

Abstract
Recently, we showed that administration of the angiotensin-converting enzyme inhibitor enalapril to aged rats attenuated muscle strength decline and mitigated apoptosis in the gastrocnemius muscle. The aim of the present study was to investigate possible mechanisms underlying the muscle-protective effects of enalapril. We also sought to discern the effects of enalapril mediated by nitric oxide (NO) from those independent of this signaling molecule. Eighty-seven male Fischer 344 × Brown Norway rats were randomly assigned to receive enalapril (n = 23), the NO synthase (NOS) inhibitor NG-nitro-l-arginine methyl ester (l-NAME; n = 22), enalapril + l-NAME (n = 19), or placebo (n = 23) from 24 to 27 months of age. Experiments were performed on the tibialis anterior muscle. Total NOS activity and the expression of neuronal, endothelial, and inducible NOS isoforms (nNOS, eNOS, and iNOS) were determined to investigate the effects of enalapril on NO signaling. Transcript levels of tumor necrosis factor-alpha (TNF-α) and peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α) were assessed to explore actions of enalapril on inflammation and mitochondrial biogenesis, respectively. Protein expression of energy-sensing and insulin signaling mediators, including protein kinase B (Akt-1), phosphorylated Akt-1 (pAkt-1), mammalian target of rapamycin (mTOR), AMP-activated protein kinase subunit alpha (AMPKα), phosphorylated AMPKα (pAMPKα), and the glucose transporter GLUT-4, was also determined. Finally, the generation of hydrogen peroxide (H2O2) was quantified in subsarcolemmal (SSM) and intermyofibrillar (IFM) mitochondria. Enalapril increased total NOS activity, which was prevented by l-NAME co-administration. eNOS protein content was enhanced by enalapril, but not by enalapril + l-NAME. Gene expression of iNOS was down-regulated by enalapril either alone or in combination with l-NAME. In contrast, protein levels of nNOS were unaltered by treatments. The mRNA abundance of TNF-α was reduced by enalapril relative to placebo, with no differences among any other group. PCG-1α gene expression was unaffected by enalapril and lowered by enalapril + l-NAME. No differences in protein expression of Akt-1, pAkt-1, AMPKα, pAMPKα, or GLUT-4 were detected among groups. However, mTOR protein levels were increased by enalapril compared with placebo. Finally, all treatment groups displayed reduced SSM, but not IFM H2O2 production relative to placebo. Our data indicate that enalapril induces a number of metabolic adaptations in aged skeletal muscle. These effects result from the concerted modulation of NO and angiotensin II signaling, rather than from a dichotomous action of enalapril on the two pathways. Muscle protection by enalapril administered late in life appears to be primarily mediated by mitigation of oxidative stress and pro-inflammatory signaling.
Keywords: Aging, Nitric oxide synthase (NOS) isoforms, Mitochondria, mTOR, Glucose tolerance, l-NAME, Inflammation, ACE inhibitors
Introduction
With age, several derangements occur in the skeletal muscle that ultimately contribute to physical performance impairment (Buford et al. 2010). Among the biological factors involved in the disabling process, alterations in the renin–angiotensin system (RAS) are indicated as potential contributors (Carter et al. 2004). Indeed, the RAS is involved in the promotion of muscular inflammation, oxidative stress, and apoptosis, all of which are implicated in the deterioration of physical performance with aging (Carter et al. 2005). Notably, treatment with the angiotensin converting enzyme (ACE) inhibitor enalapril improves whole body insulin sensitivity as well as mitochondrial mass and function in the liver, kidney, and myocardium of aged rats (Inserra et al. 1995; Ferder et al. 1998, 2002; de Cavanagh et al. 2003). These adaptations are achieved independently of reductions in blood pressure and may be secondary to improvements in nitric oxide (NO) signaling (de Cavanagh et al. 2011).
NO is a pleiotropic cell signaling molecule synthesized from l-arginine, NADPH, and oxygen by the NO synthase (NOS) class of enzymes (Alderton et al. 2001). NOS exists in at least three isoforms: neuronal (nNOS), endothelial (eNOS), and inducible (iNOS) (Alderton et al. 2001). All three isoforms are expressed in skeletal muscle, and each of them may produce specific effects on insulin sensitivity, oxidative stress, apoptosis, and inflammatory processes (Kobzik et al. 1994). The biological actions of NO are dependent upon its steady-state tissue concentrations and subcellular site of production, which in turn are linked to the isoform(s) of NOS expressed (Villanueva and Giulivi 2010).
Canonical signaling by NO involves the activation of soluble guanylate cyclase, the generation of cGMP, and the activation of specific kinases (Martínez-Ruiz et al. 2011). NO can also induce posttranslational modifications (e.g., S-nitrosylation, S-glutathionylation, and tyrosine nitration) in target molecules (Martínez-Ruiz et al. 2011). Finally, NO can modulate mitochondrial oxygen consumption, with important implications for cell respiration, redox homeostasis, and intermediary metabolism (Martínez-Ruiz et al. 2011). Within mitochondria, NO competes with oxygen for the substrate-binding site of cytochrome c oxidase (complex IV), the final electron acceptor of the electron transport chain (ETC) (Carreras and Poderoso 2007). Low levels of NO-mediated ETC inhibition induced by eNOS and or nNOS may be beneficial, by dispensing oxygen to cells at varying distances from blood vessels and by reducing oxidant generation (Thomas et al. 2010). Within skeletal myofibers, this mechanism could optimize oxygen repartition between subsarcolemmal (SSM) and intermyofibrillar mitochondria (IFM). In contrast, sustained NO production by iNOS is detrimental by causing extensive ETC inhibition, enhanced free radical production, nitrosative damage, and induction of apoptosis via cytochrome c release (Haynes et al. 2003).
The complexity of NO signaling is reflected in mice with disrupted levels of both eNOS and nNOS (Shankar et al. 2000). These rodents exhibit insulin resistance, weight gain, and reduced mitochondrial biogenesis and energy expenditure. Conversely, targeted disruption of iNOS appears to be protective, in that iNOS-null mice do not become insulin resistant, while displaying increased adiposity (Perreault and Marette 2001). Interestingly, amelioration of nNOS activity improves muscle strength and exercise tolerance in a mouse model of muscular dystrophy (Lai et al. 2009). Finally, NOS inhibition via the administration of NG-nitro-l-arginine methyl ester (l-NAME) in rats results in the loss of skeletal muscle mass and reduction of myofiber cross-sectional area, with subsequent decrease in walking speed (Wang et al. 2001). Based on these findings, it is therefore proposed that NO signaling possesses a dual effect, such that excessive NO production through iNOS leads to insulin resistance/cell death, while lower amounts of NO generated by eNOS and/or nNOS promote cell survival and metabolic control.
