighted sum of the reclassification events.
Table 1.
Top drug repositioning predictions
| Drug name | Original ATC therapeutic class | Predicted ATC therapeutic class |
|---|---|---|
| Carbamazepine |
Antiepileptics (N03) |
Cardiac therapy (C01) |
| Chlorphenamine |
Antihistamines for systemic use (R06) |
Psychoanaleptics (N06) |
| Dobutamine |
Cardiac therapy (C01) |
Beta blocking agents (C07) |
| Gefitinib |
Antineoplastic agents (L01) |
Antibacterials for systemic use (J01) |
| Hydroxyzine |
Psycholeptics (N05) |
Antihistamines for systemic use (R06) |
| Ivermectin |
Anthelmintics (P02) |
Antibacterials for systemic use (J01) |
| Levobunolol |
Ophthalmologicals (S01) |
Beta blocking agents (C07) |
| Niclosamide |
Anthelmintics (P02) |
Antineoplastic agents (L01) |
| Oxamniquine |
Anthelmintics (P02) |
Antineoplastic agents (L01) |
| Spironolactone |
Diuretics (C03) |
Sex hormones and modulators of the genital system (G03) |
| Sulfacetamide |
Ophthalmologicals (S01) |
Antibacterials for systemic use (J01) |
| Thiethylperazine | Antihistamines for systemic use (R06) | Psycholeptics (N05) |
The top 12 repositioned drugs (classification score =1) are shown in rows. The drug name, the original and the predicted therapeutic class are reported. The level 2 ATC codes are also reported in brackets.
Conclusions
In summary, we report a novel computational approach to predict drug repositioning based on a machine-learning algorithm and data integration. The novelty of our approach relies on the purposeful interpretation of classification mismatches as genuine reclassifications opportunities. Our procedure also gains from integrating different layers of information and maximizing their efficacy through computational procedures based on dimensionality reduction. Our results showed high accuracy levels, which were consistent with several literature reports. We believe our work offers new directions towards repositioning of known drugs and also for the development of novel drug discovery programs.
Methods/Experimental
Microarray data processing
The processing pipeline for the Microarray data is illustrated in Figure 4. A total of 7056 Affymetrix GeneChip raw data files (.CEL files) belonging to two chipsets (HG-U133A and HTHG-U133A) were collected from the Connectivity Map (CMap) [3] website and imported into R v. 2.12.1 [29]. Raw data files were quality checked using the R package affy v.1.32.0 [30] and affyQCReport v.1.32.0 [31] to exclude the poor quality data points, resulting in a set of usable 6736 CEL files. The probes of each chipset were re-annotated according to NCBI Entrez Gene database [32]. For this, the CDF packages v.14.1.0 were downloaded from brainarray website [33]. The background estimation and the probe summarization were done on the raw data from each chipset separately according to the RMA algorithm [34]. The two data matrices were then combined for 12139 common probe-sets obtaining an expression matrix of dimensions 12139×6736. Consequently, this matrix was normalized with the quantile method. Next, the ComBat algorithm was used to estimate and remove the technical bias (array type, scanner and vehicle) from the normalized data matrix [35]. Linear models followed by moderated t-test statistic were used to compute the p-values and the fold-changes in each drug-control pairs, by the limma package v.3.10.0 [36].
Figure 4.

Flowchart of the CMap gene expression data analysis. Green boxes indicate data, red boxes indicate processes.
