

Abstract
Cellular senescence is a state of irreversible cell cycle arrest that has been documented to both suppress cancer and promote aging. Although not well understood, extensive nuclear changes, including the remodeling of chromatin, take place as cells become senescent. In this issue, Ivanov et al. (2013. J. Cell Biol. http://dx.doi.org/jcb.201212110) report that chromatin fragments are released from the nuclei of senescent cells and are subsequently targeted for processing through the autophagy/lysosomal pathway.
More than 50 years ago, Leonard Hayflick and Paul Moorhead observed that fibroblasts from healthy human donors had a finite proliferation capacity in cell culture. When these cells reached their “Hayflick limit” they became irreversibly arrested, or “replicatively senescent” (Hayflick and Moorhead, 1961). A similar irreversible cell cycle arrest, now designated as “cellular senescence,” can be induced by a variety of stresses, including (but not limited to) activation of oncogenes, telomere maintenance defects, oxidative stress, and excessive DNA damage (Campisi and d’Adda di Fagagna, 2007; Collado et al., 2007; Kuilman et al., 2010).
Senescent cells are believed to be very stable over long periods of time, both in culture as well as in tissues (although evidence for the latter is very limited). In this issue, Ivanov et al. are challenging this dogma by providing evidence that senescence may be a more dynamic process than we have previously appreciated. They show that some senescent cells contain cytoplasmic chromatin fragments (CCFs) that apparently bud off from the nuclei. CCFs were found to be devoid of lamin A/C, but were highly positive for the histone variant γ-H2AX (a mark of DNA double-strand breaks) and enriched for the histone H3 lysine 27 trimethyl modification (H3K27me3; a mark of heterochromatin). They also observed down-regulation of lamin B1 and a striking loss of nuclear envelope integrity; features that they speculate may permit CCF formation and release. Once in the cytoplasm, CCFs were engaged by the autophagy pathway and ultimately proteolytically degraded in lysosomes. A decrease in the nuclear content of core histones was also noted, the obvious implication being that this is causally connected with the generation and destruction of CCFs. Taken together, these results represent a challenge to the classical view of cellular senescence as a single end point, and point to a more fluid situation where important changes take place after the initial cell cycle arrest, and in all likelihood progress and evolve over extended periods of time (Fig. 1).
Figure 1.
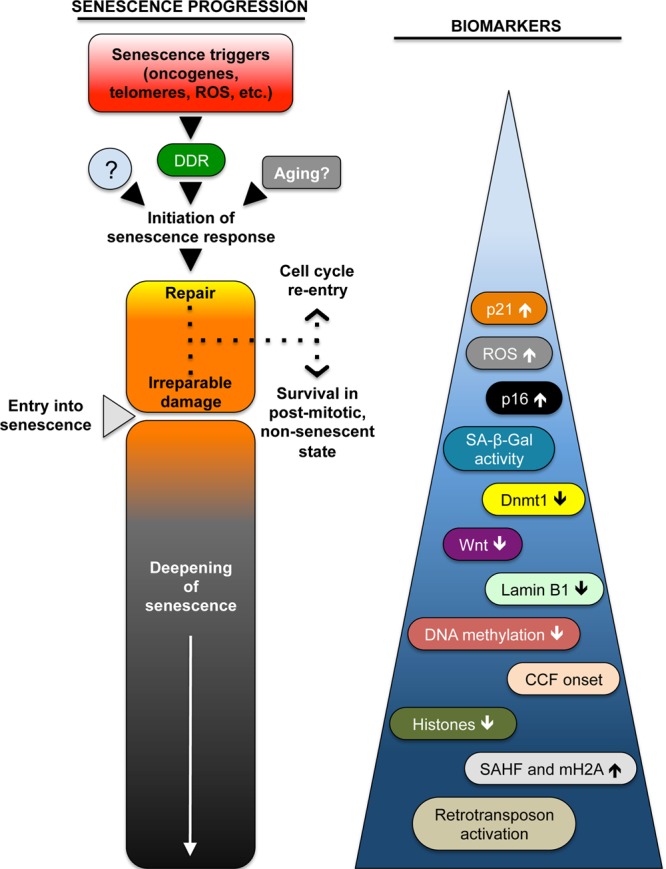
A schematic representation of the processes that lead to the establishment of cellular senescence. The progression of senescence has been separated into several components: (1) triggering events; (2) initiation of the senescence response; (3) entry into senescence; and (4) a further deepening of senescence phenotypes. Stages 2 and 3 can be separated by a period of attempted repair, which may result in recovery and survival in a healthy postmitotic state, or even resumption of cell proliferation. Entry into senescence is likely the result of the acquisition of irreparable damage, followed by an extended period during which additional degenerative changes can take place, evolve, and accumulate. On the right are illustrated some of the many molecular phenotypes, or biomarkers, that have been associated with cellular senescence. This listing is not meant to be comprehensive and similarly, the order is not meant to imply the chronological acquisition of these features. The construction of such a timeline we believe constitutes an important challenge for the field going forward. DDR, DNA damage response.