Besides their effects on NO signaling, ACE inhibitors may also preserve skeletal muscle viability and function through the abrogation of angiotensin (ANG) II-dependent activation of nuclear factor κB (NF-κB) (Wei et al. 2008). This effect has been attributed to reduced production of ANG II or increased generation of ANG-(1-7) (Iyer et al. 1998; Benter et al. 2011). Indeed, exposure of L6 myotubes to ANG II increases the activity of NF-κB, resulting in enhanced production of tumor necrosis factor-alpha (TNF-α) and decreased insulin sensitivity (Wei et al. 2008). Notably, TNF-α, besides impairing insulin signaling, also promotes muscular protein breakdown and myocyte apoptosis, thereby playing a central role in age-related muscle wasting (Marzetti et al. 2010). It is worth noting that NO and TNF-α signaling can influence one another, with TNF-α being a potent inducer of iNOS (Nussler and Billiar 1993), while elevated NO concentrations exacerbate TNF-α-mediated cellular injury (Horton et al. 2000). These findings link inflammation, oxidative stress, and NOS isoform expression with both cell death and cell survival pathways.
In a recent study, we reported that late-life administration of enalapril reduced whole body adiposity and attenuated both the extent of apoptotic DNA fragmentation and the activation of mitochondrial caspase-dependent apoptotic signaling in rat gastrocnemius muscle (Carter et al. 2011). These biochemical changes were accompanied by attenuation of muscle strength decline, in the absence of any sizeable hypertrophic effect, suggesting that enalapril improves muscle quality rather than increasing muscle mass. Therefore, the present investigation was undertaken to identify possible mechanisms underlying the muscle-protective effects of enalapril. We also sought to discern the effects of enalapril mediated by NO from those independent of this signaling molecule. We postulated that enalapril would ameliorate insulin sensitivity, stimulate mitochondrial biogenesis, reduce the generation of oxidants by mitochondria, and decrease pro-inflammatory signaling in skeletal muscle of aged rats. Furthermore, we hypothesized that enalapril would modulate the activity of NOS in muscle by favoring the expression of isoforms that promote a lower steady-state concentration of NO (i.e., nNOS and eNOS) and by concomitantly reducing iNOS expression. The NOS inhibitor l-NAME was employed to identify actions of enalapril mediated by NO-dependent and independent mechanisms.
Materials and methods
Animals
Eighty-seven male Fischer 344 × Brown Norway (F344BN) F1 hybrid rats were purchased from the National Institute on Aging Colony at Harlan Industries (Indianapolis, IN, USA). This rat strain was chosen because of its increased longevity and decreased cumulative lesion incidence as compared with other breeds (Lipman et al. 1996). Furthermore, F344BN rats develop age-related body composition changes (i.e., increase in adiposity and decrease in lean body mass) resembling those occurring during human aging (Rice et al. 2005). Animals were received at 22 months of age and housed individually on a 12-h light/dark cycle in a specific pathogen-free facility accredited by the American Association for Accreditation of Laboratory Animal Care. Health status, body weight (BW), and food intake were monitored daily. All experimental protocols were approved by the University of Florida’s Institutional Animal Care and Use Committee.
Experimental design
Rats were randomly assigned to receive 20 mg kg−1 day−1 enalapril (n = 23), 1 mg kg−1 day−1 l-NAME (n = 22), 20 mg kg−1 day−1 enalapril + 1 mg kg−1 day−1 l-NAME (n = 19), or placebo (n = 23) from 24 to 27 months of age. This timeframe corresponds to a critical window in the aging process, during which males of this strain continue to gain fat mass while simultaneously losing muscle mass (Rice et al. 2005). Drug delivery was accomplished by compounding treatments into bacon-flavored food tablets (Bio-Serv, Frenchtown, NJ, USA). Placebo-containing food tablets were identical to those delivering enalapril and/or l-NAME, except that the drug was omitted. Drug- and placebo-containing tablets were administered separately from standard chow. Drug doses were adjusted daily according to the animal’s weight. All rats consumed the whole treatment tablet at each meal.
Determination of plasma ANG I and ANG II and serum ACE activity
Two randomly chosen rats from different groups were sacrificed daily by rapid decapitation. Trunk blood was immediately collected in pre-chilled tubes containing peptidase inhibitors (25 mM EDTA, 0.44 mM 1,2-orthophenanthroline monohydrate, 1 mM sodium para-chloromercuribenzoate, and 3 μM of the rat renin inhibitor WFML-1). These inhibitors were used to prevent ANG I generation and the conversion of ANG I into ANG II (Kohara et al. 1991). Angiotensin peptides were extracted from plasma using C18 Sep-Pak® columns (Waters, Milford, MA, USA), and the eluate analyzed by radioimmunoassay for ANG I and ANG II (Kohara et al. 1991). The intra- and inter-assay coefficients of variability were 12 and 22 % for ANG I and 8 and 20 % for ANG II, respectively. Serum ACE activity was determined according to the method described by Lieberman (1989) with modifications. Briefly, serum samples (10 μL) were incubated with 100 μL HEPES buffer containing the synthetic ACE substrate [3H]-Hip-Gly-Gly (pH 8.0) for 60 min at 37 °C. The reaction was terminated by acidification with 50 μL 1 N HCl, and [3H]-hippuric acid released by ACE separated from unreacted substrate by extraction with the Ultima Gold™ F scintillation cocktail (PerkinElmer, Waltham, MA, USA). This separation procedure relies on the fact that the reaction product is soluble in the scintillation fluid, whereas the substrate is captured in the aqueous phase. The quantity of [3H]-hippuric acid released, corresponding to the enzyme activity, was calculated from the radioactivity of a measured known mass of unhydrolyzed peptide and expressed as nanomole per milliliter per minute. The intra-assay variation was 3.9 % and the inter-assay variation averaged 5.9 %.
Determination of whole body glucose tolerance
Rats were fasted overnight before receiving an intraperitoneal glucose load of 3.0 g kg−1. Blood was sampled by tail nick prior to glucose loading and 30, 60, and 120 min after. Blood glucose levels were determined using an Accu-Chek® glucose meter (Roche Diagnostics, Indianapolis, IN, USA), while serum insulin was measured via an ELISA kit (Linco Research, Inc., St. Louis, MO, USA) with a sensitivity of 0.2 ng mL-1 and an inter-assay coefficient of variability of 6.9 %. Glucose and insulin values are reported as area under the time–concentration curve for 120 min following glucose loading (AUC0–120). The homeostasis model of assessment – insulin resistance (HOMA-IR) index of insulin sensitivity was calculated by applying the formula: pg fasting serum insulin mL-1 × mg fasting glucose dL-1 × 0.424.