Drug similarities
The Jaccard Index JI, the Cosine Similarity CS and the Dice Coefficient DC similarity measurements were used to calculate the similarity between drugs based on their molecular structure in form of fingerprint of simplified molecular-input line-entry specification (SMILES [37]) retrieved for 6594 small molecules from the DrugBank database [38] and processed by the package rcdk v.3.1.3 [39]. Let be as set of drugs {D1,D2,…,Dn}. Let be such that is the binary fingerprint representation of the drug Di, i∈{1,…n}. Note that binary vectors can be seen as representations of sets, where included elements are indicated as 1-s in the vector. With abuse of notation, here we use as its corresponding set. We defined the dissimilairty measure between any two drugs as:
The same measure was used to compute drug similarities based on molecular targets, obtained for 1571 drugs from the DrugBank database. Given a drug , let be the set of targets associated with the drug Di in DrugBank. Let be defined like KCHEM, but with . We used as the dissimilarity value between two drugs Di and Dj. To cope with the scarce granularity of this measure, we also defined a finer dissimilarity measure based on on the PPI database [40,41] as follows. Let Pi,j be the set of shortest paths from each target in Ti to each target in Tj according to the PPI network. We used the range-normalized length of the shortest among the paths in Pi,j as the dissimilarity value between drug i and drug j. As final molecular target dissimilarity measure we used:
Finally, we used a weighted Spearman’s Footrule (WSF) as a drug dissimilarity measure based on gene expression profiles from the Cmap as ranked and weighted according to their p-values. Given the gene expression profiles associated with two drugs Di and Dj, we ranked the genes in ascending order of their signed p-values, where the sign is given by the opposite of the sign of their fold change. This way, top-ranking genes were those over-expressed with low p-values and bottom-ranking genes were those under-expressed with low p-values. Genes associated with high p-values tend to stay in the middle of the ranked lists. Let’s define RD(g) as such rank for the gene g in the expression profile of drug D. Analogously, let WD(g) be 1 minus the p-value of the gene g in the expression profile of drug D. The gene expression profile dissimilarity measure between drug i nd j was then defined as:
where, in order to simplify notation, we used to indicate range normalization.
Data integration
Data were collected in the form of dissimilarity matrices in order to easily integrate information over the three datasets (gene expression, chemical structure and molecular targets). The three databases had 410 drugs in common. Let be the set of such drugs. We simply defined the joint kernel matrix K as:
was a 410×410 symmetric matrix representing our training set for the prediction of level 2 ATC codes. Since the number of different ATC codes at level 2 is large compared with the number of drugs in our dataset, a number of classes appeared empty or highly under-represented at that level. For this reason we removed all the drugs falling into ATC classes with less than 8 exemplars, obtaining a final 281×281 kernel K. We made this choice on the basis of a required lower bound on the classification performance of 75%, which is verified a posteriori with a simpler version (without bootstrap) of the approach described in the next Section.
Noise reduction and classification
Let K be the 281×281 kernel as defined in the previous Section. The ATC classification codes for such drugs were retrieved from the World Health Organization (WHO). In this system, the active compounds are grouped according to relevant pharmacological and clinical properties. The classification is organized in five hierarchical levels: the first level describes the anatomical site where the compound is active; the second level refers to pharmacological/therapeutic subgroups; the third and fourth levels define chemical/pharmacological/therapeutic subgroups. In the fifth level, the individual substances are identified. We used ATC codes (at second level: therapeutic subgroup) as targets for our classifier.
In order to maximize the efficiency of this kernel, we exploited Classical Multidimensional Scaling (cMDS, or Principal Coordinate Analysis [10]) to search for an optimal Euclidean embedding of the 281 drugs in the following way. The number of eigenvalues larger than 0 was 241. Let Mi∀i∈{1,…,241} be the 241×i matrix representing the cMDS projection of K into the sub-space spanned by the first i Principal Components of K. Let e(Mi) be the classification error of a Support Vector Machine (native Multiclass SVM [42] using Gaussian kernel) on Mi by 6-fold cross validation. Cross-validation was used to reduce the risk of over-fitting the data during the assessment of the model. K-fold is chosen because of the low number of samples. Note that in our context the minimization of the validation error is only needed to obtain reliable classifications for the given data and not to assess the performance of the classifier on new data.
The validation error e(Mi)∀i∈{1,…,241}, when plotted versus the number of dimensions, suggests the existence of a hypothetical smooth latent function between the two variables (See Figure 5). We choose arg i∈{1,…,241}mine(Mi)=107 as a reasonable approximation of the theoric minimum of such hypothetical function. On the 281 drugs at ATC level 2 projected on the 107-dimensional MDS subspace, using 10,000 bootstrap iterations with 10% holdout (see also next Section), we reached a final classification performance of 78%.