The concepts of “deepening” and “late” senescence have been suggested by a number of previous studies. γ-H2AX–positive DNA damage foci become very abundant as cells approach and enter senescence, but greatly diminish as the cultures are maintained for extended periods of time (Chen and Ozanne, 2006). Passos et al. (2010) proposed that a persistent DNA damage response triggers continued production of reactive oxygen species, forming a dynamic feedback loop that actively maintains a deep senescent state. De Cecco et al. (2013) reported that after cells become fully senescent by all conventional criteria, the expression of the long interspersed nuclear element (LINE) retrotransposon L1 increased dramatically during extended culture, culminating in active transposition. This is consistent with previous observations that Alu retrotransposon expression also increases in senescent cells (Wang et al., 2011). Retrotransposon transcripts are assembled in the cytoplasm with reverse transcriptase and integrase (along other essential proteins) into ribonucleoprotein particles that subsequently have to enter the nucleus in order to insert into new genomic locations. The loss of nuclear integrity observed by Ivanov et al. (2013) could be one explanation why deeply senescent cells are supportive of retrotransposition.
Depletion of core histones, without concomitant loss of DNA, has been noted to accompany aging in yeast and nematodes (Feser et al., 2010; Ni et al., 2012), and a reduction in the biosynthesis of histones was reported in human fibroblasts during entry into senescence (O’Sullivan et al., 2010). Ivanov et al. (2013) observed that steady-state levels of all core histones progressively decreased as cells were maintained in a senescent state. As a consequence, one would anticipate a decompression of chromatin and a more “open” epigenome. This view is consistent with emerging literature from multiple species and model systems, suggesting that proper maintenance of heterochromatin has anti-aging effects (O’Sullivan and Karlseder, 2012).
The loss of histones in senescent cells is, however, complicated by the fact that total nuclear protein content actually increases (De Cecco et al., 2011). Histone loss during senescence is accompanied by recruitment of high mobility group (HMG) proteins to heterochromatic foci (Funayama et al., 2006; Narita et al., 2006). It is therefore likely that the chromatin of senescent cells undergoes a transition from being packed with histones to nonhistone proteins. It will be interesting to determine the identity of these nonhistone proteins in future studies.
One perplexing observation made by Ivanov et al. (2013) is that relatively few cells exhibit CCFs, even in fully senescent cultures. Furthermore, the number of CCFs formed is also quite low, usually less than two foci per cell. Hence, CCF formation alone would not appear to represent a robust biomarker for defining deeply senescent cells. The lysosomal processing of CCFs raises the interesting issue of whether these processes in any way reinforce other known senescence phenotypes. For example, one could ask whether the proteolytically cleaved products can serve as input material to promote the senescence-associated secretory phenotype (SASP; Coppé et al., 2008). It would also be of great interest to know the in vivo occurrence of CCFs, examining a variety of tissues, ages, or pathological states.
It is currently unclear whether the low frequency of CCFs may be explained by a dynamic steady state, where cells continuously produce and turn over CCFs (like a conveyor belt), or if CCF formation occurs only in a distinct subpopulation of senescent cells. However, given that CCFs appear to contain DNA as well as histones, neither alternative readily explains the relative stability of nuclear DNA levels in the face of significantly declining levels of core histones. It may be that histones can also be depleted by a CCF-independent process. Further characterization of the CCFs, their content, and turnover should shed light on these issues.
