

Abstract
Objective
The adverse effects of metabolic disorders in obesity have been extensively studied; however, the pathologic effects of hyperphosphatemia or phosphate toxicity in obesity have not been studied in similar depth and detail, chiefly because such an association is thought to be uncommon. Studies have established that the incidence of obesity-associated nephropathy is increasing. Because hyperphosphatemia is a major consequence of renal impairment, this study determines the in vivo effects of hyperphosphatemia in obesity.
Methods and results
We genetically induced hyperphosphatemia in leptin-deficient obese (ob/ob) mice by generating ob/ob and klotho double knockout [ob/ob-klotho−/−] mice. As a control, we made ob/ob mice with hypophosphatemia by generating ob/ob and 1-alpha hydroxylase double knockout [ob/ob-1α(OH)ase−/−] mice. Compared to the wild-type mice, all three obese background mice, namely ob/ob, ob/ob-klotho−/−, and ob/ob-1α(OH)ase−/− mice developed hypercholesterolemia. In addition, the hyperphosphatemic, ob/ob-klotho−/− genetic background induced generalized tissue atrophy and widespread soft-tissue and vascular calcifications, which led to a shorter lifespan; no such changes were observed in the hypophosphatemic, ob/ob-1α(OH)ase−/− mice. Significantly, in contrast to the reduced survival of the ob/ob-klotho−/− mice, lowering serum phosphate levels in ob/ob-1α(OH)ase−/− mice showed no such compromised survival, despite both mice being hypercholesterolemic.
Conclusion
These genetic manipulation studies suggest phosphate toxicity is an important risk factor in obesity that can adversely affect survival.
Keywords: Obese mice, Vitamin D, Phosphate, Calcium, Longevity
1. Introduction
Despite the well-known, adverse effects of obesity, it is still one of the major nutritional disorders influencing both quality of life and survival of the affected individuals. In contrast to the well-characterized effects of obesity in metabolic and cardiovascular diseases, the mechanism of obesity-induced renal structural damage, and the consequences of such damage, are not clearly defined [1,2]. In a previous study, experimental animals developed glomerulomegaly following a high-fat diet, whereas reducing body weight by providing a low-calorie diet to the obese Zucker rat not only improved glomerulopathy but also reduced proteinuria [3].
Various human studies have documented an association between obesity and renal disease progression [1,4]. As early as 1974, Weisinger et al. reported an association between severe obesity and nephrotic-range proteinuria [4]. Subsequent studies have documented obesity-associated glomerulopathy as a direct complication of obesity [5,6]. For instance, focal-segmental glomerulosclerosis with proteinuria was detected in obese patients without presence of hypertension or other systemic diseases [1,2,7,8]. Obesity was also determined to be an important risk factor in the progression of IgA nephropathy [9,10], and is associated with an increased risk of chronic graft dysfunction following renal transplantation [11,12].
Because chronic reduction of renal function is a major cause of hyperphosphatemia [13,14], we used leptin-deficient obese (ob/ob) mice to examine the effects of hyperphosphatemia in obesity. We and others have genetically induced phosphate toxicity in mice by eliminating the function of klotho [15–17], and we reduced serum phosphate levels by genetically eliminating 1α(OH)ase activity [18,19] or providing a vitamin D-deficient diet [20]. We used klotho knockout mice [21,22] to induce phosphate toxicity in ob/ob mice by generating ob/ob and klotho double knockout mice [ob/ob-klotho−/−]. Double knockout [ob/ob-1α(OH)ase−/−] mice, which have low serum phosphate levels, were used as controls. These genetically engineered, double mutant mice provided us with the unique tools to study the in vivo effects of different concentrations of phosphate in obesity.
2. Materials and methods
2.1. Generation of double mutant mice
We crossbred the ob/ob and klotho mutant mice, or ob/ob and 1α(OH)ase mutant mice, to obtain compound heterozygous animals [18,23]. These animals were interbred to generate the desired double homozygous mutants [ob/ob-klotho−/− or ob/ob-1α(OH)ase−/−], as a part of an ongoing project, as reported earlier [23]. Mouse genotyping was performed by routine PCR using genomic DNA extracted from tail clips [17,24]. Mice were maintained in accordance with the NIH Guide for the Care and Use of Laboratory Animals, and all protocols were approved by the institution’s subcommittee on animal care (IACUC).