Figure 5.

Kernel noise reduction through MDS projections. Values on the x axis correspond to SVM classifiers trained with kernel K (see text) as projected into the subspace spanned by its first x Principal Components. The y axis reports the corresponding 6-fold cross validation error. The green dot indicates minimum error.
From misclassifications to repositioning
For each drug we assigned a probability distribution to the outcome of the classier basing on the frequencies collected during bootstrap. Let be the subsets of obtained during bootstrap iteration i. Each contained 90% of the drugs of and was used to train an SVM. Different sets induce different predictions for each drug in general, with the level of variability depending on the sensitivity of each data point to perturbations in the overall distribution. Given a drug , and m predictions Pj={p1, …,pm}, pi∈{1,…,c}, where c is the number of classes, we defined the repositioning scoreSi,j of each prediction pi for the drug Dj as the frequency of pi in Pj. While the final prediction for the drug Dj was assumed to be the most frequent one (which gives overall 78% accuracy as stated in the previous Section), we used all the other predictions as repositioning suggestions and their Si,j as a corresponding index of reliability. By definition, Si,j∈[0,1], with higher values indicating higher reliability. Table 1 reports all the obtained misclassifications having Si,j=1.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
DG and MDA conceived the study; FN, YZ and DG designed and performed the computational pipeline; R.T. supervised the machine learning and pattern recognition aspects; F.N., Y.Z., V.M.M., R.T., J.K., M.D.A. and D.G. wrote the manuscript. All authors read and approved the final manuscript.
Supplementary Material
Complete list of the drug repositioning predictions
Contributor Information
Francesco Napolitano, Email: franapoli@gmail.com.
Yan Zhao, Email: yan.zhao@helsinki.fi.
Vânia M Moreira, Email: vania.moreira@helsinki.fi.
Roberto Tagliaferri, Email: robtag@unisa.it.
Juha Kere, Email: juha.kere@ki.se.
Mauro D’Amato, Email: mauro.damato@ki.se.
Dario Greco, Email: dario.greco@ki.se.
Acknowledgements
The authors are grateful to Dr. Antonello Di Lieto, Prof. Vincenzo Cerullo and Dr. Roberto Pagliarini for critical comments on the manuscript. F.N. was supported by HPC-Europa2 (EU FP7 grant n.228398); Y.Z. was supported by the Finnish Cultural Foundation; V.M.M. was supported by the Magnus Ehrnrooth Foundation; J.K. was supported by the Swedish Research Council, Novo Nordisk Foundation, Sigrid Juslius Foundation and the Academy of Finland, and D.G. was supported by the Ella and Georg Ehrnrooth Foundation and the Paulo Foundation.
References
- Walters WP, Green J, Weiss JR, Murcko MA. What do medicinal chemists actually make? A 50-year retrospective. J Med Chem. 2011;54(19):6405–6416. doi: 10.1021/jm200504p. [DOI] [PubMed] [Google Scholar]
- O’Connor KA, Roth BL. Finding new tricks for old drugs: an efficient route for public-sector drug discovery. Nat Rev Drug Discov. 2005;4(12):1005–1014. doi: 10.1038/nrd1900. [DOI] [PubMed] [Google Scholar]
- Lamb J, Crawford ED, Peck D, Modell JW, Blat IC, Wrobel MJ, Lerner J, Brunet JP, Subramanian A, Ross KN, Reich M, Hieronymus H, Wei G, Armstrong SA, Haggarty SJ, Clemons PA, Wei R, Carr SA, Lander ES, Golub TR. The connectivity map: using gene-expression signatures to connect small molecules, genes, and disease. Science. 2006;313(5795):1929–1935. doi: 10.1126/science.1132939. [ http://www.sciencemag.org/cgi/content/abstract/sci;313/5795/1929] [DOI] [PubMed] [Google Scholar]
