

Summary
Identification of somatic rearrangements in cancer genomes has accelerated through analysis of high-throughput sequencing data. However, characterization of complex structural alterations and their underlying mechanisms remains inadequate. Here, applying an algorithm to predict structural variations from short reads, we report a comprehensive catalog of somatic structural variations and the mechanisms generating them, using high-coverage whole-genome sequencing data from 140 patients across ten tumor types. We characterize the relative contributions of different types of rearrangements and their mutational mechanisms, find that ~20% of the somatic deletions are complex deletions formed by replication errors, and describe the differences between the mutational mechanisms in somatic and germline alterations. Importantly, we provide detailed reconstructions of the events responsible for loss of CDKN2A/B and gain of EGFR in glioblastoma, revealing that these alterations can result from multiple mechanisms even in a single genome and that both DNA double-strand breaks and replication errors drive somatic rearrangements.
Introduction
Cancer is a disease driven by genetic alterations, which include single nucleotide variations (SNVs), structural variations (SVs) and aneuploidy. The spectrum of somatic SNVs studied suggests that mismatch repair deficiency and specific mutagenic exposure such as smoking, UV light and chemotherapy can be inferred from the mutational signatures for specific tumor types (Greenman et al., 2007; Lee et al., 2010; Pleasance et al., 2009a; Pleasance et al., 2009b). Larger scale SVs including deletions, insertions, inversions, tandem duplications, translocations and more complex rearrangements constitute another frequent type of alterations that could alter normal gene function in tumors. Somatic SVs have been characterized in several previous studies (Bass et al., 2011; Berger et al., 2011; Campbell et al., 2010; Hillmer et al., 2011; Stephens et al., 2009); however, which driving forces exist for SV formation is still unclear.
Three main types of mechanisms known to cause SVs are homologous recombination, non-replicative non-homologous repair and replication-based mechanisms (Gu et al., 2008; Hastings et al., 2009b). Homologous recombination is the most common DNA repair mechanism and is generally accurate, except when the pairing is between incorrect homologous regions, as in non-allelic homologous recombination (NAHR). Deficiency in homologous recombination is believed to be a major source of cancer genome instability (Hoeijmakers, 2001). For example, BRCA1 and BRCA2 are required for the homology-directed repair of chromosomal breaks, and loss of these two genes often results in genome instability and cancer (Venkitaraman, 2002). Non-homologous end-joining (NHEJ) is a non-replicative non-homologous repair mechanism that requires no homology and sometimes can generate very short microhomology or small insertions at the breakpoint (Mahaney et al., 2009). In addition, alternative end joining (alt-EJ) mechanism, also called microhomology-mediated end joining (MMEJ), can generate blunt ends, small insertions, and, more frequently, microhomology at the deletion breakpoints (Bennardo et al., 2008; McVey and Lee, 2008). Which factors are involved in alt-EJ is much less clear than it is for NHEJ, and alt-EJ appears to be independent of the canonical NHEJ factors such as Ku70 and XRCC4 (Arlt et al., 2012; Bennardo et al., 2008). NHEJ has often been implicated in tumor genomes, based on the very few overlapping sequences at breakpoints (Stephens et al., 2011; Stephens et al., 2009). For complex rearrangements, a replicative mechanism called fork stalling and template switching (FoSTeS) has been described (Lee et al., 2007), which later was generalized to microhomology-mediated break-induced repair (MMBIR). It has been proposed that the replication at the fork can stall and the polymerase can shift template via microhomology to any nearby single stranded DNA, resulting in inversion, tandem duplication, translocation, or more complex rearrangements (Hastings et al., 2009a; Zhang et al., 2009). A recent assessment of SV formation (mostly focusing on deletions) in a normal human population by the 1000 Genomes Project (Mills et al., 2011) did not address the role of replication-based mechanisms. In cancer, a recent study hypothesized that complex genomic rearrangements can arise from a single catastrophic event (Stephens et al., 2011) driven by NHEJ; similar complex rearrangements have also been observed in pathogenic germline alterations (Chiang et al., 2012). Another study suggested that multiple amplifications in developmental delay and cognitive anomalous patients are generated by a replication-based mechanism (Liu et al., 2011) since microhomology is frequently observed at the breakpoints. However, there is no comprehensive study of the mechanisms that underlie somatic SVs in cancer genomes, and many aspects are still poorly characterized, including the forces that drive SV formation, relative contribution of different mechanisms across tumor types, and whether additional mechanisms play a role.
In our analysis of somatic SVs across ten tumor types, non-homology-based and microhomology-based mechanisms are consistently the dominant mutational mechanisms responsible for genomic rearrangements, driven by DNA double-strand breaks and replication errors. Importantly, multiple mechanisms sometimes act on a single gene in a genome and two driving forces can act together on different part of a genome to create mutations and promote tumorigenesis.
Results
Identification of germline complex deletion events using Meerkat
To characterize the mutational spectrum of somatic SVs in cancer, it is important to identify both simple (e.g., deletion, insertion, and inversion) and complex SVs at base-pair resolution. The most common type of complex SV is a deletion with an insertion or inversion at the breakpoint (Conrad et al., 2010; Kidd et al., 2010) generated by FoSTeS/MMBIR. Previously, the identification of such events involved capturing and sequencing of the segments adjacent to the deletion breakpoints (Conrad et al., 2010; Kidd et al., 2010). Here, we predict both germline and somatic SVs directly from short read data, focusing on complex events such as those generated by FoSTeS/MMBIR (an example shown in Figure 1A-D). This is made possible by a new algorithm called Meerkat (see Experimental Procedures and Supplemental Experimental Procedures). With the base-pair resolution of the breakpoints identified by our method, the mechanisms forming SVs are inferred based on sequence homology at the breakpoints (Kidd et al., 2010; Lam et al., 2009; Mills et al., 2011) (Figure 1E, see also Discussion). Identification of somatic SVs from short read data is challenging due to several factors, including sequencing errors, GC content and other biases in sequencing, ambiguous alignments due to repetitive sequences, a large number of germline SVs, chimeric molecules generated during library construction, normal cell contamination in tumor samples, and heterogeneity within tumor cell populations. The distinguishing feature of our method is that it considers the configuration of multiple clusters of discordant read pairs (read pairs in which the mapped reads are at unexpected distance or orientation) to recognize complex events with high accuracy, in addition to efficiently utilizing split, clipped, and multiple-aligned reads (Supplemental Experimental Procedures).
Figure 1. Example of a complex deletion generated by FoSTeS/MMBIR and a pipeline for predicting SV mechanisms.