2.2. Gross phenotype and survival
Total body weight of the wild-type, ob/ob, ob/ob-klotho−/− and ob/ob-1α(OH)ase−/− mice were measured every week from 3 to 25 weeks. Survival through 25 weeks was also recorded.
2.3. Biochemical measurements
For all mice, blood was collected at various time points via cheek pouch bleeding. Serum was isolated by centrifugation at 3000g for 10 min and stored at −80 °C. Serum cholesterol, 1,25-dihydroxyvitamin D [1,25(OH)2D3], calcium and phosphate levels were measured at different time points. Serum phosphorus and calcium levels were measured using the Stanbio Phosphorus Liqui-UV Test or the Calcium LiquiColor Test (Arsenazo), respectively. Serum cholesterol levels were also measured using enzymatic assays (Wako Chemicals, Japan). Serum levels of 1,25(OH)2D3 were measured using an ELISA-based kit purchased from Immunodiagnostic Systems Ltd., Fountain Hills, AZ.
2.4. Histological analyses
Histological sections were prepared from various soft tissues and fixed in either a 10% buffered formalin solution, or Carnoy’s solution. Tissue sections were stained with hematoxylin and eosin, periodic acid-Schiff stain (PAS), periodic acid-Schiff methenamine silver stain (PAM), Masson’s trichrome stain, and von Kossa stain [25,26]. Tissue visualization under light microscopy was used to observe histological changes in the various organs.
2.5. Calcification analyses
To determine the effects of phosphate toxicity on ectopic and vascular calcification, sections were prepared from the heart, lung, aorta and kidneys of wild-type, ob/ob, ob/ob-klotho−/− and ob/ob-1α(OH)ase−/− mice and stained with von Kossa stain to visualize mineralized tissues. The von Kossa staining procedure has been previously published [19].
2.6. Statistics
Statistically significant differences between groups were determined using the Student’s t-test. All values were expressed as the mean ± SD or ±SE. A p-value of less than 0.05 was considered statistically significant. All analyses were performed using Microsoft Excel or Prism Software (GraphPad Software Inc., San Diego, CA). Kaplan–Meier survival curves were generated using Prism Software.
3. Results and discussion
3.1. Measuring obesity in the ob/ob mutant mice
The leptin-deficient obese mice, commonly referred to as ob/ob mice, are extensively used to study the underlying mechanisms of obesity. We used these mice to determine the effects of various concentrations of serum phosphate levels in obesity. Compared to wild-type mice, ob/ob mice rapidly gained body weight starting from 3 weeks of age, with massive body fat accumulation including in the abdomen (Supplementary Fig. 1). With age by 12 weeks, the ob/ob mice gained almost two times more weight compared to the wild-type controls (Supplementary Fig. 1). We studied the pathological consequences of phosphate dysregulation in obesity by generating two new ob/ob mouse models with high and low serum phosphate levels. In earlier mouse model studies, we and others have shown that genetic inactivation of klotho can induce hyperphosphatemia [15–17,27], while inactivation of 1a(OH)ase can lead to hypophosphatemia [18]. Here, we used both the klotho and the 1a(OH)ase mutant mice as model systems to manipulate serum phosphate levels in the ob/ob background by crossing the ob/ob mice with either the klotho or the 1a(OH)ase mutant strains.
3.2. Measuring serum cholesterol levels in the ob/ob mutant mice
We measured serum cholesterol levels in the various mouse genetic backgrounds (Fig. 1). By 9–11 weeks, ob/ob mice displayed elevated serum cholesterol levels (266 ± 36 mg/dl) compared to the wild-type mice (136 ± 3 mg/dl). A similar increase in serum cholesterol levels were detected in both the ob/ob-1α(OH)ase−/− (241 ± 13 mg/dl) and the ob/ob-klotho−/− (205 ± 15 mg/dl) double mutant mice.
Fig. 1.