- Iorio F, Bosotti R, Scacheri E, Belcastro V, Mithbaokar P, Ferriero R, Murino L, Tagliaferri R, Brunetti-Pierri N, Isacchi A. di Bernardo D. Discovery of drug mode of action and drug repositioning from transcriptional responses. Proc Nat Acad Sci. 2010;107(33):14621–6. doi: 10.1073/pnas.1000138107. http://dx.doi.org/10.1073/pnas.1000138107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iskar M, Campillos M, Kuhn M, Jensen LJ, van Noort V, Bork P. Drug-induced regulation of target expression. PLoS Comput Bio. 2010;6(9):e1000925. doi: 10.1371/journal.pcbi.1000925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudley JT, Sirota M, Shenoy M, Pai RK, Roedder S, Chiang AP, Morgan AA, Sarwal MM, Pasricha PJ, Butte AJ. Computational repositioning of the anticonvulsant topiramate for inflammatory bowel disease. Sci Trans Med. 2011;3(96):96ra76–96ra76. doi: 10.1126/scitranslmed.3002648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanseau P, Agarwal P, Barnes MR, Pastinen T, Richards JB, Cardon LR, Mooser V. Use of genome-wide association studies for drug repositioning. Nat Biotech. 2012;30(4):317–320. doi: 10.1038/nbt.2151. [DOI] [PubMed] [Google Scholar]
- Gottlieb A, Stein GY, Ruppin E, Sharan R. PREDICT: a method for inferring novel drug indications with application to personalized medicine. Mol Syst Biol. 2011;7:496. doi: 10.1038/msb.2011.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shawe-Taylor J, Cristianini N. Kernel Methods for Pattern Analysis. Cambridge University Press; 2004. [Google Scholar]
- Borg I, Groenen PJF. Modern Multidimensional Scaling: Theory and Applications, 2nd edition. 2005.
- Chen L, Zeng WM, Cai YD, Feng KY, Chou KC. Predicting Anatomical Therapeutic Chemical (ATC) classification of drugs by integrating chemical-chemical interactions and similarities. PLoS ONE. 2012;7(4):e35254. doi: 10.1371/journal.pone.0035254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omura S, Crump A. The life and times of ivermectin - a success story. Nature Rev Microbiol. 2004;2(12):984–989. doi: 10.1038/nrmicro1048. [DOI] [PubMed] [Google Scholar]
- Pourgholami MH, Szwajcer M, Chin M, Liauw W, Seef J, Galettis P, Morris DL, Links M. Phase I clinical trial to determine maximum tolerated dose of oral albendazole in patients with advanced cancer. Cancer Chemother Pharmacol. 2009;65(3):597–605. doi: 10.1007/s00280-009-1157-8. [DOI] [PubMed] [Google Scholar]
- Doudican N, Rodriguez A, Osman I, Orlow SJ. Mebendazole induces apoptosis via Bcl-2 inactivation in chemoresistant melanoma cells. Mol Cancer Res. 2008;6(8):1308–1315. doi: 10.1158/1541-7786.MCR-07-2159. [DOI] [PubMed] [Google Scholar]
- Martarelli D, Pompei P, Baldi C, Mazzoni G. Mebendazole inhibits growth of human adrenocortical carcinoma cell lines implanted in nude mice. Cancer Chemother Pharmacol. 2007;61(5):809–817. doi: 10.1007/s00280-007-0538-0. [DOI] [PubMed] [Google Scholar]
- Bai RY, Staedtke V, Aprhys C, MGallia GL, Riggins GJ. Antiparasitic mebendazole shows survival benefit in 2 preclinical models of glioblastoma multiforme. 2011. [ http://neuro-oncology.oxfordjournals.org] [DOI] [PMC free article] [PubMed]
- Mukhopadhyay T, Sasaki Ji, Ramesh R, Roth JA. Mebendazole elicits a potent antitumor effect on human cancer cell lines both in vitro and in vivo. 2002. [ http://clincancerres.aacrjournals.org] [PubMed]