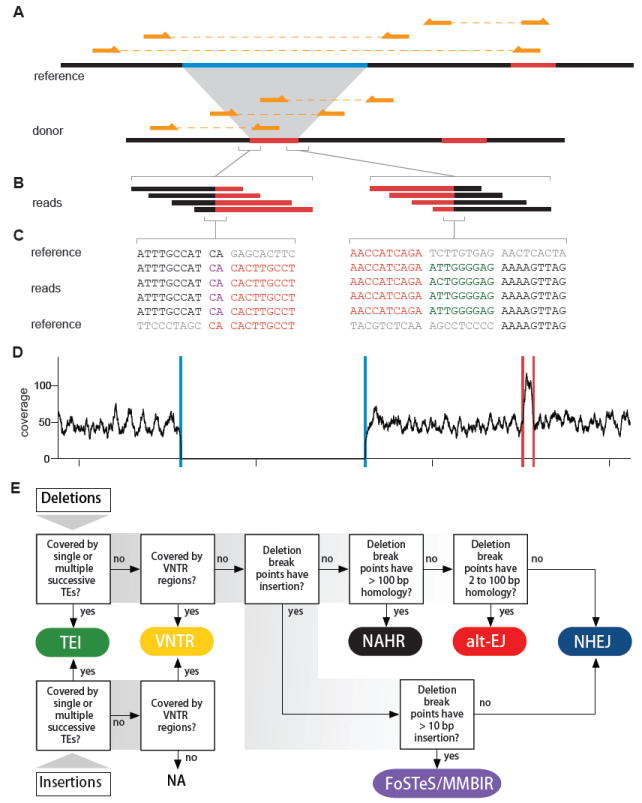
(A) A complex deletion is predicted by three discordant clusters. The sequence in light blue on the reference is deleted; the sequence in red on the reference is duplicated and inserted into the deletion breakpoints. Three read pairs from the donor are shown above the donor sequence. Three discordant read pairs mapped to the reference are shown above the reference sequence.
(B) Reads covering the breakpoints of insertion. The breakpoints are covered by 27 and 11 reads, respectively (only four are shown for each). Reads matching different parts of the reference genome are shown in the corresponding colors.
(C) Nucleotide sequences of the reads covering the breakpoints of insertion. Black and red colors indicate the reads and the reference sequences that match each other and the grey sequences indicate unmatched references. There are a 2 bp microhomology (shown in purple) at the breakpoint on the left and a 9 bp insertion of unknown source (shown in dark green) at the breakpoint on the right.
(D) Sequencing depth. Blue and red lines denote the predicted deletion and the predicted insertion donor sites, respectively, showing that the copy number is consistent with the SV call.
(E) This flowchart, adapted mainly from Kidd et al. 2010, shows the breakpoint features for determining the mechanism that is likely to have generated the observed SV. Six types of mechanisms are assigned: transposable element insertion (TEI), variable number of tandem repeats (VNTR), non-homologous end joining (NHEJ), alternative end joining (alt-EJ), non-allelic homologous recombination (NAHR) and fork stalling and template switching/microhomology mediated break induced repair (FoSTeS/MMBIR).
See also Figure S1 and Table S1 and S2.
To verify the accuracy of our method, we applied Meerkat to two HapMap genomes (NA18507 and NA12878) that have been sequenced at high coverage on the Illumina platform and for which complex deletions have been previously reported (Kidd et al., 2010) based on the sequencing of fosmid library with 40-kb inserts. We identified a total of 3,508 and 2,327 SVs, respectively (Table S1). Of our simple deletion predictions from NA18507 and NA12878, 91.4% (2102/2301) and 93.7% (1304/1391) were reported in the Database of Genomic Variants (DGV10) (Iafrate et al., 2004) or the 1000 Genomes Project (Mills et al., 2011), respectively, suggesting that the vast majority of our predictions were true events (Figure S1). To further validate the events we detected, we randomly selected 49 events across different types in NA18507 including 24 complex deletion events. We were able to validate 48 events by PCR, including all complex deletions (Table S1). We identified a total of 379 and 253 complex deletions in NA18507 and NA12878, demonstrating that our method is far more sensitive than the previous effort, which reported 2 and 17 complex deletions, respectively (Kidd et al., 2010). Therefore, with the Meerkat algorithm, we can provide a more comprehensive spectrum of mechanisms of SVs in a genome. An example for a complex deletion in NA18507 identified by Meerkat is shown in Figure 1A-D (the same event was reported by Kidd et al. but in a different individual NA18956). Comparing our predictions to the simple and complex deletions reported in Kidd et al., most of the events we failed to identify occur in repetitive regions of the genome (Figure. S1 and Table S2). This is expected since events reported by Kidd et al. were based on Sanger sequencing. The examination of repetitive elements with short reads is a challenging problem that we have addressed in a separate study (Lee et al., 2012).
Somatic structural variations across tumor types
We analyzed high-coverage whole-genome sequencing data from 140 individuals across ten tumor types, including 14 colorectal adenocarcinoma (Bass et al., 2011; Lee et al., 2012) (CRC), 7 multiple myeloma (Chapman et al., 2011) (MM), 7 prostate adenocarcinoma (Berger et al., 2011) (PR), 9 ovarian serous cystadenocarcinoma (Lee et al., 2012) (OV), 16 glioblastoma multiforme (Lee et al., 2012) (GBM), 19 hepatocellular carcinoma (Sung et al., 2012) (HCC), 35 breast invasive carcinoma (The Cancer Genome Atlas Research Network, 2012a) (BRCA), 19 lung squamous cell carcinoma (The Cancer Genome Atlas Research Network, 2012b) (LUSC), 10 uterine corpus endometrioid carcinoma (UCEC) and 4 kidney renal clear cell carcinoma (KIRC) patients. With data from both tumor and germline samples for each patient to distinguish germline and somatic variants, a total of 140 pairs of genomes consisting of about half trillion (458 billion, ~35X coverage per genome on average) paired-end reads (75-101 bp) were analyzed.
A total of 25,874 high-confidence somatic SVs (Table S3) were identified from the 140 cancer genomes (Table S4), ranging from 0 to 3160 per genome with an average of 185 (Figure 2A). To assess the accuracy of somatic SV predictions, we randomly selected 78 out of 138 SVs in an ovarian tumor (OV0725), including two complex events with two breakpoints each, and examined them in both tumor and normal tissues. By PCR, we were able to validate 73 out of 80 (91%) breakpoints (Table S3) as somatic events.
Figure 2. Spectrum of somatic SV types and mechanisms.
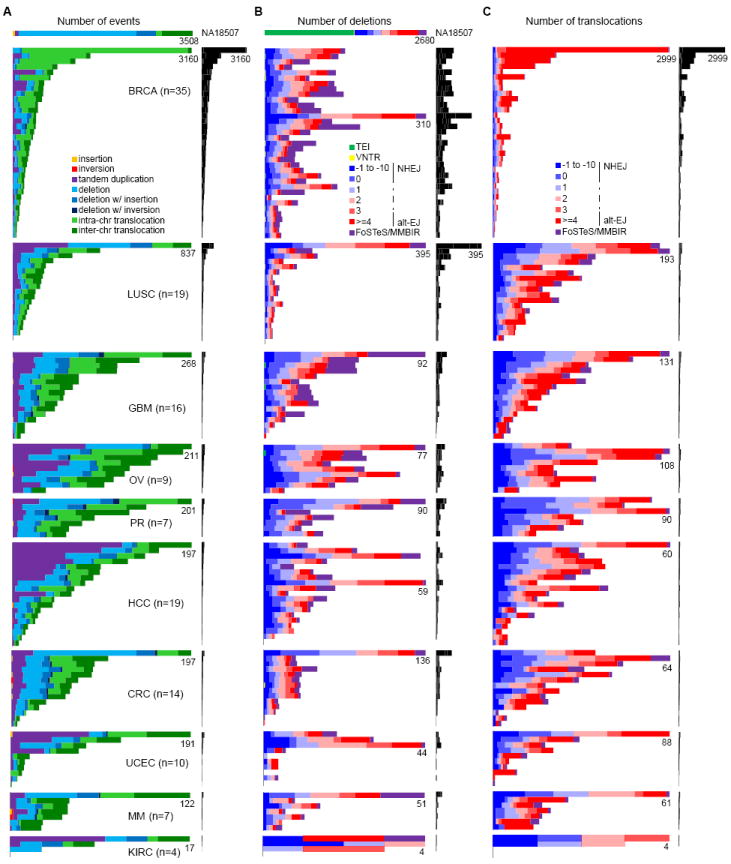
(A) Frequencies of types of somatic SVs identified in each patient. Each horizontal bar displays the number of SVs for one sample. The colored bar charts on the left show the number of events scaled by the maximum number of events (as noted) in each tumor type. The black bar charts on the right show the number of events for all patients on the same scale. A HapMap genome (NA18507) is shown at the top as an example of germline events; see Figure S2 for germline events for all patients. Most (59%) of the translocations in NA18507 are TE insertions, as described previously (Lee et al., 2012), 18% are repeat-related events including TE insertions not identified by Lee et al. 2012, and the remaining ones might be events too complex to be identified by Meerkat.