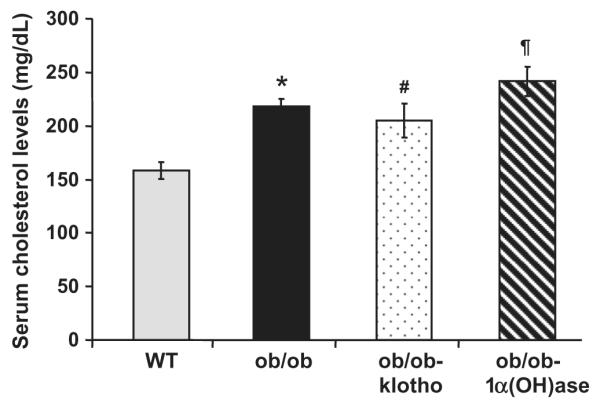
Biochemical measurement of serum cholesterol in the mouse models. Compared to the wild-type (WT) mice (136 ± 3; n = 4), serum cholesterol levels were markedly increased in the ob/ob mice (266 ± 36; n = 6). A similar increase in serum cholesterol levels were observed in the ob/ob-klotho−/− (205 ± 15, n = 12) and the ob/ob-1α(OH)ase−/− (241 ± 13; n = 12) double mutant mice [23]. (*p < 0.01 vs. WT; #p < 0.05 vs. WT; ¶p < 0.01 vs. WT).
The above observations suggest that manipulating serum phosphate levels in the ob/ob mice does not affect their serum cholesterol levels. Furthermore, all three obese mouse models display increased subcutaneous fat accumulation compared to the wild-type mice (Supplementary Fig. 2). The leptin-deficient obese (ob/ob) mice do not show the obesity-associated renal injury observed in other animal models, such as the leptin receptor-deficient obese (db/db) mice. Here, we chose the ob/ob mouse model instead of the db/db mouse model to minimize the systemic effects of renal impairments and to focus on the in vivo effects of hyperphosphatemia in obesity, independent of other uremic toxins.
3.3. Estimation of serum 1,25(OH)2D3 in the ob/ob mutant mice
Serum 1,25(OH)2D3 levels were measured in wild-type, ob/ob, ob/ob-klotho−/− and ob/ob-1α(OH)ase−/− mice. The serum 1,25(OH)2D3 levels in ob/ob-klotho−/− mice were higher than those observed in the wild-type control mice (Fig. 2). The serum 1,25(OH)2D3 level in wild-type mice was 105 ± 15 pmol/L, whereas the ob/ob-klotho−/− mice had a serum level of 664 ± 46 pmol/L. In contrast to the in ob/ob-klotho−/− mice, the serum 1,25(OH)2D3 level in ob/ob-1α(OH)ase−/− mice was undetectable, while in ob/ob mice the level was 316 ± 96 pmol/L.
Fig. 2.
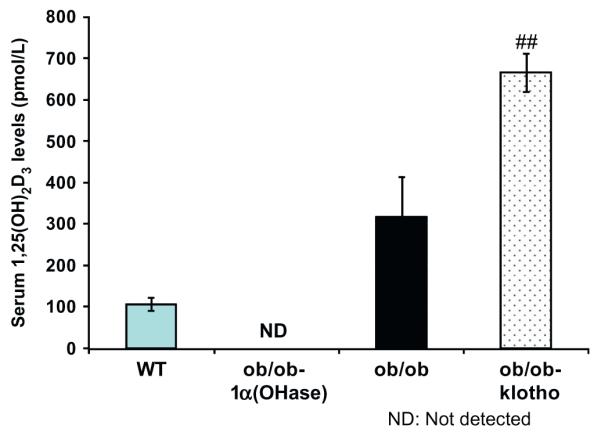
Biochemical measurements of serum 1,25(OH)2D3 in various genotypes. Compared with the wildtype (WT) mice (105 ± 15 pmol/L; n = 12), markedly increased serum levels are noted in ob/ob-klotho−/− mice (664 ± 46 pmol/L; n = 4). In contrast to the in ob/ob-klotho−/− mice, the serum 1,25(OH)2D3 level in ob/ob-1α(OH)ase−/− mice was not detectable (n = 3), while in ob/ob mice the level was 316 ± 96 pmol/L (n = 4). (##p < 0.0001, WT vs. ob/ob-klotho−/−).