Isolation of skeletal muscle mitochondrial subpopulations
IFM and SSM were extracted from the tibialis anterior (TA) muscle according to the procedure described by Servais et al. (2003). As previously shown by our group (Hofer et al. 2009), this protocol allows for the isolation of high-quality, well-functioning mitochondria. The TA muscle was chosen because it undergoes significant atrophy over the course of aging (Clavel et al. 2006). Protein concentration was determined using the method developed by Bradford (1976), and freshly isolated IFM and SSM immediately utilized for analyses.
Measurement of mitochondrial hydrogen peroxide (H2O2) production
H2O2 production was quantified in both IFM and SSM according to the method described by Barja (2002), adapted to a 96-well microplate format, as detailed elsewhere (Hofer et al. 2009). This method is based on the conversion of homovanillic acid (HVA) into a fluorescent dimer (312 nm excitation/420 nm emission) by horseradish peroxidase (HRP) in the presence of H2O2. Briefly, freshly isolated IFM and SSM (0.1 mg mL-1) were incubated in assay buffer (145 mM KCl, 30 mM HEPES, 5 mM KH2PO4, 3 mM MgCl2, 0.1 mM EGTA, 0.3 % fatty acid-free BSA, pH 7.4 at 37 °C), followed by the addition of HRP (1.2 U), HVA (100 mM), and 2.5 mM glutamate/2.5 mM malate. Samples were incubated for 15 min at 37 °C in a light-protected microplate incubator placed on a microplate rotator. Fluorescence was determined with a SpectraMax Gemini XS fluorometer (Molecular Devices, Sunnyvale, CA, USA). Fluorescence units were converted into H2O2 concentrations using a glucose oxidase standard curve, with results expressed as nanomole per minute per milligram of mitochondrial protein.
Determination of muscle NOS activity
NOS activity was determined by measuring the formation of l-[3H]-citrulline from l-[3H]-arginine, as described by Weissman and Gross (2001). Briefly, frozen TA muscle samples were powdered in liquid nitrogen and homogenized in 50 mM Tris–HCl buffer containing 0.1 mM EDTA, 0.1 mM EGTA, 1 mM phenylmethylsulfonyl fluoride, 1.0 μg mL-1 leupeptin, and 10 μM calpain inhibitor (pH 7.4). Sample aliquots (100–200 μg of protein) were incubated in 50 mM Tris–HCl buffer (pH 7.4) containing 0.1 mM EDTA, 0.1 mM EGTA, 0.5 mM DTT, 1 mM NADPH, 100 nM calmodulin, 10 μM tetrahydrobiopterin (BH4), 1.25 mM CaCl2, and 5 μM combined l-arginine and purified l-[3H]-arginine (0.6 μCi). Total volume of the reaction was 200 μL. After 15 min of incubation at 37 °C, reactions were terminated by adding 800 μL of ice-cold 50 mM HEPES and 5 mM EDTA buffer (pH 5.5). l-[3H]-citrulline was separated from l-[3H]-arginine by cationic exchange chromatography (Dowex AG50W-X8; Na+ form; Bio-Rad Laboratories, Hercules, CA, USA). The amount of l-[3H]-citrulline eluted was quantified by liquid scintillation counting. NOS activity determined in the gastrocnemius muscle from the first placebo control rat was used to normalize TA NOS activity in all samples. Procedures for protein extraction and enzymatic activity measurement for the internal control sample were identical to those described for TA. The specific activity of NOS is expressed as l-[3H]-citrulline formed and reported as percent of the placebo group, the value of which was set to 100 %.
Western blot analysis
TA muscle whole tissue extracts were obtained as described previously (Marzetti et al. 2008b) and used to determine the protein expression of nNOS, eNOS, protein kinase B (Akt-1), phosphorylated Akt-1 (pAkt-1), AMP-activated protein kinase subunit alpha (AMPKα), phosphorylated AMPKα (pAMPKα), mammalian target of rapamycin (mTOR), and the glucose transporter GLUT-4, by western blot analysis. Electrophoresis and immunoblotting were carried out as detailed elsewhere (Marzetti et al. 2008b). Primary antibodies employed were mouse monoclonal anti-eNOS (1:1,000; BD Transduction Laboratories, San Jose, CA, USA), mouse monoclonal anti-nNOS (1:500; BD Transduction Laboratories), and mouse monoclonal anti-Akt-1, pAkt-1, AMPKα, pAMPKα, GLUT-4, and mTOR (1:1,000; all from Cell Signaling Technology, Beverly, MA, USA). An anti-mouse HRP-conjugated antibody (1:2,500; Affinity Bioreagents, Golden, CO, USA) was used as the secondary antibody. Generation of the chemiluminescent signal, digital acquisition, and densitometry analysis were performed as described elsewhere (Marzetti et al. 2009).
Quantitative real-time polymerase chain reaction (qRT-PCR)
To determine the relative gene expression of iNOS, peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α), and TNF-α in TA muscle samples, qRT-PCR analysis was performed. Isolation of total RNA and cDNA synthesis were carried out as detailed elsewhere (Marzetti et al. 2008a). qRT-PCR was performed using an ABI 7300 real-time PCR system (ABI, Foster City, CA, USA). Primers were designed with the on-board software Primer Express 3.0 (Table 1). The Power SYBR green PCR Master Mix (ABI) and ABI universal cycling conditions were employed, as previously described (Xu et al. 2012). All samples were examined in triplicate, with the placebo group used as the calibrator. Negative controls (no template and no reverse transcriptase) were also included and run in triplicate. Differences in gene expression were determined by the 2−ΔΔCT method (Livak and Schmittgen 2001), with β-actin as the housekeeping gene. The expression of β-actin was unvarying among the experimental groups (data not shown).
Table 1.
Accession number and primer sequences of the investigated genes
Statistical analysis
All analyses were performed using Sigma Plot/Stat 11.0 (San Jose, CA, USA). For each variable, one-way analysis of variance (ANOVA) was employed to assess the effects of treatment across the four groups (placebo, l-NAME, enalapril, and enalapril + l-NAME). For the glucose tolerance test, a two-way ANOVA for repeated measures was used to determine both the main effect of treatment and the interaction between treatment and time (post-glucose injection—:0, 30, 60, and 120 min). When needed, post hoc analyses were performed using the Student–Newman–Keuls test. All data are presented as mean ± standard error of the mean (SEM), with significance set at p < 0.05.
Results
Whole body measurements
Morphological and physiological characteristics of experimental animals
Changes in BW, expressed as percent difference between 27 and 24 months of age, were assessed to determine if l-NAME itself affected BW and if NOS blockade prevented weight-lowering by enalapril. As previously reported (Carter et al. 2011), enalapril reduced BW by approximately 4 % (p = 0.045 vs. placebo), which was modestly attenuated when combined with l-NAME (p = 0.07 vs. placebo; Fig. 1). TA muscle wet weight was not different across groups (data not shown). All rats experienced a decrease in food intake over time (p = 0.03 vs. baseline), with no differences among treatments (data not shown).