(B) Frequencies of somatic deletion mechanisms. The order of the samples is the same as in (A).
(C) Frequencies of somatic translocation mechanisms. The order of the samples is the same as in (A).
See also Figure S2 and Table S3, S4 and S5.
The frequency of different types of somatic SVs in each sample is shown in Figure 2A. We first note the remarkable variation in the number of SVs among individuals within and across tumor types (the x-axis for each tumor type is scaled differently in Figure 2A). Some genomes contain no SV (e.g., LUSC1078 and KIRC4856), while others show thousands of SVs (e.g., BRCAA0J6); in a single tumor type, the number of SVs can vary by an order of magnitude between individuals. Among the tumor types, breast tumors and lung squamous cell tumors have significantly more SVs than any other tumors (P = 4.70e-23 and 6.68e-5, respectively, ANOVA tests using a negative binomial model). The number of SVs identified in breast cancer patients here (16,125 in 35 patients, ~461 per patient on average) is much larger than those in previous studies (2,166 in 24 patients, ~90 per patient on average (Stephens et al., 2009) and 2,476 in 22 patients, ~113 per patient on average (Banerji et al., 2012)), due to increased sequencing coverage, sensitivity in the detection method, and sample variation. Kidney cancer patients have significantly less SVs than other tumor types (P = 1.74e-5, ANOVA test using a negative binomial model). In terms of event types, translocations (57%) are the most abundant SV type, while deletions and tandem duplications make up 25% and 17%, respectively. The proportions of different types of SVs across different tumor types are highly variable (Figure 2A). For instance, the breast tumors have significantly more intra-chromosomal translocations than other tumors types do (P = 1.22e-53, ANOVA test using a negative binomial model). There are also considerable differences between individual genomes. For example, all rearrangements in a kidney sample (KIRC5010) are deletions, while there are no deletions in a liver cancer (HCC13). In non-tumor samples, each individual has about 3,000 germline SVs with deletions always being the most abundant (~60%) (Figure S2A), similar to what we find in the HapMap individual NA18507 (Figure 2A).
By pairing multiple clusters of discordant reads to predict complex events, we achieved a better description of the nature of SVs than previously obtained. For example, in the PR0581 genome, a “close chain” pattern had been described to form the TMPRSS2-ERG fusion gene, involving C21orf45 (Berger et al., 2011). We identified two related events in this genome. The first event is a 3 Mb deletion that causes the TMPRSS2-ERG fusion. The second event is a 74 bp deletion in the first intron of C21orf45 at which the 3 Mb deletion from the first event was inserted. The copy numbers of the aforementioned regions were unchanged, supporting the two events we predicted. Detailed descriptions of the events involving CDKN2A/B, EGFR, and CDK4 are provided later.
Certain pathways, such as DNA replication, DNA repair, and cell cycle pathways, are likely to malfunction in order for the cell to generate and maintain the genomic rearrangements. To investigate this, we identified mutations in genes that in above pathways caused by SVs as well as single nucleotide variants (Bass et al., 2011; Berger et al., 2011; Chapman et al., 2011; The Cancer Genome Atlas Research Network, 2012a; The Cancer Genome Atlas Research Network, 2012b). As expected, almost all patients have at least one gene altered in at least one of these pathways (Table S5); nearly half of the mutations are caused by SVs.
Mutational mechanisms for somatic SVs
The number of deletions per genome ranges from 0 to 395, with an average of 46 (Figure 2B). Deletions are usually a result of DNA double-strand break repair. The mechanisms of deletion formation are predicted as shown in Figure 1E (see the Figure 1E legend for information on how mechanistic categories were assigned). In the cancer genomes we studied, NHEJ (39%) and alt-EJ (41%) are the dominant mechanisms. This is in contrast to the mechanisms in the HapMap genome NA18507 (Figure 2B) and non-tumor genomes (Figure S2B), in which TEI is always the dominant mechanism, and the frequencies of NHEJ and alt-EJ in germline deletions are about ~15% and ~22%, respectively. The increased ratio of NHEJ to alt-EJ in somatic deletions compared to that in germline is statistically significant (P = 1.41e-12, Wilcoxon’s paired rank sum test). We also find that about 20% of the somatic deletions are complex deletions formed by FoSTeS/MMBIR (Figure 2B), in contrast to ~5% for the germline deletions (Figure 2B and Figure S2B). The mechanisms of somatic deletions are also variable across different tumor types and between samples. For instance, some genomes have a notable portion of FoSTeS/MMBIR in somatic deletions, while others have none (Figure 2B).
We identified between 0 and 2,999 inter- and intra-chromosomal translocations in the cancer genomes with an average of 106 translocations per genome (Figure 2C). Again, NHEJ and alt-EJ are the dominant mechanisms with alt-EJ being more abundant in most cases. A small number of translocations are formed by FoSTeS/MMBIR with variable frequencies across genomes. Breast cancer patients have significantly more translocations formed by alt-EJ than other tumor types (P = 4.21e-19, ANOVA test using a negative binomial model). We note that the proportion of translocations formed by NHEJ and alt-EJ are comparable in most tumor types, but alt-EJ is much more prominent in breast tumors that have a large number of translocations (>500). The reason for this difference in translocation formation in those samples is not clear, but it may be due to an alteration in a specific pathway that induces translocations.
Tandem duplications are known to result from unequal crossing-over (Edlund and Normark, 1981) or by FoSTeS/MMBIR (Hastings et al., 2009a). Short sequence homologies are required for FoSTeS/MMBIR—the microhomology can be as short as 2 bp to allow new DNA synthesis to start (Zhang et al., 2009)—while a larger degree of homology is required for unequal crossing-over. Complex rearrangements, especially ones involving dosage gains, are often driven by FoSTeS/MMBIR (Hastings et al., 2009a; Liu et al., 2011) as evidenced by the microhomology frequently observed at the breakpoints. Events with no more than 10 bp insertions at breakpoints were classified as NHEJ since NHEJ is known to generate small insertions at breakpoints (Haviv-Chesner et al., 2007). In HapMap (Figure 3) and other non-tumor samples (Figure S3A and S3B), the majority of the breakpoints of tandem duplications (73%) and complex deletions (71%) have microhomology that support the MMBIR models. In contrast, the fraction of breakpoints with microhomology is significantly less in somatic tandem duplications (46%) and complex deletions (52%) (P = 6.78e-19 and P = 1.09e-13, respectively, using Wilcoxon’s paired rank sum test; Figure 3, Figure S3C and S3D). Although most of the germline tandem duplications and complex deletions were generated by FoSTeS/MMBIR (Figure S3A and S3C), a small number have no homology at the breakpoints. In somatic tandem duplications and complex deletions, we do not observe homology at many breakpoints (Figure 3). Thus, we suspect that a template-switching mechanism that does not require microhomology or another non-homology-based mechanism is often utilized in somatic cells to form tandem duplications and complex deletions.