3.4. Estimation of serum phosphate and calcium levels in the ob/ob mutant mice
Serum phosphate and calcium levels were measured in 3-, 6- and 9-week-old wild-type, ob/ob, ob/ob-klotho−/− and ob/ob-1α(OH)ase−/− mice. By 3 weeks, the ob/ob-1α(OH)ase−/− double mutants were hypophosphatemic (5.6 ± 0.3 mg/dl) compared to the wild-type mice (7.3 ± 0.3 mg/dl). Indeed, these mutant mice remained hypophosphatemic at all measured time points (Fig. 3). In contrast, the ob/ob-klotho−/− mice developed severe hyperphosphatemia by 3 weeks (10.7 ± 0.2 mg/dl) and remained hyperphosphatemic throughout their life (Fig. 3).
Fig. 3.

Biochemical measurements of both serum phosphate and calcium in the mouse models. Serum phosphate levels (upper panel) were higher in the ob/ob-klotho−/− mice compared to the wild-type (WT) mice, but they were lower in the ob/ob-1α(OH)ase−/− mice. Compared to the ob/ob mice (n = 16; 8.7 ± 0.2), the serum phosphate levels were significantly higher in the ob/ob-klotho−/− mice (n = 10; 10.7 ± 0.2) by 3 weeks. By 9 weeks, higher serum phosphate levels were observed in ob/ob-klotho−/− mice (n = 11; 11.6 ± 0.2). In contrast, the serum phosphate levels were significantly reduced in ob/ob-1α(OH)ase−/− mice at both 3 weeks (n = 5; 5.6 ± 0.3) and 9 weeks (n = 6; 6.6 ± 0.2). Serum phosphate levels of WT mice were 7.3 ± 0.3 (n = 6) at 9 weeks. Serum calcium levels (lower panel) were significantly higher in the ob/ob-klotho−/− mice (n = 10; 9.8 ± 0.5) at 3 weeks compared to the WT mice (n = 11; 7.5 ± 0.2). Higher levels of calcium were observed in the ob/ob-klotho−/− mice (n = 6; 9.6 ± 0.3) at 9 weeks compared to the WT mice (n = 11; 7.5 ± 0.2 at 3 weeks and n = 9; 7.5 ± 0.2 at 9 weeks). In contrast, the serum calcium levels were significantly reduced in the ob/ob-1α(OH)ase−/− mice at both 3 weeks (n = 5; 6.6 ± 0.1) and 9 weeks (n = 6; 5.6 ± 0.2). Serum calcium levels in the ob/ob mice were 9.2 ± 0.2 (n = 17) and 9.2 ± 0.2 (n = 9) at 3 and 9 weeks, respectively. (*p < 0.001 vs. WT; ¶p < 0.001 vs. ob/ob; #p < 0.001 vs. ob/ob-klotho−/−) [23].
The serum phosphate level in 3-week-old ob/ob mice was 8.7 ± 0.2 mg/dl, and remained stable at both weeks 6 and 9. These findings suggest that inactivating klotho function in the ob/ob mice induced phosphate toxicity, while inactivating 1α(OH)ase function in the ob/ob mice led to hypophosphatemia. These mutant mice provide us with the appropriate in vivo tools to study the effects of different concentrations of phosphate in obese mouse models.
Serum calcium levels were also measured in all mice (Fig. 3). At 3 weeks, the ob/ob-klotho−/− mice displayed elevated serum calcium levels (9.8 ± 0.5 mg/dl) compared to the wild-type mice (7.5 ± 0.2 mg/dl). This remained true for all measured time points. In contrast to the ob/ob-klotho−/− mice, the serum calcium levels were significantly reduced in the ob/ob-1α(OH)ase−/− double mutants, which had a level of 6.6 ± 0.1 mg/dl at 3 weeks. However, the serum calcium levels in ob/ob mice were higher at 3 weeks (9.2 ± 0.2 mg/dl) compared to the wild-type mice. These results suggest that our hypercholesterolemic mouse models, with opposing serum phosphate balance, can be useful in determining the pathologic effects of phosphate on various organs in an obese mouse model.
3.5. Effects of phosphate toxicity on ectopic calcification in the ob/ob mutant mice
We examined the effects of opposing serum phosphate balance on calcification in the ob/ob mutants by von Kossa staining. We detected extensive soft tissue and vascular calcification in the kidney, aorta, lung and other organs in the hyperphosphatemic ob/ob-klotho−/− mice (Fig. 4). The extensive calcification noted in the ob/ob-klotho−/− mice was absent in the hypophosphatemic ob/ob-1α(OH)ase−/− mice. Of relevance, no ectopic calcification was observed in 11-week-old wild-type or ob/ob mice. These observations clearly suggest a crucial role for phosphate in vascular and soft tissue calcification in an obese mouse model.