Fig. 1.

Percent changes in body weight relative to baseline across experimental groups. P placebo, Ll-NAME, E enalapril, E+L enalapril + l-NAME. *p < 0.05 vs. placebo and l-NAME. n = 19–23/group. Data are mean ± SEM
Plasma ANG I and ANG II levels and serum ACE activity
Circulating RAS peptide levels and ACE activity were determined to verify if l-NAME interfered with the ability of enalapril to act systemically. As shown in Fig. 2a, serum ACE activity was reduced by enalapril independent of l-NAME (p < 0.05 vs. placebo and l-NAME). As expected with long-term ACE inhibition, a significant increase in ANG I (p < 0.001 vs. placebo and l-NAME; Fig. 2b) and marginal changes in ANG II levels were detected in the circulation (Fig. 2c). l-NAME alone or in combination with enalapril did not significantly impact any of these measures.
Fig. 2.
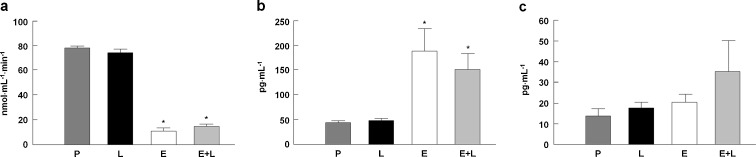
Serum ACE activity (a) and plasma ANG I (b) and ANG II (c) levels across experimental groups. P placebo, Ll-NAME, E enalapril, E+L enalapril + l-NAME. n = 15/group. *p < 0.05 vs. placebo and l-NAME. n = 19–23/group. Data are mean ± SEM
Glucose tolerance
Glucose homeostasis is marginally impaired in aged F344BN rats (Larkin et al. 2001). In the present study, glucose tolerance was not altered by any treatment. Neither peak blood glucose concentrations nor AUC0–120 was affected by enalapril and/or l-NAME (p = 0.57; Fig. 3). Fasting serum insulin is elevated in aged F344BN rats (Larkin et al. 2001), but was not affected by any treatment in the present study (data not shown). Similarly, the HOMA-IR index was unaltered by the interventions (p = 0.69; data not shown).
Fig. 3.

Glucose tolerance across experimental groups. n = 19–23/group. Data are mean ± SEM
Whole tissue analyses
NOS activity
A significant treatment effect was detected (p < 0.0001; Fig. 4). The Student–Newman–Keuls post hoc analysis revealed that NOS activity was increased by enalapril relative to placebo (p < 0.01). This effect was prevented by l-NAME co-treatment. NOS activity in rats that received l-NAME either alone or in combination with enalapril did not significantly differ from the placebo group. These data show for the first time that in vivo administration of enalapril increases NOS activity in skeletal muscle, an effect that is prevented by l-NAME co-treatment.
Fig. 4.

NOS activity in the tibialis anterior muscle across experimental groups. P placebo, Ll-NAME, E enalapril, E+L enalapril + l-NAME. *p < 0.05 vs. placebo, l-NAME, and enalapril + l-NAME. n = 9–10/group. Data are mean ± SEM and are presented as percent of the placebo group (100 %)
Analysis of NOS isoforms
Western blot analysis was used to determine protein levels of nNOS and eNOS in TA muscle samples. For iNOS, which is mainly regulated at transcription, qRT-PCR was employed to explore differences in gene expression among experimental groups. Protein levels of nNOS tended to be higher in rats treated with enalapril compared with placebo, but the effect was not statistical significant (p = 0.13; Fig. 5a). eNOS protein expression was increased by enalapril (p = 0.016 vs. placebo; Fig. 5b), which was prevented by l-NAME co-treatment. Administration of l-NAME alone slightly reduced eNOS protein content relative to placebo (p = 0.011). Finally, iNOS gene expression was down-regulated by enalapril either alone or in combination with l-NAME (p < 0.05 vs. placebo and l-NAME; Fig. 5c). l-NAME alone tended to increase the mRNA abundance of iNOS relative to placebo, but this change was not statistically significant (p = 0.068). These data show for the first time that in vivo enalapril administration affects the expression of NOS isoforms in muscle of aged rats by reducing iNOS and simultaneously up-regulating eNOS expression.
Fig. 5.
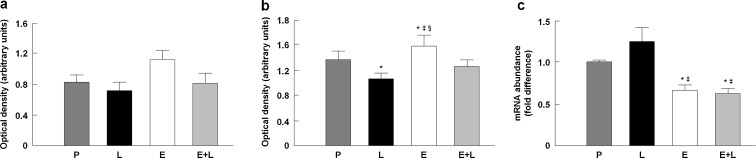
Protein levels of nNOS (a) and eNOS (b) and relative gene expression of iNOS (c) in the tibialis anterior muscle across experimental groups. P placebo, L l-NAME, E enalapril, E+L enalapril + l-NAME. *p < 0.05 vs. placebo; ‡p < 0.05 vs. l-NAME; §p < 0.05 vs. enalapril + l-NAME. n = 12/group. Data are mean ± SEM
Markers of inflammation and mitochondrial biogenesis
qRT-PCR was employed to determine relative gene expression levels of TNF-α and PCG-1α in TA muscle samples. Specifically, the mRNA abundance of TNF-α was quantified to assess levels of inflammation. PCG-1α transcript levels were measured as an index of mitochondrial biogenesis. As shown in Fig. 6a, TNF-α gene expression was down-regulated by enalapril relative to placebo (p = 0.042), with no differences among any other treatment group, suggesting a NO-dependent anti-inflammatory effect of this ACE inhibitor. The mRNA abundance of PCG-1α was unaltered by enalapril and l-name individually, but decreased when the two compounds were administered in combination relative to all other groups (p < 0.05; Fig. 6b).
Fig. 6.