Notwithstanding these uncertainties, the work of Ivanov et al. (2013) presents a picture of very substantial loss of nuclear composition and integrity, especially if extrapolated over long periods of time. It is hard to reconcile these degenerative changes with the many historical observations of the apparent long-term stability and viability of senescent cells. Recent studies have suggested that senescent cells can be manipulated to regain proliferative capacity by modulating the interaction with the extracellular matrix (Choi et al., 2011), or by reprogramming using induced pluripotent stem cell (iPSC) technologies (Lapasset et al., 2011). At face value, one would surely expect the processes described by Ivanov et al. (2013) to have profoundly negative effects on cell survival, and make cell cycle reentry unlikely for cells that have entered deeper stages of senescence.
It will also be important to develop a coherent nomenclature for describing the apparently dynamic state of cellular senescence. The classical criterion of irreversible proliferation arrest thus likely represents only an early, or “shallow” step in the process. These shallow steps may not even be necessary, given a recent report that differentiated, postmitotic cells can acquire some features of senescence (Jurk et al., 2012). It has been speculated that “senescent after differentiation” (SAD) cells may significantly affect the development of a variety of age-related diseases in humans (Naylor et al., 2013).
In a more mechanistic vein, it will be critically important to define the molecular and cellular markers of a deeply senescent cell versus an early senescent cell. Furthermore, it will be necessary to determine the chronology, and most importantly functional relevance, of these steps during the “deepening” process. Such knowledge would address the possibility of turning back cells, at specific points during the progression of senescence, into potentially normally functioning cells. It will also be relevant to compare these features in vitro and in vivo. For example, if “deepening” is observed in vivo, do the markers of this process vary between tissues and organisms? Do they vary between oncogene-induced senescence and other forms? There are obviously many, many interesting questions to investigate, but Ivanov et al. (2013) have provided us with an intriguing first look into where these paths may take us.
Footnotes
Abbreviation used in this paper:
- CCF
- cytoplasmic chromatin fragment
References
- Campisi J., d’Adda di Fagagna F. 2007. Cellular senescence: when bad things happen to good cells. Nat. Rev. Mol. Cell Biol. 8:729–740 10.1038/nrm2233 [DOI] [PubMed] [Google Scholar]
- Chen J.H., Ozanne S.E. 2006. Deep senescent human fibroblasts show diminished DNA damage foci but retain checkpoint capacity to oxidative stress. FEBS Lett. 580:6669–6673 10.1016/j.febslet.2006.11.023 [DOI] [PubMed] [Google Scholar]
- Choi H.R., Cho K.A., Kang H.T., Lee J.B., Kaeberlein M., Suh Y., Chung I.K., Park S.C. 2011. Restoration of senescent human diploid fibroblasts by modulation of the extracellular matrix. Aging Cell. 10:148–157 10.1111/j.1474-9726.2010.00654.x [DOI] [PubMed] [Google Scholar]
- Collado M., Blasco M.A., Serrano M. 2007. Cellular senescence in cancer and aging. Cell. 130:223–233 10.1016/j.cell.2007.07.003 [DOI] [PubMed] [Google Scholar]
- Coppé J.P., Patil C.K., Rodier F., Sun Y., Muñoz D.P., Goldstein J., Nelson P.S., Desprez P.Y., Campisi J. 2008. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 6:2853–2868 10.1371/journal.pbio.0060301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Cecco M., Jeyapalan J., Zhao X., Tamamori-Adachi M., Sedivy J.M. 2011. Nuclear protein accumulation in cellular senescence and organismal aging revealed with a novel single-cell resolution fluorescence microscopy assay. Aging (Albany NY). 