Fig. 4.

Macroscopic and microscopic changes in the kidney and aorta. Gross morphology of the kidneys from the wild-type (WT), ob/ob, ob/ob-klotho−/− and ob/ob-1α(OH)ase−/− mice. Compared to the ob/ob and ob/ob-1α(OH)ase−/− mice, the kidney sizes were smaller in the hyperphosphatemic ob/ob-klotho−/− mice. All animals were age-matched (11 weeks) (upper panel). Histological sections of kidneys were prepared from WT, ob/ob, ob/ob-klotho−/− and ob/ob-1α(OH)ase−/− mice (middle panel). Inducing phosphate toxicity in the ob/ob mice resulted in extensive calcifications (dark black staining, arrows) in the kidneys of the ob/ob-klotho−/− mice. In contrast, reducing the serum phosphate levels in ob/ob mice did not show any calcification in the ob/ob-1α(OH)ase−/− mice (20× magnification). Aortic sections were prepared from all mice (lower panel). Inducing phosphate toxicity in the ob/ob mice resulted in extensive aortic calcifications (dark black staining, arrows) compared to the ob/ob-klotho−/− mice. In contrast, reducing serum phosphate levels in the ob/ob mice did not lead to any calcification (von Kossa staining; 20× magnification).
3.6. Effects of phosphate toxicity on soft tissue injury in the ob/ob mutant mice
In contrast to the wild-type and the ob/ob-1α(OH)ase−/− mice, the hyperphosphatemic ob/ob-klotho−/− mice displayed generalized tissue atrophy. The genital organs in both sexes of the ob/ob-klotho−/− mice were severely atrophic (Supplementary Fig. 3); similar atrophic changes were also observed in lymphoid organs, including the spleen (Supplementary Fig. 3). In addition, the hyperphosphatemic ob/ob-klotho−/− mice displayed typical features of lung emphysema (Supplementary Fig. 4), appearing as early as 6 weeks. This observation is consistent with similar emphysematous changes documented in other experimental animal models for phosphate toxicity [19,28]. No pulmonary emphysematous change was detected in the hypophosphatemic ob/ob-1α(OH)ase−/− mice (Supplementary Fig. 4), suggesting a role for phosphate toxicity in lung injury [29].
3.7. Effects of phosphate toxicity on the survival of the ob/ob mutant mice
We studied the effects of opposing serum phosphate balance on the survival of the ob/ob mutant mice. The survival of mice from all four genotypes was recorded weekly for 25 weeks. None of the wild-type or the ob/ob mutant mice died by week 25 (Supplementary Fig. 5), whereas 100% of the ob/ob-klotho−/− mice died by week 20. This suggests that various organ injuries inflicted by phosphate toxicity can significantly reduce the survival of these mice. In contrast, despite hypercholesterolemia and obesity in the ob/ob-1α(OH)ase−/− mice, reducing serum phosphate levels did not affect their survival (Supplementary Fig. 5).
In the ob/ob-klotho−/− mice, both serum phosphate and 1,25(OH)2D3 levels were significantly increased compared to wild-type mice. In contrast to the ob/ob-klotho−/− mice, the ob/ob-1α(OH)ase−/− mice had both lower serum phosphate and 1,25(OH)2D3 levels (Fig. 2). It is unclear if some of the observed pathologies in the ob/ob-klotho−/− mice are due to increased vitamin D activities; however, we have shown that vascular calcification and other related complications can be delayed or eliminated by reducing serum phosphate levels even in the presence of high serum calcium and vitamin D levels [27,30]. Although we cannot completely rule out the role of vitamin D, most of our documented pathologies in the ob/ob-klotho−/− mice are likely due to phosphate toxicity [14,31,32].