Relative gene expression of TNF-α (a) and PCG-1α (b) in the tibialis anterior muscle across experimental groups. P placebo, Ll-NAME, E enalapril, E+L enalapril + l-NAME. *p < 0.05 vs. placebo; ‡p < 0.05 vs. l-NAME; §p < 0.05 vs. enalapril. n = 12/group. Data are mean ± SEM
Muscular energy-sensing and insulin signaling pathways
Western immunoblotting was employed to quantify the expression of several proteins associated with myocyte energy-sensing and insulin signaling (phosphorylated and total Akt-1, phosphorylated and total AMPK-α, and GLUT-4). These mediators are responsive to changes in inflammation, oxidative stress, and NOS activity (Jornayvaz and Shulman 2010). As show in Table 2, no significant differences in the expression of Akt-1, pAkt-1, AMPKα, pAMPKα, or GLUT-4 were detected among experimental groups. Subsequently, we measured the protein expression of mTOR; however, due to tissue limitation, this determination was only possible in enalapril and placebo groups. mTOR is down-regulated in skeletal muscle with age and up-regulated by exercise or chronic overload in old rats and humans (Chale-Rush et al. 2009; Mayhew et al. 2009; Rennie 2009; Pasini et al. 2012). Furthermore, mTOR is a negative regulator of insulin signaling in myocytes (Ge et al. 2011). In our study, mTOR protein levels were significantly increased by enalapril (p = 0.048 vs. placebo; Fig. 7).
Table 2.
Expression levels of proteins associated with myocyte energy-sensing and insulin signaling in the tibialis anterior muscle across experimental groups
| Experimental groups | |||||
|---|---|---|---|---|---|
| Placebo (n = 15) | l–NAME (n = 15) | Enalapril (n = 15) | Enalapril + l–NAME (n = 15) | p value | |
| Akt–1 | 2.03 ± 0.41 | 1.64 ± 0.37 | 1.76 ± 0.42 | 1.65 ± 0.32 | 0.87 |
| pAkt–1 | 0.85 ± 0.17 | 1.01 ± 0.23 | 0.58 ± 0.11 | 1.12 ± 0.30 | 0.36 |
| AMPKα | 1.27 ± 0.17 | 0.84 ± 0.09 | 1.04 ± 0.10 | 1.24 ± 0.26 | 0.17 |
| pAMPKα | 1.13 ± 0.13 | 1.15 ± 0.25 | 1.13 ± 0.22 | 1.15 ± 0.21 | 1.00 |
| GLUT–4 | 0.73 ± 0.15 | 1.92 ± 0.24 | 1.55 ± 0.41 | 1.59 ± 0.43 | 0.06 |
Densitometry data are expressed in arbitrary optical density units. Values are mean ± SEM
Fig. 7.

mTOR protein levels in the tibialis anterior muscle. P placebo, E enalapril. *p = 0.048 vs. placebo. n = 12/group. Data are mean ± SEM
Isolated mitochondrial measures: H2O2 production
No differences in IFM H2O2 production were observed among groups (data not shown). As depicted in Fig. 8, all treatment groups displayed reduced SSM H2O2 production relative to placebo (p < 0.05). These findings are similar to those previously reported in kidney mitochondria of aged rats after enalapril administration (de Cavanagh et al. 2003) and highlight the complicated interplay between NO bioavailability and mitochondrial H2O2 production.
Fig. 8.

SSM H2O2 production in the tibialis anterior muscle across experimental groups. P placebo, Ll-NAME, E enalapril, E+L enalapril + l-NAME. *p < 0.05 vs. placebo. n = 19–23/group. Data are mean ± SEM
Discussion
In the current set of studies, we tested the hypothesis that treatment with the ACE inhibitor enalapril started late in life would mitigate several adverse age-related changes in skeletal muscle, including increased oxidative stress and pro-inflammatory signaling, reduced insulin sensitivity, and diminished mitochondrial biogenesis. In addition, we sought to establish whether these effects occur through NO-dependent or independent mechanisms. Results are discussed in the context of systemic, mitochondrial, and whole tissue effects.
Systemic effects
As expected with long-term enalapril treatment, decreased ACE activity and increased ANG I levels were observed in the circulation, with modest changes in ANG II levels (Fig. 2a–c). l-NAME had no effect on any of these measures, indicating that NOS inhibition did not interfere with the upstream RAS peptide synthesis pathway. Circulating ANG II levels remain substantially unchanged after 3 months of enalapril treatment (Fig. 2c). This finding is consistent with the “ACE escape” phenomenon, that is the restoration of baseline ANG II levels after protracted ACE inhibition through alternative pathways (van de Wal et al. 2006). Circulating ANG II levels were higher, although nonsignificantly different, in rats treated with enalapril + l-NAME relative to all other groups. This finding, together with the higher scattering of ANG II data under enalapril + l-NAME, may imply that the “ACE escape” could have occurred to a greater extent and with higher variability across animals with chronic NOS blockade. The possibility that NO signaling might be involved in the “ACE escape” phenomenon deserves further investigation.
The effect of long-term ACE inhibition on other components of the RAS such as ANG II receptor subtypes is less predictable. Indeed, unchanged (Wu et al. 1994), decreased (Wilson et al. 1988; Schmeisser et al. 2004; Reuter et al. 2006), or increased levels (Wilson et al. 1988; Amiri et al. 1999) of either type 1 or type 2 ANG II receptors have been reported, depending on the tissue and cell type studied (e.g., brain, vascular endothelium or smooth muscle, kidney, circulating monocytes), ACE inhibitor tested, duration of treatment, and experimental model. However, treatment effects on ANG II receptor expression were not evaluated in the present study. Further investigation is needed to address the effect of chronic ACE inhibition on the distribution of ANG II receptors in skeletal muscle at old age.
No changes in whole body glucose tolerance were observed following enalapril administration (Fig. 3), despite opposite reports in the literature, albeit mostly in young animals (Santos et al. 2009; Weisinger et al. 2009). The failure of enalapril to improve insulin responsiveness in our system may derive from the fact that insulin sensitivity is only modestly impaired in aged F344BN rats. Indeed, in this rat strain, the glucose infusion rate during hyperinsulinemic–euglycemic clamping is reduced by 20 % between 8 and 18 months of age, with no further decrease by 28 months (Larkin et al. 2001). Moreover, fasting plasma insulin is elevated by 43 % at 18 months, with no further increase by 28 months (Larkin et al. 2001). Finally, GLUT-4 expression in the gastrocnemius muscle is decreased by 25 % between 18 and 28 months of age (Larkin et al. 2001). Collectively, these observations suggest that age-related changes in insulin/glucose homeostasis in the circulation do not always correlate with those observed at the tissue level.