Figure 3. Proportion of homologies at the breakpoints of somatic tandem duplications and complex deletions compared with NA18507.
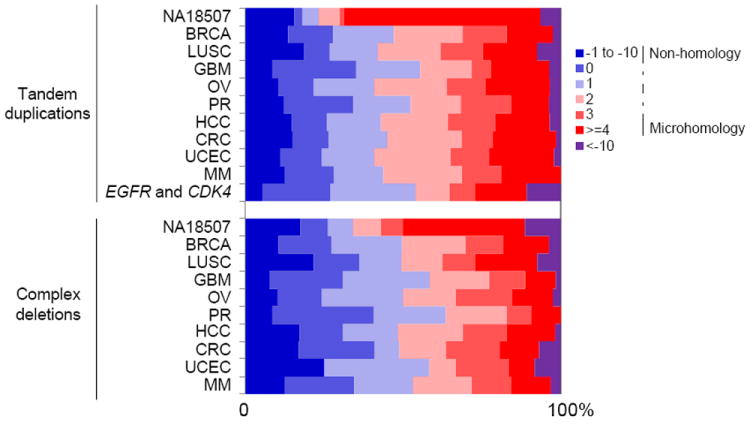
Homologies in base pairs are shown for each breakpoint as a positive number. A blunt end has a homology of 0 bp. Small insertions with unknown source are shown as negative numbers. Somatic tandem duplications and complex tandem duplications that are responsible for EGFR and CDK4 amplifications in GBM patients are shown in a separate category.
See also Figure S3.
Reconstruction of complex rearrangements in GBM patients
We are particularly interested in GBM genomes since several recurrent copy number alterations we found are known to play an important role in tumorigenesis (The Cancer Genome Atlas Research Network, 2008). In our 16 GBM whole-genome datasets, 15 genomes have loss of heterozygosity (LOH) of chromosome 10, 12 have homozygous deletions of CDKN2A/B, 14 have EGFR amplifications, and 5 have CDK4 amplifications (Table 1). We tested 26 SVs involving loss or gain of CDKN2A/B, EGFR and CDK4 and validated 25 as somatic SVs by PCR (Table S3).
Table 1.
CDKN2A/B loss, chromosome 10 loss, and EGFR, CDK4 amplification in GBM samples.
| ID |
CDKN2A/B event types
|
EGFR
|
CDK4
|
||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| AL | Del/Del_ins
|
CP | Event types
|
Allelic amplification | Copy ratio | Event types
|
Allelic amplification | Copy ratio | |||||||||
| NHEJ | MMEJ | AG | Del | Del_ins | Dup | CDup | CP | AG | CDup | CP | |||||||
| GBM0145 | 1 | 1 | 1 | 1 | 1 | 1 | bi | 66.9 | 1 | mono | 6.0 | ||||||
| GBM0185 | 1 | 1 | 1 | 1 | 1 | bi | 24.8 | 1.0 | |||||||||
| GBM0188 | 1 | 1 | 1 | mono | 1.3 | 1.2 | |||||||||||
| GBM0208 | 1 | 1 | 1 | 1 | mono | 19.1 | 1.1 | ||||||||||
| GBM0214 | 1 | 1 | 1 | 1 | 1 | mono | 8.9 | 1 | mono | 1.5* | |||||||
| GBM0152 | 1 | mono | 61.8 | 1 | mono | 38.8 | |||||||||||
| GBM0155 | 1 | 1 | 1 | 3 | bi | 5.7 | 0.9 | ||||||||||
| GBM0648 | 1 | 1 | 1 | mono | 1.5* | 1.2 | |||||||||||
| GBM0786 | 1 | 1 | 1 | 1 | bi | 70.0 | 1.1 | ||||||||||
| GBM0877 | 1 | 1 | 1 | 1 | mono | 17.9 | 1 | mono | 1.3 | ||||||||
| GBM0881 & | 1 | mono | 4.2 | 1.0 | |||||||||||||
| GBM1086 | 2 | 1.2 | 1.3 | ||||||||||||||
| GBM1401 | 1 § | 1 | 1 | bi | 25.5 | 1.5* | |||||||||||
| GBM1438 | 1 | bi | 1.4 | 1 | mono | 11.6 | |||||||||||
| GBM1454 | 1.1* | 0.8 | |||||||||||||||
| GBM1459 | 1 | 1 | 1 | 1 | 1 | mono | 33.3 | 0.8 | |||||||||
AL: arm level loss; Del: deletion; Del_ins: deletion with insertion in the breakpoints; CP: complex events; AG: arm level gain; Dup: tandem duplication, CDup: complex tandem duplication.
denotes the only sample without chromosome 10 loss.
denotes a deletion that is also involved in a copy-neutral loss of heterozygosity event which caused the loss of both copies of CDKN2A/B.
denotes inconsistent copy ratio estimates between the read-depth and Affymetrix SNP Array 6.0 (http://tcga-data.nci.nih.gov/tcga/) data. Samples with IDs in bold are the ones in which experimental validation of CDKN2A/B loss, EGFR and CDK4 amplifications has been performed. Copy loss/gain were predicted jointly from copy ratios and allele ratios of germline heterozygous SNPs. See also Supplemental Experimental Procedures and Figure S4.
Although the copy number changes in these regions have been documented previously based on array data, the exact configuration of the rearrangements and the mechanisms underlying those events are largely unknown. Using Meerkat, we not only ascertained the types of events that generated the observed configuration but also gained insights into the mechanisms by analyzing sequence homology at the breakpoints. It is interesting to note that in both CDKN2A/B loss and EGFR gain, most tumor genomes have both arm-level and focal loss/gain (Table 1). Out of twelve patients harboring CDKN2A/B loss, six have arm-level loss and focal deletions (Figure 4A), two have two independent focal deletions (Figure 4B), and four have complex rearrangements (Figure 4C). SVs responsible for CDKN2A/B loss in other patients are displayed in Figure S5. Most (11 of 13) of the focal deletions were generated by NHEJ, which suggests these alterations are mostly formed through erroneous repair of DNA double-strand breaks.
Figure 4. CDKN2A/B losses in GBM patients.