- Dobrosotskaya IY, Hammer GD, Schteingart DE, Maturen KE, Worden FP. Mebendazole monotherapy and long-term disease control in metastatic adrenocortical carcinoma. Endocr Pract. 2011;17(3):e59—e62. doi: 10.4158/EP10390.CR. [DOI] [PubMed] [Google Scholar]
- Yo YT, Lin YW, Wang YC, Balch C, Huang RL, Chan MWY, Sytwu HK, Chen CK, Chang CC, Nephew KP, Huang T, Yu MH, Lai HC. Growth inhibition of ovarian tumor-initiating cells by Niclosamide. Mol Cancer Ther. 2012;11(8):1703–1712. doi: 10.1158/1535-7163.MCT-12-0002. [DOI] [PubMed] [Google Scholar]
- Lu W, Lin C, Roberts MJ, Waud WR, Piazza GA, Li Y. Niclosamide suppresses cancer cell growth by inducing Wnt co-receptor LRP6 degradation and inhibiting the Wnt/-Catenin pathway. PLoS ONE. 2011;6(12):e29290. doi: 10.1371/journal.pone.0029290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khanim FL, Merrick BAME, Giles HV, Jankute M, Jackson JB, Giles LJ, Birtwistle J, Bunce CM, Drayson MT. Redeployment-based drug screening identifies the anti-helminthic niclosamide as anti-myeloma therapy that also reduces free light chain production. Blood Cancer J. 2011;1(10):e39. doi: 10.1038/bcj.2011.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SJ, Kang H, Cho DH. Niclosamide induces mitochondria fragmentation and promotes both apoptotic and autophagic cell death. Biochem Mol Bio Rep. 2011;44(8):517–522. doi: 10.5483/bmbrep.2011.44.8.517. [DOI] [PubMed] [Google Scholar]
- Sack U, Walther W, Scudiero D, Selby M, Kobelt D, Lemm M, Fichtner I, Schlag PM, Shoemaker RH, Stein U. Novel effect of Antihelminthic Niclosamide on S100A4-mediated metastatic progression in colon cancer. 2011. [ http://Jnci.Oxfordjournals.Org] [DOI] [PubMed]
- Jin Y, Lu Z, Ding K, Li J, Du X, Chen C, Sun X, Wu Y, Zhou J, Pan J. Antineoplastic mechanisms of Niclosamide in acute myelogenous leukemia stem cells: inactivation of the NF-B pathway and generation of reactive oxygen species. 2010. [ http://cancerres.aacrjournals.org] [DOI] [PubMed]
- Chen M, Wang J, Lu J, Bond MC, Ren XR, Lyerly HK, Barak LS, Chen W. The anti-helminthic Niclosamide inhibits Wnt/Frizzled1 signaling. J Med Chem. 2009;48(43):10267–10274. doi: 10.1021/bi9009677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonseca BD, Diering GH, Bidinosti MA, Dalal K, Alain T, Balgi AD, Forestieri R, Nodwell M, Rajadurai CV, Gunaratnam C, Tee AR, Duong F, Andersen RJ, Orlowski J, Numata M, Sonenberg N, Roberge M. Structure-activity analysis of Niclosamide reveals potential role for cytoplasmic pH in control of mammalian target of Rapamycin complex 1 (mTORC1) signaling. J Biol Chem. 2012;287(21):17530–17545. doi: 10.1074/jbc.M112.359638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren X, Duan L, He Q, Zhang Z, Zhou Y, Wu D, Pan J, Pei D, Ding K. Identification of Niclosamide as a new small-molecule inhibitor of the STAT3 signaling pathway. J Med Chem. 2010;1(9):454–459. doi: 10.1021/ml100146z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pica-Mattoccia L, Cioli D, Archer S. Binding of oxamniquine to the DNA of schistosomes. Trans R Soc Trop Med Hyg. 1989;83(3):373–376. doi: 10.1016/0035-9203(89)90508-7. [DOI] [PubMed] [Google Scholar]
- R Development Core Team. R: A language and environment for statistical computing. Vienna: R Foundation for Statistical Computing. 2009. [ISBN 3-900051-07-0]