Muscular mitochondrial effects
H2O2 production by SSM was reduced with both ACE and NOS inhibition (Fig. 8), indicating a NO-dependent event. A similar adaptation was observed in renal mitochondria of rats treated with enalapril (de Cavanagh et al. 2003) and may be attributed to a complex interaction between ACE inhibition and NO signaling affecting the function of the ETC, the expression of uncoupling protein-2, and oxidant scavenging by manganese-dependent superoxide dismutase (Nègre-Salvayre et al. 1997; Casteilla et al. 2001; de Cavanagh et al. 2003; Nilakantan et al. 2005; Piotrkowski et al. 2007). No changes in H2O2 generation by IFM were detected across treatments. Contrary to SSM, IFM isolated from old rats do not produce larger amount of H2O2 compared with younger animals (Judge et al. 2005a). This difference has been attributed to a higher bioenergetic efficiency and a richer repertoire of enzymatic and non-enzymatic antioxidant defenses of IFM relative to SSM (Servais et al. 2003; Judge et al. 2005a, b). It is therefore conceivable that the age-related increase in oxidant generation by SSM is amenable to interventions such as ACE inhibition. In contrast, IFM may be intrinsically protected against excessive ROS production and, thus, less susceptible to pharmacological improvements. Alternatively, it may be hypothesized that the subcellular topology of the two mitochondrial populations could itself impact their responsiveness to treatments. Indeed, SSM, being located directly underneath the sarcolemma, may experience greater exposure to pharmacological agents as well as to eNOS-derived NO compared with IFM, which are situated between the myofibers.
It was unexpected that the mRNA abundance of PCG-1α (Fig. 6b), the master regulator of mitochondrial biogenesis, was not up-regulated by enalapril. PCG-1α functions as a coactivator of transcription factors, including nuclear respiratory factor 1 and mitochondrial transcription factor A (TFAM), which in turn regulate the expression of several genes crucial for mitochondrial function and for the replication of mitochondrial genome (Kim et al. 2008). The expression of PGC-1α is regulated by eNOS, such that eNOS-null mice present markedly reduced mitochondrial mass (Nisoli et al. 2003). ACE inhibition was shown to rescue mitochondrial function and biogenesis in rat skeletal muscle after the induction of myocardial infarction (Zoll et al. 2006). These findings have been extended to the heart, liver, and kidney in F344 rats, where enalapril treatment attenuates age-related structural and functional mitochondrial abnormalities and prevents the decrease in mitochondrial mass (de Cavanagh et al. 1995, 1997, 2000, 2003; Inserra et al. 1996; Piotrkowski et al. 2007). The lack of increase in PGC-1α gene expression in our system may be linked to the “ACE escape” phenomenon. Indeed, ANG II reduces the mitochondrial mass in C2C12 cells in a dose-dependent manner by directly inhibiting the expression of genes involved in mitochondrial biogenesis, including PGC-1α and TFAM (Mitsuishi et al. 2009). Similar effects were observed in skeletal muscle of mice after ANG II infusion (Mitsuishi et al. 2009). It may therefore be speculated that the lack of reduction of plasma ANG II levels in enalapril-treated rats might have counterbalanced the stimulatory effect of NO on PGC-1α expression. This hypothesis is supported by the reduction of PGC-1α transcript levels observed under enalapril + l-NAME. In this condition, ANG II levels tended to be higher which, together with the lack of NO stimulation, might have down-regulated the expression of PGC-1α. Moreover, in L6 myotubes, NO stimulates mitochondrial biogenesis by inducing the phosphorylation of AMPKα, which in turn up-regulates PCG-1α (Lira et al. 2010). Hence, the lack of elevation of pAMPKα in enalapril-treated rats could further explain why PCG-1α was not up-regulated despite increased NOS activity.
It should also be considered that impairment of muscular mitochondrial function may not be the primary mediator of declining physical performance. Betik et al. (2009) reported that 29-month-old F344BN rats exposed to 5 or 7 months of treadmill training exhibited greater running capacity, survival, and body fat, while aerobic capacity, muscle mass, and mitochondrial enzyme activities declined. Furthermore, perindopril administration in conjunction with endurance training increased exercise tolerance without any discernible impact on oxidative capacity in the plantaris and soleus muscles of female Wistar rats (Habouzit et al. 2009). In addition, improvements in strength and physical performance in male F344BN rats after enalapril treatment (Carter et al. 2011) or treadmill training (Marzetti et al. 2008a) started at advanced age were accompanied by reduced inflammatory and apoptotic signaling in skeletal muscle. Collectively, these findings suggest that amelioration of physical performance elicited by pharmacological or behavioral interventions initiated late in life may not be mediated by improvements in muscular mitochondrial bioenergetics. Rather, modulation of cell death/survival pathways may be required to preserve skeletal muscle function during aging (Marzetti et al. 2012a, b).
Whole tissue effects
As predicted, overall TA muscle NOS activity was increased by enalapril, an effect prevented by l-NAME co-administration (Fig. 4). Gene expression levels of iNOS were decreased (Fig. 5c), whereas protein levels of nNOS and eNOS were increased by enalapril (Fig. 5a, b); however, statistical significance was observed for eNOS only. The effects of l-NAME, alone or in combination with enalapril, on the expression of NOS isoforms were negligible; yet, a small but significant effect was observed for eNOS. This is not surprising given the fact that l-NAME may potentiate iNOS gene expression, whereas it is more selective for both nNOS and eNOS (Miller et al. 1996). It should be noted that the pattern of NOS activity across experimental groups does not recapitulate that observed for individual NOS isoform expression. While iNOS is mainly controlled at transcription, the activity of eNOS and nNOS is also regulated at the translational and posttranslational levels. For instance, nNOS and eNOS activities are influenced by interaction with heterologous proteins (e.g., heat shock protein-90, calmodulin, and caveolin), phosphorylation/dephosphorylation, and dimerization (both isoforms are active as dimers) (Zhou and Zhu 2009; Rafikov et al. 2011). It follows that for similar levels of expression, NOS activity can be substantially different depending on the interaction among the abovementioned factors. However, specific mechanisms responsible for the lack of convergence of NOS expression and activity were not investigated in the present study.
Central to the effects described in this report is the down-regulation of TNF-α gene expression elicited by enalapril (Fig. 6a). TNF-α is a potent inducer of iNOS in skeletal muscle (Torres et al. 2004). Accordingly, a decrease in iNOS expression was observed in rats treated with enalapril, which was unaffected by l-NAME co-administration. While this finding may suggest a NO-independent regulation of iNOS by enalapril, it should be considered that chronic l-NAME administration can induce a compensatory up-regulation of this NOS isoform (Miller et al. 1996). Accumulating evidence indicates that the TNF-α/iNOS axis is centrally involved in the pathogenesis of muscle atrophy and functional decline during aging as well as in the setting of various disease conditions (Hall et al. 2011). Indeed, iNOS is proposed to be a primary effector of cytokine-mediated muscular degeneration (Hall et al. 2011). TNF-α acts primarily through the transcription factor NF-κB, which is responsible for the regulation of a wide variety of genes, including iNOS (Ghosh and Karin 2002). Excessive NO generation by iNOS can result in the formation of highly toxic peroxynitrite (ONOO−) through the reaction of NO with superoxide anions (O•−2). ONOO− in turn can down-regulate MyoD, a transcription factor involved in myogenesis and myocyte maintenance (Guttridge et al. 2000; Di Marco et al. 2005). The loss of MyoD causes myosin heavy chain depletion (Di Marco et al. 2005), which compromises the integrity of myofilaments, leading to reduced muscle strength independently of atrophy (Reid et al. 2002). In addition, increased protein levels of iNOS have been detected in skeletal muscle of old rats in conjunction with Akt S-nitrosylation and dysfunction and reduced levels of the Akt downstream target mTOR (Wu et al. 2009). In the present study, the reduction of iNOS mRNA in enalapril-treated animals was concomitant with increased protein content of mTOR (Fig. 7). This adaptation could have been mediated by improvements in Akt function in spite of unaltered protein levels.