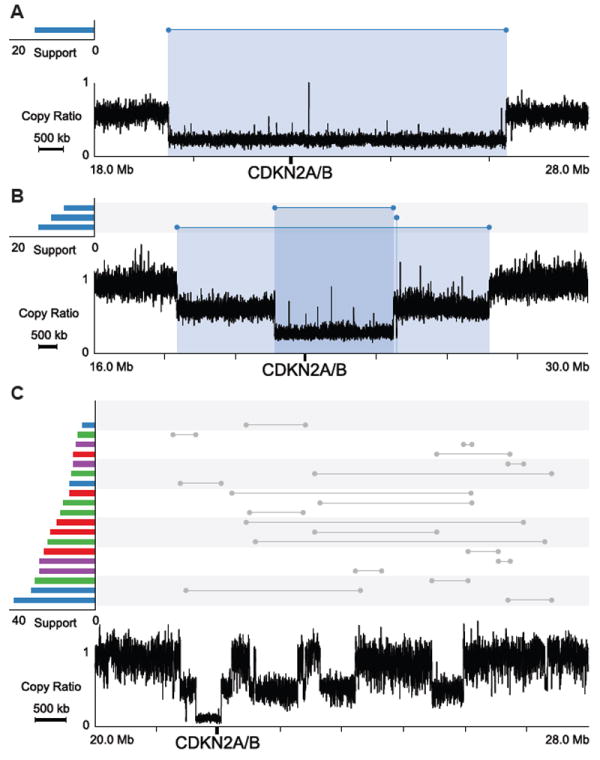
Profiles in the lower part of the plots show copy ratios (tumor vs. matched normal). Above the copy ratio profiles, predicted somatic SVs are represented by lines with the breakpoints indicated by dots. SVs corresponding to a notable copy number change are colored, with the color indicating the orientation of the breakpoints. A red cluster typically suggests a tandem duplication; a blue cluster typically suggests a deletion. The number of supporting discordant read pairs for each SV is shown on the left using the same color-coding. The copy-loss regions are highlighted with blue shades.
(A) GBM0208, an arm level loss and a focal deletion.
(B) GBM1086, two focal deletions.
(C) GBM0648, complex rearrangements.
See also Figure S5.
In the 14 GBM genomes with EGFR amplification, most have more than one event contributing to the copy gain: 9 with a chromosome arm gain, 8 with tandem duplication(s), 1 with a complex tandem duplication, and 8 with complex events. For GBM0155 (Figure 5A), three tandem duplications (1.6 Mb, 983 kb and 28 kb) involving EGFR were identified. In GBM0145 (Figure 5B), EGFR is amplified by a 789 kb tandem duplication, but a complex deletion was also found (deletion of a 417 kb fragment with insertion of a 50 kb fragment in the breakpoint). From the copy ratios, it appears that this deletion only affects a subset of the tandem-duplicated copies, suggesting that it happened during the tandem duplication. The complex deletion may not have been generated by FoSTeS/MMBIR since no microhomology was found at the breakpoints. Similarly, GBM0214 (Figure 5C) contains a 59 kb tandem duplication and multiple subsequent rearrangements of various types in the EGFR region that are exceedingly difficult to disentangle. SVs responsible for EGFR amplifications in other patients are displayed in Figure S6.
Figure 5. EGFR amplifications in GBM patients.
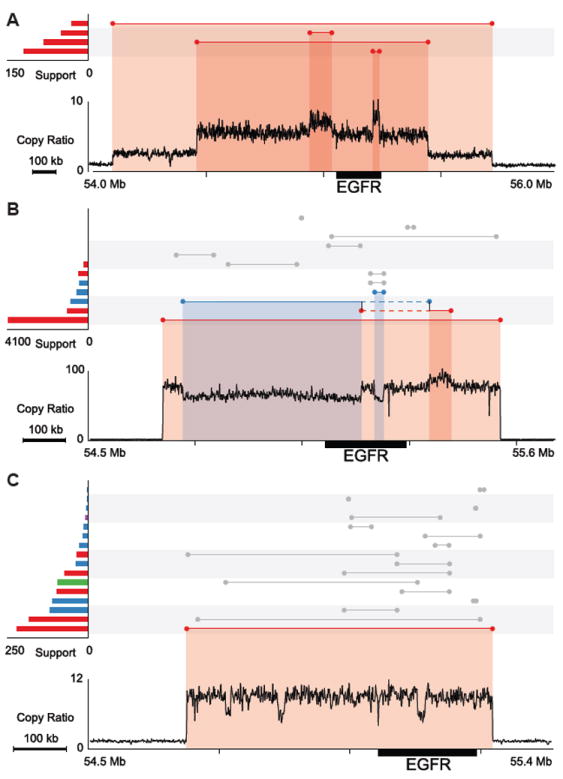
SVs and copy ratios are displayed as described in Figure 4. The copy-loss and gain regions are highlighted with blue and red shades, respectively.
(A) GBM0155, three tandem duplications.
(B) GBM0145, one tandem duplication and a deletion with insertion at the breakpoints. Two vertical black lines connecting two single events denote a complex deletion, which was predicted by combining two discordant read pair clusters. The solid blue and red lines represent segments that have been deleted and duplicated. The dashed lines denote a region of no copy number change.
(C) GBM0214, one tandem duplication and complex rearrangements.
See also Figure S6.
In GBM0152, a 923 kb fragment covering EGFR (Figure 6A) is merged with two fragments from chromosome 12 (a 5,620 bp fragment and a 286 bp fragment in inverted orientation) and then tandem-duplicated (Figure 6B). Moreover, nearly 40 regions on chromosome 12 (including CDK4) are coalesced in an elaborate complex series of events with the copy ratios of various fragments at approximately 40-fold, 75-fold and 110-fold gain (Figure 6C). In this case, the three prominent copy ratios and all the amplified segments being connected by discordant read pair clusters make it possible to disentangle the underlying events (Figure 6D). Based on the pattern of segments connected by discordant read pairs and the corresponding copy ratio for each segment (Figure 6E), we present one possible co-amplified unit (Figure 6F) that is consistent with all the observed copy ratios and discordant read pairs while other compatible configurations are also possible. This single unit (Figure 6F), composed of dozens of fragments, was tandem-duplicated to reach a copy ratio of about 40.
Figure 6. Amplifications of EGFR and chromosome 12 in GBM0152.

(A) Copy ratio and rearrangements involving EGFR. Colored boxes with arrows denote the amplified regions and their orientations.
(B) Diagram of the resulting rearrangements. Three segments of DNA from chromosome 7 and chromosome 12 are merged into one and tandem-duplicated.
(C) Copy ratio and somatic rearrangements on chromosome 12. The three grey dashed lines in copy ratio panel (bottom of this figure) denote copy ratios of 40, 75 and 110. The rearrangements marked by “a”, “b”, “c” and “d” have approximately twice as many supporting discordant read pairs as other rearrangements. These rearrangements are also marked in (D), (E) and (F).
(D) The 14 Mb region of chromosome 12 shown in (C) was segmented according to copy ratios. Each segment was re-scaled and assigned an identifier from 0 to 40. The rearrangement marked with a black arrow is not involved in the amplifications of other segments on chromosome 12, but is involved in the amplification of EGFR on chromosome 7 as displayed in (A).
(E) Each segment in (D) is shown as a numbered node connected by arrows and lines. Black arrows connected by lines denote concordant connections. Ratios of segments are denoted by the number of dots above the segment IDs inside each node. Non-amplified segments are not shown. The connection marked with “e” (also marked in (F)) is a germline deletion.
(F) This diagram shows one possible solution on how segments are connected. Segments with a white background are in an inverted orientation. Colored dashed lines denote discordant connections while black lines denote concordant connections.
While the complexity of the rearrangements in Figure 6C is reminiscent of chromothripsis (Stephens et al., 2011), it is unlikely to be the case here; instead, it is likely to have been generated by a replication-based mechanism. Chromosomes that have undergone chromothripsis have copy numbers that oscillate between two levels. The complex tandem duplications in our example have several distinct copy numbers, indicating that they are more likely to be a result of a series of replication-based template-switching events. A single unit of amplification contains multiple instances of the segment (junctions 2-4, 12-14, 14-20 and 18-29 in Figure 6F)—it is unlikely that at least two copies of chromosome 12 were shattered at the same place and joined with the same segments at the exact breakpoints after a “one-off” catastrophic event. Therefore, we suspect that certain junctions (such as 14-20 in Figure 6F) were formed first by a template switching event, and then the resulting fragment served as an additional template in subsequent switching events to form more rearranged fragments.