- Gautier L, Cope L, Bolstad BM, Irizarry RA. affy–analysis of Affymetrix GeneChip data at the probe level. Bioinformatics. 2004;20(3):307–315. doi: 10.1093/bioinformatics/btg405. [ http://bioinformatics.oxfordjournals.org/content/20/3/307] [DOI] [PubMed] [Google Scholar]
- Parman C, Halling C, Gentleman R. affyQCReport: A package to generate QC reports for Affymetrix array data. 2011. [ http://www.bioconductor.org/packages/2.12/bioc/html/affyQCReport.html]
- Wheeler DL, Barrett T, Benson DA, Bryant SH, Canese K, Chetvernin V, Church DM, DiCuccio M, Edgar R, Federhen S, Geer LY, Kapustin Y, Khovayko O, Landsman D, Lipman DJ, Madden TL, Maglott DR, Ostell J, Miller V, Pruitt KD, Schuler GD, Sequeira E, Sherry ST, Sirotkin K, Souvorov A, Starchenko G, Tatusov RL, Tatusova TA, Wagner L, Yaschenko E. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2007;35(suppl 1):D5—D12. doi: 10.1093/nar/gkl1031. [ http://nar.oxfordjournals.org/content/35/suppl_1/D5] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai M, Wang P, Boyd AD, Kostov G, Athey B, Jones EG, Bunney WE, Myers RM, Speed TP, Akil H, Watson SJ, Meng F. Evolving gene/transcript definitions significantly alter the interpretation of GeneChip data. Nucleic Acids Res. 2005;33(20):e175—e175. doi: 10.1093/nar/gni179. [ http://nar.oxfordjournals.org/content/33/20/e175] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irizarry RA, Hobbs B, Collin F, BeazerŘBarclay YD Antonellis KJ. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4(2):249–264. doi: 10.1093/biostatistics/4.2.249. [ http://biostatistics.oxfordjournals.org/content/4/2/249] [DOI] [PubMed] [Google Scholar]
- Johnson WE, Li C, Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics. 2007;8:118–127. doi: 10.1093/biostatistics/kxj037. [ http://biostatistics.oxfordjournals.org/content/8/1/118] [DOI] [PubMed] [Google Scholar]
- Smyth GK. In: Bioinformatics and Computational Biology Solutions Using R and Bioconductor, Statistics for Biology and Health. Gentleman R CareyVJHuber W, editor. Springer; 2005. limma: Linear Models for Microarray Data; pp. 397–420. [ http://link.springer.com/chapter/10.1007/0-387-29362-0_23] [Google Scholar]
- Anderson E. SMILES, a line notation and computerized interpreter for chemical structures. 1987.
- Wishart DS, Knox C, Guo AC, Cheng D, Shrivastava S, Tzur D, Gautam B, Hassanali M. DrugBank: a knowledgebase for drugs, drug actions and drug targets. Nucleic Acids Res. 2008;36(Database issue):D901—D906. doi: 10.1093/nar/gkm958. [ http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2238889/] [PMID: 18048412 PMCID: PMC2238889] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guha R. Chemical informatics functionality in R. J Stat Softw. 2007;18(5):1–16. [ http://www.jstatsoft.org/v18/i05] [Google Scholar]
- Cowley MJ, Pinese M, Kassahn KS, Waddell N, Pearson JV, Grimmond SM, Biankin AV, Hautaniemi S, Wu J. PINA v2.0: mining interactome modules. Nucleic Acids Res. 2012;40(Database issue):D862–865. doi: 10.1093/nar/gkr967. [PMID: 22067443] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Vallenius T, Ovaska K, Westermarck J, MÃďkelÃď TP, Hautaniemi S. Integrated network analysis platform for protein-protein interactions. Nature Methods. 2009;6:75–77. doi: 10.1038/nmeth.1282. [PMID:19079255] [DOI] [PubMed] [Google Scholar]
- Crammer K, Singer Y. On the algorithmic implementation of multiclass kernel-based vector machines. J Mach Learn Res. 2002;2:265–292. [ http://dl.acm.org/citation.cfm?id=944790.944813] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Complete list of the drug repositioning predictions