Contrary to iNOS, constitutively expressed eNOS and nNOS are involved in the adaptation of skeletal myocytes to mechanical and metabolic stress (Stamler and Meissner 2001). Age-related alterations in eNOS signaling have been linked to defective endothelium-dependent vasodilatation in skeletal muscle (Delp et al. 2008). However, these changes are amenable to interventions. For instance, in a study by Song et al. (2009), treadmill training restored the expression and activity of eNOS and nNOS, while reducing iNOS levels in the white gastrocnemius muscle of old rats. Likewise, Spier et al. (2004) found that treadmill running increased the expression of eNOS in the soleus muscle of aged rats, thereby ameliorating endothelial-mediated vasodilatation. The up-regulation of eNOS protein expression by enalapril may therefore constitute a further means whereby ACE inhibition protects muscle function at old age. On the other hand, the lack of elevation of nNOS protein levels after enalapril administration might be linked to a fiber type-specific regulation of this NOS isoform. Indeed, nNOS is more abundant in muscles predominantly comprised of type I fibers relative to fast-twitch muscles, such as the TA (Pullen 1977; Song et al. 2009). Moreover, the age-dependent decrease of nNOS expression appears to be limited to highly oxidative muscles (Song et al. 2009). The possibility that enalapril differentially impacts the expression of nNOS in muscles with distinct fiber type composition warrants further investigation.
Limitations
Although reporting novel findings, the present study presents some limitations that deserve further discussion. First, experiments were conducted on the TA muscle which is mostly comprised of type II fibers. Therefore, our findings may not be extended to muscles with different fiber type composition. In addition, only total NOS activity was determined, which does not allow to infer about the effects of enalapril and NOS blockade on the function of individual isoforms. In our system, enalapril did not up-regulate the expression of PGC-1α, thus opening questions about the impact of ACE inhibition on muscular mitochondrial biogenesis at old age. However, no indices of mitochondrial mass were determined, which does not negate possible changes in mitochondrial number with enalapril treatment. Moreover, the effects of enalapril on mitochondrial dynamics and quality control mechanisms (e.g., macroautophagy) were not assessed. Future studies should evaluate whether enalapril (and ACE inhibitors in general) impact mitochondrial turnover in aged skeletal muscle. Finally, although pharmacodynamic properties are similar for most ACE inhibitors, individual compounds possess distinct molecular characteristics that can impact their pharmacokinetics as well as their ability to function as antioxidant and anti-inflammatory agents (Reid 1997; Van Antwerpen et al. 2006). Further investigation is necessary to establish whether single ACE inhibitors differentially affect NO signaling, inflammation, and mitochondrial function in aged muscle.
Conclusion
In summary, our data indicate that the effects of late-life enalapril administration on skeletal muscle are not merely attributable to a dichotomous action on NO or ANG II signaling, but may instead result from the convergence of the two pathways. Based on the current findings and on our previous investigations (Carter et al. 2011), enalapril appears to protect against age-related muscle deterioration mainly by attenuating mitochondrial oxidant generation, pro-inflammatory signaling, and cell death pathways, rather than via improvements in mitochondrial biogenesis and insulin sensitivity.
Acknowledgements
This work was funded through NIH grants R01-AG024526 and P30-AG028740 to Dr. Carter and HL-051952 to Dr. Diz.
References
- Alderton WK, Cooper CE, Knowles RG. Nitric oxide synthases: structure, function and inhibition. Biochem J. 2001;357:593–615. doi: 10.1042/0264-6021:3570593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amiri F, Haddad G, Garcia R. Renal angiotensin II receptor regulation and renin–angiotensin system inhibition in one-kidney, one clip hypertensive rats. J Hypertens. 1999;17:279–286. doi: 10.1097/00004872-199917020-00013. [DOI] [PubMed] [Google Scholar]
- Barja G. The quantitative measurement of H2O2 generation in isolated mitochondria. J Bioenerg Biomembr. 2002;34:227–233. doi: 10.1023/A:1016039604958. [DOI] [PubMed] [Google Scholar]
- Benter IF, Yousif MH, Al-Saleh FM, Raghupathy R, Chappell MC, Diz DI. Angiotensin-(1-7) blockade attenuates captopril- or hydralazine-induced cardiovascular protection in spontaneously hypertensive rats treated with NG-nitro-L-arginine methyl ester. J Cardiovasc Pharmacol. 2011;57:559–567. doi: 10.1097/FJC.0b013e31821324b6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betik AC, Thomas MM, Wright KJ, Riel CD, Hepple RT. Exercise training from late middle age until senescence does not attenuate the declines in skeletal muscle aerobic function. Am J Physiol Regul Integr Comp Physiol. 2009;297:R744–R755. doi: 10.1152/ajpregu.90959.2008. [DOI] [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Buford TW, Anton SD, Judge AR, Marzetti E, Wohlgemuth SE, Carter CS, et al. Models of accelerated sarcopenia: critical pieces for solving the puzzle of age-related muscle atrophy. Ageing Res Rev. 2010;9:369–383. doi: 10.1016/j.arr.2010.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carreras MC, Poderoso JJ. Mitochondrial nitric oxide in the signaling of cell integrated responses. Am J Physiol Cell Physiol. 2007;292:C1569–C1580. doi: 10.1152/ajpcell.00248.2006. [DOI] [PubMed] [Google Scholar]
- Carter CS, Cesari M, Ambrosius WT, Hu N, Diz D, Oden S, et al. Angiotensin-converting enzyme inhibition, body composition, and physical performance in aged rats. J Gerontol A Biol Sci Med Sci. 2004;59:416–423. doi: 10.1093/gerona/59.5.B416. [DOI] [PubMed] [Google Scholar]