Obesity is an important risk factor for both de novo renal disease and as a complication of existing chronic kidney disease (CKD). In a 20 year follow-up study, a single unit increase of body mass index was shown to be responsible for a 20% increase in kidney disease [33]. Obesity-associated nephropathy is clinicopathologically characterized by albuminuria, glomerulomegaly and eventual secondary focal glomerulosclerosis [1,34]. These obesity-related renal structural changes lead to impaired renal function. Because hyperphosphatemia is an important consequence of renal impairment, it is important to know the effects of phosphate dysregulation on obesity-associated nephropathy. It is likely that once obesity affects kidney function, the prognosis of the patient may be related to the consequences of renal impairment rather than obesity itself. An important to note is that our poor understanding of the underlying mechanism in the development of obesity-associated nephropathy prevents us from dissociating the intrinsic from the extrinsic effects of obesity in disease progression. Indeed, studies have suggested that obesity in patients with CKD may act as a buffer to prevent patients from wasting; however, by helping sequester uraemic toxins, it may present a false microenvironment that may lead to treatment error, including underdialysis [35].
The vascular changes in obesity and hypercholesterolemia in humans (i.e., plaque formation) differ from those in patients with chronic hyperphosphatemia (i.e., media calcification). In this study, the combinations of these two very different etiologies of vascular damaging models were incorporated in a single mouse model. This novel mouse model should prove to be useful to study how phosphate toxicity might accelerate the deleterious effects of obesity and vice versa.
In conclusion, the results of our in vivo genetic manipulation studies demonstrate that inducing phosphate toxicity in an obese mouse model can accelerate tissue injury and reduce overall survival. We further demonstrated that reducing serum phosphate levels in isogenic, obese mice could significantly reduce these types of tissue injuries, despite the underlying presence of hypercholesterolemia and obesity. Although the obesity paradox needs additional clarification, we revealed that phosphate toxicity has pronounced effects on the survival of obese, hypercholesterolemic mice, implying that once obesity-induced renal injury is established, subsequent biochemical changes may partially determine the outcome of the disease process.
Supplementary Material
Acknowledgments
Part of the research work is supported by the Grant (R01-DK077276 to Dr. Razzaque) provided by NIH. Dr. Ohnishi is the recipient of 2011 Dean’s Scholarship from Harvard School of Dental Medicine, Boston, MA. The present work is the continuation of earlier published works from the same laboratory [23]. The authors thank Dr. Rene St. Arnoud (Shriners Hospital for Children, Montreal, Quebec, Canada) for kindly providing 1α(OH)ase knockout mice. Thanks are also due to Junko Akiyoshi (MD), Satoko Osuka (MD), Yongguen Hong (PhD), Khadijah Turkistani (BDS) and Ismail Eddafali (Harvard School of Dental Medicine, Boston) for technical assistance.
Footnotes
Appendix A. Supplementary data Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.bbrc.2011.10.076.
References
- [1].Kato S, Nazneen A, Nakashima Y, Razzaque MS, Nishino T, Furusu A, Yorioka N, Taguchi T. Pathological influence of obesity on renal structural changes in chronic kidney disease. Clin. Exp. Nephrol. 2009;13:332–340. doi: 10.1007/s10157-009-0169-3. [DOI] [PubMed] [Google Scholar]
- [2].Praga M, Morales E. Obesity, proteinuria and progression of renal failure. Curr. Opin. Nephrol. Hypertens. 2006;15:481–486. doi: 10.1097/01.mnh.0000242172.06459.7c. [DOI] [PubMed] [Google Scholar]
- [3].Liao J, Richards R, Zhang R, Reisin E. Effects of a modified low calorie diet in metabolic changes and kidney histology in young obese Zucker rats. J. Am. Soc. Nephrol. 2007;18:823A. [Google Scholar]