Most of the GBM patients examined in this study have both EGFR gain, most likely through a replication-based mechanism, and CDKN2A/B loss, mostly by NHEJ repair of DNA double-strand breaks. Furthermore, in all tumor types, a significant portion of the focal deletions was generated by FoSTeS/MMBIR in addition to the dominant NHEJ and alt-EJ mechanisms. This suggests that cancer genomes are likely to have more than one driving force (e.g., replication error and erroneous repair of DNA double-strand breaks) acting together in the same individual to initiate different types of rearrangements in different parts of the genome and provide advantageous mutations for cancer progression.
Double minute chromosomes and complex rearrangements
Double minute chromosomes (DMs) are extra circular chromosomal DNA with neither centromeres nor telomeres that can duplicate autonomously. They have been found in a variety of solid tumors as well as in leukemia (Thomas et al., 2004). EGFR has been shown to be amplified by DMs in glioma and glioblastoma (Vogt et al., 2004). All of the EGFR amplifications we identified above are likely to be DMs since the amplified fragments in DM loop structures would be predicted as tandem duplications. With paired-end sequencing, we are not able to determine if the amplifications were tandem duplications on the same chromosome or circularized as double minute chromosomes. In one patient, we identified a deletion whose breakpoints matched the tandem duplication (Figure S6G), suggesting an excision of the DNA fragment followed by circularization of that fragment, similar to the excisions of amplified DM fragments reported based on FISH (Storlazzi et al., 2010; Van Roy et al., 2006).
It was previously shown that most DMs that amplified EGFR in gliomas are a single fragment circularized by a microhomology-based mechanism (Vogt et al., 2004), likely FoSTeS/MMBIR (microhomology was detected at 6 out of 7 breakpoints), and subsequently amplified by recombination to join multiple fragments into one larger circular DNA or by rolling circle replication. The copy number of the amplified region we observed is the average across many tumor cells; each cell or a sub-population of cells could have a different number of the amplified unit. Most of the initial circularizations of DMs in neuroblastoma and small cell lung carcinoma (Storlazzi et al., 2010) were also generated by FoSTeS/MMBIR, with 23 out of 32 breakpoints showing either microhomologies or large insertions. Similar to EGFR amplifications in glioma, most of the EGFR and CDK4 amplifications in GBM patients reported here can be explained by an initial circularization of a single DNA fragment resulting from replication error; others can be explained by the circularization of multiple DNA fragments. However, we observed more breakpoints without homology than with microhomology (Figure 3), suggesting that some of these initial circularizations were generated by FoSTeS/MMBIR but more were formed by non-homology-based replicative mechanisms.
Discussion
We have reported a comprehensive catalog of somatic rearrangements in cancer, revealing the diversity in the types of somatic SVs and the mechanisms that generate them across different tumor types and individuals. Given the disruptive nature of some genomic rearrangements and their role in promoting cancer progression, precise characterization of the rearrangements and their mechanisms is crucial. While much of the work on structural variations so far has focused on their impact on genes and the mutations occurring in inter-genic regions have often been considered “passenger” events, recent work by the ENCODE consortium (The ENCODE Project Consortium, 2012) has shown that the fraction of the non-coding genome that plays a role in gene regulation is much larger than previously thought. This suggests that, in addition to a direct impact on protein structure (e.g., by fusion transcripts), other, perhaps more subtle types of misregulations may result from rearrangements that involve non-coding regions (e.g., disruption of enhancer activity or binding of a non-coding RNA). Thus, it is advantageous to know not simply whether a genomic region is amplified or not but also where the amplified segments are located. At some point in the future, improved DNA sequencing technology will accommodate much longer reads (on the order of kilobases or longer) to make reconstruction of structural alterations easier; in the meantime, innovative approaches such as the one we report here are needed to dissect the evolution of the cancer genome based on short-read data.
It is important to note that assigning mechanisms to events based on sequence features at the breakpoints is an inexact process. For instance, we found that NHEJ and alt-EJ contribute the most to focal deletions and translocations. The events generated by alt-EJ tend to have more microhomology than by NHEJ (Bennardo et al., 2008; McVey and Lee, 2008), but there is no consensus cutoff for distinguishing NHEJ and alt-EJ (Arlt et al., 2012), as both mechanisms can generate rearrangements with blunt ends, microhomology, or small insertions at the breakpoints. In addition, events generated by FoSTeS/MMBIR frequently have microhomology at the breakpoints. These ambiguities in the thresholds, however, are unlikely to materially affect our comparisons of germline and somatic events or comparisons across tumor types, since we apply the same criteria to all events. We also note that SVs generated by NAHR typically require at least 100bp of homology (Liskay et al., 1987; Waldman and Liskay, 1988); therefore we could not identify these SVs generate by NAHR based on the short (≤100 bp) reads we have.
We found more microhomology-based mechanisms (alt-EJ and FoSTeS/MMBIR) for germline SVs (e.g., deletions, tandem duplications and complex events) than for somatic SVs, suggesting that those mechanisms may be suppressed in cancer cells. It is also possible that DNA breakage and replication fork stalling are more frequent in cancer cells, and a non-homology-based mechanism is the easiest way to repair. A similar trend was observed in pathogenic germline rearrangements (Chiang et al., 2012), with less microhomology at the breakpoints of pathogenic balanced translocations and inversions.
The driving forces behind large-scale genomic rearrangements have been less well characterized than those for single nucleotide alterations. In addition to the chromosome arm-level alterations (induced, e.g., by mutations in genes that maintain genome stability (Solomon et al., 2011)), focal losses and gains of CDKN2A/B and EGFR in GBM patients involved distinct mechanisms acting together on the same locus in the same genome. Why multiple mechanisms act on certain regions of the genome repeatedly remains unclear. The rearrangements we observe are a snapshot of the combined effect of bias in formation and selection of the alternations in cancer genomes. It is possible that specific regions are biased toward the formation of genomic rearrangement, driven by their genomic and epigenetic features as well as their regulatory function (De and Michor, 2011a; De and Michor, 2011b; Fudenberg et al., 2011). For example, the recruitment of specific proteins induced by androgen can trigger DNA double-strand breaks which result in TMPRSS2-ERG gene fusion in prostate cancer (Haffner et al., 2010). It is also possible that alterations occur randomly for the most part and the fitness of the cell increases with certain alterations. Further studies are needed to better understand the relationships between the driving forces and their targets and how each step of the alteration confers growth advantage to the selected clones.
Experimental Procedures
Identification of SVs using the Meerkat algorithm
In short, we predict SVs based on discordant read pairs and refine the precise breakpoints by looking for the reads that cover the SV breakpoint junctions. Mutational mechanisms are predicted based on homology and sequencing features at the breakpoints (Figure 1E) which is adapted from Kidd et al. 2010. See Supplemental Experimental Procedures for more details. The Meerkat package is available at http://compbio.med.harvard.edu/Meerkat/.