3:955–967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Cecco M., Criscione S.W., Peckham E.J., Hillenmeyer S., Hamm E.A., Manivannan J., Peterson A.L., Kreiling J.A., Neretti N., Sedivy J.M. 2013. Genomes of replicatively senescent cells undergo global epigenetic changes leading to gene silencing and activation of transposable elements. Aging Cell. 12:247–256 10.1111/acel.12047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feser J., Truong D., Das C., Carson J.J., Kieft J., Harkness T., Tyler J.K. 2010. Elevated histone expression promotes life span extension. Mol. Cell. 39:724–735 10.1016/j.molcel.2010.08.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funayama R., Saito M., Tanobe H., Ishikawa F. 2006. Loss of linker histone H1 in cellular senescence. J. Cell Biol. 175:869–880 10.1083/jcb.200604005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayflick L., Moorhead P.S. 1961. The serial cultivation of human diploid cell strains. Exp. Cell Res. 25:585–621 10.1016/0014-4827(61)90192-6 [DOI] [PubMed] [Google Scholar]
- Ivanov A., Pawlikowski J., Manoharan I., van Tuyn J., Nelson D., Rai T.S., Shah P., Hewitt G., Korolchuk V., Passos J.F., et al. 2013. Lysosome-mediated processing of chromatin in senescent cells. J. Cell Biol. 202:129–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurk D., Wang C., Miwa S., Maddick M., Korolchuk V., Tsolou A., Gonos E.S., Thrasivoulou C., Saffrey M.J., Cameron K., von Zglinicki T. 2012. Postmitotic neurons develop a p21-dependent senescence-like phenotype driven by a DNA damage response. Aging Cell. 11:996–1004 10.1111/j.1474-9726.2012.00870.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuilman T., Michaloglou C., Mooi W.J., Peeper D.S. 2010. The essence of senescence. Genes Dev. 24:2463–2479 10.1101/gad.1971610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapasset L., Milhavet O., Prieur A., Besnard E., Babled A., Ait-Hamou N., Leschik J., Pellestor F., Ramirez J.M., De Vos J., et al. 2011. Rejuvenating senescent and centenarian human cells by reprogramming through the pluripotent state. Genes Dev. 25:2248–2253 10.1101/gad.173922.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narita M., Narita M., Krizhanovsky V., Nuñez S., Chicas A., Hearn S.A., Myers M.P., Lowe S.W. 2006. A novel role for high-mobility group a proteins in cellular senescence and heterochromatin formation. Cell. 126:503–514 10.1016/j.cell.2006.05.052 [DOI] [PubMed] [Google Scholar]
- Naylor R.M., Baker D.J., van Deursen J.M. 2013. Senescent cells: a novel therapeutic target for aging and age-related diseases. Clin. Pharmacol. Ther. 93:105–116 10.1038/clpt.2012.193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni Z., Ebata A., Alipanahiramandi E., Lee S.S. 2012. Two SET domain containing genes link epigenetic changes and aging in Caenorhabditis elegans. Aging Cell. 11:315–325 10.1111/j.1474-9726.2011.00785.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Sullivan R.J., Karlseder J. 2012. The great unravelling: chromatin as a modulator of the aging process. Trends Biochem. Sci. 37:466–476 10.1016/j.tibs.2012.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Sullivan R.J., Kubicek S., Schreiber S.L., Karlseder J. 2010. Reduced histone biosynthesis and chromatin changes arising from a damage signal at telomeres. Nat. Struct. Mol. Biol. 17:1218–1225 10.1038/nsmb.1897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passos J.F., Nelson G., Wang C., Richter T., Simillion C., Proctor C.J., Miwa S., Olijslagers S., Hallinan J., Wipat A., et al. 2010. Feedback between p21 and reactive oxygen production is necessary for cell senescence. Mol. Syst. Biol. 6:347 10.1038/msb.2010.5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J., Geesman G.J., Hostikka S.L., Atallah M., Blackwell B., Lee E., Cook P.J., Pasaniuc B., Shariat G., Halperin E., et al. 2011. Inhibition of activated pericentromeric SINE/Alu repeat transcription in senescent human adult stem cells reinstates self-renewal. Cell Cycle. 10:3016–3030 10.4161/cc.10.17.17543 [DOI] [PMC free article] [PubMed] [Google Scholar]