- [4].Weisinger JR, Kempson RL, Eldridge L, Swenson RS. The nephrotic syndrome: a complication of massive obesity. Ann. Intern. Med. 1974;81:440–447. doi: 10.7326/0003-4819-81-4-440. [DOI] [PubMed] [Google Scholar]
- [5].Verani RR. Obesity associated focal segmental glomerulosclerosis: pathological features of the lesion and relationship with cardiomegaly and hyperlipidemia. Am. J. Kidney Dis. 1992;20:629–634. doi: 10.1016/s0272-6386(12)70230-5. [DOI] [PubMed] [Google Scholar]
- [6].Warnke RA, Kempson RL. The nephrotic syndrome in massive obesity. A study by light, immunofluorescence and electron microscopy. Arch. Pathol. Lab. 1978;102:431–438. [PubMed] [Google Scholar]
- [7].Pausova Z. From big fat cells to high blood pressure: a pathway to obesity-associated hypertension. Curr. Opin. Nephrol. Hypertens. 2006;15:173–178. doi: 10.1097/01.mnh.0000214775.42103.a5. [DOI] [PubMed] [Google Scholar]
- [8].Peralta CA, Kurella M, Lo JC, Chertow GM. The metabolic syndrome and chronic kidney disease. Curr. Opin. Nephrol. Hypertens. 2006;15:361–365. doi: 10.1097/01.mnh.0000232875.27846.7e. [DOI] [PubMed] [Google Scholar]
- [9].Tanaka M, Yamada S, Iwasaki Y, Sugishita T, Yonemoto S, Tsukamoto T, Fukui S, Takasu K, Muso E. Impact of obesity on IgA nephropathy: comparative ultrastructural study between obese and non-obese patients. Nephron Clin. Pract. 2009;112:c71–c78. doi: 10.1159/000213084. [DOI] [PubMed] [Google Scholar]
- [10].Tsuboi N, Kawamura T, Ishii T, Hosoya T. Obesity-related nephropathy associated with a history of IgA nephropathy. Intern. Med. 2008;47:1713–1718. doi: 10.2169/internalmedicine.47.1196. [DOI] [PubMed] [Google Scholar]
- [11].Potluri K, Hou S. Obesity in kidney transplant recipients and candidates. Am. J. Kidney Dis. 2010;56:143–156. doi: 10.1053/j.ajkd.2010.01.017. [DOI] [PubMed] [Google Scholar]
- [12].Sampaio MS, Reddy PN, Kuo HT, Poommipanit N, Cho YW, Shah T, Bunnapradist S. Obesity was associated with inferior outcomes in simultaneous pancreas kidney transplant. Transplantation. 2010;89:1117–1125. doi: 10.1097/TP.0b013e3181d2bfb2. [DOI] [PubMed] [Google Scholar]
- [13].Fukagawa M, Hamada Y, Nakanishi S, Tanaka M. The kidney and bone metabolism: nephrologists’ point of view. J. Bone Miner. Metab. 2006;24:434–438. doi: 10.1007/s00774-006-0719-7. [DOI] [PubMed] [Google Scholar]
- [14].Razzaque MS. Phosphate toxicity: new insights into an old problem. Clin. Sci. (Lond.) 2011;120:91–97. doi: 10.1042/CS20100377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Kuro-o M, Matsumura Y, Aizawa H, Kawaguchi H, Suga T, Utsugi T, Ohyama Y, Kurabayashi M, Kaname T, Kume E, Iwasaki H, Iida A, Shiraki-Iida T, Nishikawa S, Nagai R, Nabeshima YI. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature. 1997;390:45–51. doi: 10.1038/36285. [DOI] [PubMed] [Google Scholar]
- [16].Nakatani T, Bara S, Ohnishi M, Densmore MJ, Taguchi T, Goetz R, Mohammadi M, Lanske B, Razzaque MS. In vivo genetic evidence of klotho-dependent functions of FGF23 in regulation of systemic phosphate homeostasis. FASEB J. 2009;23:433–441. doi: 10.1096/fj.08-114397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Nakatani T, Ohnishi M, Razzaque MS. Inactivation of klotho function induces hyperphosphatemia even in presence of high serum fibroblast growth factor 23 levels in a genetically engineered hypophosphatemic (Hyp) mouse model. FASEB J. 2009;23:3702–3711. doi: 10.1096/fj.08-123992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Ohnishi M, Nakatani T, Lanske B, Razzaque MS. Reversal of mineral ion homeostasis and soft-tissue calcification of klotho knockout mice by deletion of vitamin D 1alpha-hydroxylase. Kidney Int. 2009;75:1166–1172. doi: 10.1038/ki.2009.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Razzaque MS, Sitara D, Taguchi T, St.-Arnaud R, Lanske B. Premature ageing-like phenotype in fibroblast growth factor 23 null mice is a vitamin-D mediated process. FASEB J. 2006;20:720–722. doi: 10.1096/fj.05-5432fje. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Tsujikawa H, Kurotaki Y, Fujimori T, Fukuda K, Nabeshima Y. Klotho, a gene related to a syndrome resembling human premature aging, functions in a negative regulatory circuit of vitamin D endocrine system. Mol. Endocrinol. 2003;17:2393–2403. doi: 10.1210/me.2003-0048. [DOI] [PubMed] [Google Scholar]