Experimental validation of SVs
A set of SVs predicted by Meerkat was validated by PCR. PCR primers were designed using Primer3 (http://frodo.wi.mit.edu/primer3/) to amplify the predicted SV breakpoints. The primer pairs were designed to produce a product under 10 kb for SVs of NA18507 or about 200 bp long for somatic SVs predicted in cancer samples. For NA18507, PCRs were run on genomic DNA. For somatic SVs, PCRs were run on whole-genome amplified DNA of both tumor and matched normal samples to ensure that the somatic SVs were found only in tumor but not in the matched normal sample. Whole-genome DNA amplification was performed with Sigma/Rubicon’s WGA kit per manufacturer’s instructions. For NA18507, five to ten SVs were randomly selected from each event type and an SV was considered validated if the predicted product across breakpoints was detectable in genomic DNA. For deletions with a large insertion in the breakpoints, large insertions, deletions with an inversion in the breakpoints, three PCRs were performed. Two PCRs were aimed at amplifying across the two breakpoints and the third PCR was targeted to amplify the entire insertion or inversion event if the insertion or inversion was <10 kb. An event was considered validated if all three PCRs yielded products of expected sizes. If a PCR validation was not successful, two more pairs of primers were attempted.
Supplementary Material
Highlights.
A cross-tumor analysis of whole-genome sequencing data from 140 patients.
An algorithm that identifies complex deletions from short-read data.
A comprehensive spectrum of structural variation types and mechanisms reported.
Main driving forces of cancer genome rearrangements proposed.
Acknowledgments
This work was supported by grants from NIH (RC1HG005482 to P.J.P., U24CA144025 to R.K. and U24CA143845 to L.C.). We thank The Cancer Genome Atlas project for providing the whole-genome data, the Information Technology Department at Harvard Medical School for providing computational resources, and Michael Talkowski and members of the Park laboratory for a careful reading of the manuscript.
Footnotes
Supplemental Information
Supplemental Information includes Supplemental Experimental Procedures, six figures and five tables.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Arlt MF, Rajendran S, Birkeland SR, Wilson TE, Glover TW. De novo CNV formation in mouse embryonic stem cells occurs in the absence of Xrcc4-dependent nonhomologous end joining. PLoS Genetics. 2012;8:e1002981. doi: 10.1371/journal.pgen.1002981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerji S, Cibulskis K, Rangel-Escareno C, Brown KK, Carter SL, Frederick AM, Lawrence MS, Sivachenko AY, Sougnez C, Zou L, et al. Sequence analysis of mutations and translocations across breast cancer subtypes. Nature. 2012;486:405–409. doi: 10.1038/nature11154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bass AJ, Lawrence MS, Brace LE, Ramos AH, Drier Y, Cibulskis K, Sougnez C, Voet D, Saksena G, Sivachenko A, et al. Genomic sequencing of colorectal adenocarcinomas identifies a recurrent VTI1A-TCF7L2 fusion. Nature Genetics. 2011;43:964–968. doi: 10.1038/ng.936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennardo N, Cheng A, Huang N, Stark JM. Alternative-NHEJ is a mechanistically distinct pathway of mammalian chromosome break repair. PLoS Genetics. 2008;4:e1000110. doi: 10.1371/journal.pgen.1000110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger MF, Lawrence MS, Demichelis F, Drier Y, Cibulskis K, Sivachenko AY, Sboner A, Esgueva R, Pflueger D, Sougnez C, et al. The genomic complexity of primary human prostate cancer. Nature. 2011;470:214–220. doi: 10.1038/nature09744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell PJ, Yachida S, Mudie LJ, Stephens PJ, Pleasance ED, Stebbings LA, Morsberger LA, Latimer C, McLaren S, Lin M-L, et al. The patterns and dynamics of genomic instability in metastatic pancreatic cancer. Nature. 2010;467:1109–1113. doi: 10.1038/nature09460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman MA, Lawrence MS, Keats JJ, Cibulskis K, Sougnez C, Schinzel AC, Harview CL, Brunet J-P, Ahmann GJ, Adli M, et al. Initial genome sequencing and analysis of multiple myeloma. Nature. 2011;471:467–472. doi: 10.1038/nature09837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang C, Jacobsen JC, Ernst C, Hanscom C, Heilbut A, Blumenthal I, Mills RE, Kirby A, Lindgren AM, Rudiger SR, et al. Complex reorganization and predominant non-homologous repair following chromosomal breakage in karyotypically balanced germline rearrangements and transgenic integration. Nature Genetics. 2012;44:390–397. doi: 10.1038/ng.2202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad DF, Bird C, Blackburne B, Lindsay S, Mamanova L, Lee C, Turner DJ, Hurles ME. Mutation spectrum revealed by breakpoint sequencing of human germline CNVs. Nature Genetics. 2010;42:385–391. doi: 10.1038/ng.564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De S, Michor F. DNA replication timing and long-range DNA interactions predict mutational landscapes of cancer genomes. Nature Biotechnology. 2011a;29:1103–1108. doi: 10.1038/nbt.2030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De S, Michor F. DNA secondary structures and epigenetic determinants of cancer genome evolution. Nature Structural & Molecular Biology. 2011b;18:950–955. doi: 10.1038/nsmb.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edlund T, Normark S. Recombination between short DNA homologies causes tandem duplication. Nature. 1981;292:269–271. doi: 10.1038/292269a0. [DOI] [PubMed] [Google Scholar]
- Fudenberg G, Getz G, Meyerson M, Mirny LA. High order chromatin architecture shapes the landscape of chromosomal alterations in cancer. Nature Biotechnology. 2011;29:1109–1113. doi: 10.1038/nbt.2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenman C, Stephens P, Smith R, Dalgliesh GL, Hunter C, Bignell G, Davies H, Teague J, Butler A, Stevens C, et al. Patterns of somatic mutation in human cancer genomes. Nature. 2007;446:153–158. doi: 10.1038/nature05610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu W, Zhang F, Lupski JR. Mechanisms for human genomic rearrangements. PathoGenetics. 2008;1:4. doi: 10.1186/1755-8417-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haffner MC, Aryee MJ, Toubaji A, Esopi DM, Albadine R, Gurel B, Isaacs WB, Bova GS, Liu W, Xu J, et al. Androgen-induced TOP2B-mediated double-strand breaks and prostate cancer gene rearrangements. Nature Genetics. 2010;42:668–675. doi: 10.1038/ng.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hastings P, Ira G, Lupski JR. A microhomology-mediated break-induced replication model for the origin of human copy number variation. PLoS Genetics. 2009a;5:e1000327. doi: 10.1371/journal.pgen.1000327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hastings PJ, Lupski JR, Rosenberg SM, Ira G. Mechanisms of change in gene copy number. Nature Reviews Genetics. 2009b;10:551–564. doi: 10.1038/nrg2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haviv-Chesner A, Kobayashi Y, Gabriel A, Kupiec M. Capture of linear fragments at a double-strand break in yeast. Nucleic Acids Research. 2007;35:5192–5202. doi: 10.1093/nar/gkm521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillmer AM, Yao F, Inaki K, Lee WH, Ariyaratne PN, Teo ASM, Woo XY, Zhang Z, Zhao H, Ukil L, et al. Comprehensive long-span paired-end-tag mapping reveals characteristic patterns of structural variations in epithelial cancer genomes. Genome Research. 2011;21:665–675. doi: 10.1101/gr.113555.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeijmakers JHJ. Genome maintenance mechanisms for preventing cancer. Nature. 2001;411:366–374. doi: 10.1038/35077232. [DOI] [PubMed] [Google Scholar]
- Iafrate AJ, Feuk L, Rivera MN, Listewnik ML, Donahoe PK, Qi Y, Scherer SW, Lee C. Detection of large-scale variation in the human genome. Nature Genetics. 2004;36:949–951. doi: 10.1038/ng1416. [DOI] [PubMed] [Google Scholar]
- Kidd JM, Graves T, Newman TL, Fulton R, Hayden HS, Malig M, Kallicki J, Kaul R, Wilson RK, Eichler EE. A human genome structural variation sequencing resource reveals insights into mutational mechanisms. Cell. 2010;143:837–847. doi: 10.1016/j.cell.2010.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee E, Iskow R, Yang L, Gokcumen O, Haseley P, Luquette LJ, Lohr JG, Harris CC, Ding L, Wilson RK, et al. Landscape of somatic retrotransposition in human cancers. Science. 2012 doi: 10.1126/science.1222077. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JA, Carvalho C, Lupski JR. A DNA replication mechanism for generating nonrecurrent rearrangements associated with genomic disorders. Cell. 2007;131:1235–1247. doi: 10.1016/j.cell.2007.11.037. [DOI] [PubMed] [Google Scholar]
- Lee W, Jiang Z, Liu J, Haverty PM, Guan Y, Stinson J, Yue P, Zhang Y, Pant KP, Bhatt D, et al. The mutation spectrum revealed by paired genome sequences from a lung cancer patient. Nature. 2010;465:473–477. doi: 10.1038/nature09004. [DOI] [PubMed] [Google Scholar]
- Liskay RM, Letsou A, Stachelek JL. Homology requirement for efficient gene conversion between duplicated chromosomal sequences in mammalian cells. Genetics. 1987;115:161–167. doi: 10.1093/genetics/115.1.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P, Erez A, Nagamani Sandesh CS, Dhar Shweta U, Kolodziejska Katarzyna E, Dharmadhikari Avinash V, Cooper ML, Wiszniewska J, Zhang F, Withers Marjorie A, et al. Chromosome catastrophes involve replication mechanisms generating complex genomic rearrangements. Cell. 2011;146:889–903. doi: 10.1016/j.cell.2011.07.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahaney BL, Meek K, Lees-Miller SP. Repair of ionizing radiation-induced DNA double strand breaks by non-homologous end-joining. The Biochemical journal. 2009;417:639. doi: 10.1042/BJ20080413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McVey M, Lee SE. MMEJ repair of double-strand breaks (director’s cut): deleted sequences and alternative endings. Trends in Genetics. 2008;24:529–538. doi: 10.1016/j.tig.2008.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills RE, Walter K, Stewart C, Handsaker RE, Chen K, Alkan C, Abyzov A, Yoon SC, Ye K, Cheetham RK, et al. Mapping copy number variation by population-scale genome sequencing. Nature. 2011;470:59–65. doi: 10.1038/nature09708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pleasance ED, Cheetham RK, Stephens PJ, McBride DJ, Humphray SJ, Greenman CD, Varela I, Lin M-L, Ordóñez GR, Bignell GR, et al. A comprehensive catalogue of somatic mutations from a human cancer genome. Nature. 2009a;463:191–196. doi: 10.1038/nature08658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pleasance ED, Stephens PJ, O’Meara S, McBride DJ, Meynert A, Jones D, Lin M-L, Beare D, Lau KW, Greenman C, et al. A small-cell lung cancer genome with complex signatures of tobacco exposure. Nature. 2009b;463:184–190. doi: 10.1038/nature08629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomon DA, Kim T, Diaz-Martinez LA, Fair J, Elkahloun AG, Harris BT, Toretsky JA, Rosenberg SA, Shukla N, Ladanyi M. Mutational inactivation of STAG2 causes aneuploidy in human cancer. Science. 2011;333:1039–1043. doi: 10.1126/science.1203619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens PJ, Greenman CD, Fu B, Yang F, Bignell GR, Mudie LJ, Pleasance ED, Lau KW, Beare D, Stebbings LA, et al. Massive genomic tearrangement acquired in a single catastrophic event during cancer development. Cell. 2011;144:27–40. doi: 10.1016/j.cell.2010.11.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens PJ, McBride DJ, Lin M-L, Varela I, Pleasance ED, Simpson JT, Stebbings LA, Leroy C, Edkins S, Mudie LJ, et al. Complex landscapes of somatic rearrangement in human breast cancer genomes. Nature. 2009;462:1005–1010. doi: 10.1038/nature08645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storlazzi CT, Lonoce A, Guastadisegni MC, Trombetta D, D’Addabbo P, Daniele G, L’Abbate A, Macchia G, Surace C, Kok K. Gene amplification as double minutes or homogeneously staining regions in solid tumors: Origin and structure. Genome Research. 2010;20:1198–1206. doi: 10.1101/gr.106252.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung WK, Zheng H, Li S, Chen R, Liu X, Li Y, Lee NP, Lee WH, Ariyaratne PN, Tennakoon C. Genome-wide survey of recurrent HBV integration in hepatocellular carcinoma. Nature Genetics. 2012;44:765–769. doi: 10.1038/ng.2295. [DOI] [PubMed] [Google Scholar]
- The Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–1068. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Cancer Genome Atlas Research Network. Comprehensive molecular portraits of human breast tumours. Nature. 2012a;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Cancer Genome Atlas Research Network. Comprehensive genomic characterization of squamous cell lung cancers. Nature. 2012b;489:519–525. doi: 10.1038/nature11404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas L, Stamberg J, Gojo I, Ning Y, Rapoport AP. Double minute chromosomes in monoblastic (M5) and myeloblastic (M2) acute myeloid leukemia: two case reports and a review of literature. American journal of hematology. 2004;77:55–61. doi: 10.1002/ajh.20151. [DOI] [PubMed] [Google Scholar]
- Van Roy N, Vandesompele J, Menten B, Nilsson H, De Smet E, Rocchi M, De Paepe A, Påhlman S, Speleman F. Translocation–excision–deletion–amplification mechanism leading to nonsyntenic coamplification of MYC and ATBF1. Genes, Chromosomes and Cancer. 2006;45:107–117. doi: 10.1002/gcc.20272. [DOI] [PubMed] [Google Scholar]
- Venkitaraman AR. Cancer susceptibility and the functions of BRCA1 and BRCA2. Cell. 2002;108:171–182. doi: 10.1016/s0092-8674(02)00615-3. [DOI] [PubMed] [Google Scholar]
- Vogt N, Lefèvre SH, Apiou F, Dutrillaux AM, Cör A, Leuraud P, Poupon MF, Dutrillaux B, Debatisse M, Malfoy B. Molecular structure of double-minute chromosomes bearing amplified copies of the epidermal growth factor receptor gene in gliomas. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:11368. doi: 10.1073/pnas.0402979101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldman A, Liskay R. Dependence of intrachromosomal recombination in mammalian cells on uninterrupted homology. Molecular and cellular biology. 1988;8:5350–5357. doi: 10.1128/mcb.8.12.5350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Khajavi M, Connolly AM, Towne CF, Batish SD, Lupski JR. The DNA replication FoSTeS/MMBIR mechanism can generate genomic, genic and exonic complex rearrangements in humans. Nature Genetics. 2009;41:849–853. doi: 10.1038/ng.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.