- [21].Razzaque MS. The FGF23-klotho axis: endocrine regulation of phosphate homeostasis. Nat. Rev. Endocrinol. 2009;5:611–619. doi: 10.1038/nrendo.2009.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Razzaque MS. FGF23-mediated regulation of systemic phosphate homeostasis: is klotho an essential player? Am. J. Physiol. Renal Physiol. 2009;296:F470–F476. doi: 10.1152/ajprenal.90538.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Ohnishi M, Kato S, Akiyoshi J, Atfi A, Razzaque MS. Dietary and genetic evidence for enhancing glucose metabolism and reducing obesity by inhibiting klotho functions. FASEB J. 2011;25:2031–2039. doi: 10.1096/fj.10-167056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Dardenne O, Prud’homme J, Arabian A, Glorieux FH, St.-Arnaud R. Targeted inactivation of the 25-hydroxyvitamin D(3)-1(alpha)-hydroxylase gene (CYP27B1) creates an animal model of pseudovitamin D-deficiency rickets. Endocrinology. 2001;142:3135–3141. doi: 10.1210/endo.142.7.8281. [DOI] [PubMed] [Google Scholar]
- [25].Zha Y, Le VT, Higami Y, Shimokawa I, Taguchi T, Razzaque MS. Life-long suppression of growth hormone-insulin-like growth factor I activity in genetically altered rats could prevent age-related renal damage. Endocrinology. 2006;147:5690–5698. doi: 10.1210/en.2006-0302. [DOI] [PubMed] [Google Scholar]
- [26].Razzaque MS, Taguchi T. Collagen-binding heat shock protein (HSP) 47 expression in anti-thymocyte serum (ATS)-induced glomerulonephritis. J. Pathol. 1997;183:24–29. doi: 10.1002/(SICI)1096-9896(199709)183:1<24::AID-PATH1106>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- [27].Ohnishi M, Nakatani T, Lanske B, Razzaque MS. In vivo genetic evidence for suppressing vascular and soft-tissue calcification through the reduction of serum phosphate levels, even in the presence of high serum calcium and 1,25-dihydroxyvitamin D levels. Circ. Cardiovasc. Genet. 2009;2:583–590. doi: 10.1161/CIRCGENETICS.108.847814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Lanske B, Razzaque MS. Premature aging in klotho mutant mice: cause or consequence? Ageing Res. Rev. 2007;6:73–79. doi: 10.1016/j.arr.2007.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Sato A, Hirai T, Imura A, Kita N, Iwano A, Muro S, Nabeshima Y, Suki B, Mishima M. Morphological mechanism of the development of pulmonary emphysema in klotho mice. Proc. Natl. Acad. Sci. USA. 2007;104:2361–2365. doi: 10.1073/pnas.0607882104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Razzaque MS. The dualistic role of vitamin D in vascular calcifications. Kidney Int. 2011;79:708–714. doi: 10.1038/ki.2010.432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Razzaque MS. Osteo-renal regulation of systemic phosphate metabolism. IUBMB Life. 2011;63:240–247. doi: 10.1002/iub.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Ohnishi M, Razzaque MS. Dietary and genetic evidence for phosphate toxicity accelerating mammalian aging. FASEB J. 2010;24:3562–3571. doi: 10.1096/fj.09-152488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Fox C, Larson M, Leip E, Culleton B, Wilson P, Levy D. Predictors of new-onset kidney disease in a community-based population. JAMA. 2004;291:844–850. doi: 10.1001/jama.291.7.844. [DOI] [PubMed] [Google Scholar]
- [34].Mathew AV, Okada S, Sharma K. Obesity related kidney disease. Curr. Diabetes Rev. 2011;7:41–49. doi: 10.2174/157339911794273928. [DOI] [PubMed] [Google Scholar]
- [35].Speakman JR, Westerterp KR. Reverse epidemiology, obesity and mortality in chronic kidney disease: modelling mortality expectations using energetics. Blood Purif. 2010;29:150–157. doi: 10.1159/000245642. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.