- Carter CS, Onder G, Kritchevsky SB, Pahor M. Angiotensin-converting enzyme inhibition intervention in elderly persons: effects on body composition and physical performance. J Gerontol A Biol Sci Med Sci. 2005;60:1437–1446. doi: 10.1093/gerona/60.11.1437. [DOI] [PubMed] [Google Scholar]
- Carter CS, Giovannini S, Seo DO, Dupree J, Morgan D, Chung HY, et al. Differential effects of enalapril and losartan on body composition and indices of muscle quality in aged male Fischer 344 × Brown Norway rats. Age (Dordr) 2011;33:167–183. doi: 10.1007/s11357-010-9196-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casteilla L, Rigoulet M, Penicaud L. Mitochondrial ROS metabolism: modulation by uncoupling proteins. IUBMB Life. 2001;52:181–188. doi: 10.1080/15216540152845984. [DOI] [PubMed] [Google Scholar]
- Chale-Rush A, Morris EP, Kendall TL, Brooks NE, Fielding RA. Effects of chronic overload on muscle hypertrophy and mTOR signaling in young adult and aged rats. J Gerontol A Biol Sci Med Sci. 2009;64:1232–1239. doi: 10.1093/gerona/glp146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clavel S, Coldefy AS, Kurkdjian E, Salles J, Margaritis I, Derijard B. Atrophy-related ubiquitin ligases, atrogin-1 and MuRF1 are up-regulated in aged rat Tibialis Anterior muscle. Mech Ageing Dev. 2006;127:794–801. doi: 10.1016/j.mad.2006.07.005. [DOI] [PubMed] [Google Scholar]
- de Cavanagh EM, Inserra F, Ferder L, Romano L, Ercole L, Fraga CG. Superoxide dismutase and glutathione peroxidase activities are increased by enalapril and captopril in mouse liver. FEBS Lett. 1995;361:22–24. doi: 10.1016/0014-5793(95)00137-X. [DOI] [PubMed] [Google Scholar]
- de Cavanagh EM, Fraga CG, Ferder L, Inserra F. Enalapril and captopril enhance antioxidant defenses in mouse tissues. Am J Physiol. 1997;272:R514–R518. doi: 10.1152/ajpregu.1997.272.2.R514. [DOI] [PubMed] [Google Scholar]
- de Cavanagh EM, Inserra F, Ferder L, Fraga CG. Enalapril and captopril enhance glutathione-dependent antioxidant defenses in mouse tissues. Am J Physiol Regul Integr Comp Physiol. 2000;278:R572–R577. doi: 10.1152/ajpregu.2000.278.3.R572. [DOI] [PubMed] [Google Scholar]
- de Cavanagh EM, Piotrkowski B, Basso N, Stella I, Inserra F, Ferder L, et al. Enalapril and losartan attenuate mitochondrial dysfunction in aged rats. FASEB J. 2003;17:1096–1098. doi: 10.1096/fj.02-0063fje. [DOI] [PubMed] [Google Scholar]
- de Cavanagh EM, Inserra F, Ferder L. Angiotensin II blockade: a strategy to slow ageing by protecting mitochondria? Cardiovasc Res. 2011;89:31–40. doi: 10.1093/cvr/cvq285. [DOI] [PubMed] [Google Scholar]
- Delp MD, Behnke BJ, Spier SA, Wu G, Muller-Delp JM. Ageing diminishes endothelium-dependent vasodilatation and tetrahydrobiopterin content in rat skeletal muscle arterioles. J Physiol. 2008;586:1161–1168. doi: 10.1113/jphysiol.2007.147686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Marco S, Mazroui R, Dallaire P, Chittur S, Tenenbaum SA, Radzioch D, et al. NF-kappa B-mediated MyoD decay during muscle wasting requires nitric oxide synthase mRNA stabilization, HuR protein, and nitric oxide release. Mol Cell Biol. 2005;25:6533–6545. doi: 10.1128/MCB.25.15.6533-6545.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferder L, Romano LA, Ercole LB, Stella I, Inserra F. Biomolecular changes in the aging myocardium: the effect of enalapril. Am J Hypertens. 1998;11:1297–1304. doi: 10.1016/S0895-7061(98)00152-6. [DOI] [PubMed] [Google Scholar]
- Ferder LF, Inserra F, Basso N. Advances in our understanding of aging: role of the renin–angiotensin system. Curr Opin Pharmacol. 2002;2:189–194. doi: 10.1016/S1471-4892(02)00139-X. [DOI] [PubMed] [Google Scholar]
- Ge Y, Yoon MS, Chen J. Raptor and Rheb negatively regulate skeletal myogenesis through suppression of insulin receptor substrate 1 (IRS1) J Biol Chem. 2011;286:35675–35682. doi: 10.1074/jbc.M111.262881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh S, Karin M. Missing pieces in the NF-kappaB puzzle. Cell. 2002;109(Suppl):S81–S96. doi: 10.1016/S0092-8674(02)00703-1. [DOI] [PubMed] [Google Scholar]
- Guttridge DC, Mayo MW, Madrid LV, Wang CY, Baldwin AS., Jr NF-kappaB-induced loss of MyoD messenger RNA: possible role in muscle decay and cachexia. Science. 2000;289:2363–2366. doi: 10.1126/science.289.5488.2363. [DOI] [PubMed] [Google Scholar]
- Habouzit E, Richard H, Sanchez H, Koulmann N, Serrurier B, Monnet R, et al. Decreased muscle ACE activity enhances functional response to endurance training in rats, without change in muscle oxidative capacity or contractile phenotype. J Appl Physiol. 2009;107:346–353. doi: 10.1152/japplphysiol.91443.2008. [DOI] [PubMed] [Google Scholar]
- Hall DT, Ma JF, Marco SD, Gallouzi IE. Inducible nitric oxide synthase (iNOS) in muscle wasting syndrome, sarcopenia, and cachexia. Aging (Albany NY) 2011;3:702–715. doi: 10.18632/aging.100358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haynes V, Elfering SL, Squires RJ, Traaseth N, Solien J, Ettl A, et al. Mitochondrial nitric-oxide synthase: role in pathophysiology. IUBMB Life. 2003;55:599–603. doi: 10.1080/15216540310001628681. [DOI] [PubMed] [Google Scholar]
- Hofer T, Servais S, Seo AY, Marzetti E, Hiona A, Upadhyay SJ, et al. Bioenergetics and permeability transition pore opening in heart subsarcolemmal and interfibrillar mitochondria: effects of aging and lifelong calorie restriction. Mech Ageing Dev. 2009;130:297–307. doi: 10.1016/j.mad.2009.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton JW, Maass D, White J, Sanders B. Nitric oxide modulation of TNF-alpha-induced cardiac contractile dysfunction is concentration dependent. Am J Physiol Heart Circ Physiol. 2000;278:H1955–H1965. doi: 10.1152/ajpheart.2000.278.6.H1955. [DOI] [PubMed] [Google Scholar]
- Inserra F, Romano L, Ercole L, de Cavanagh EM, Ferder L. Cardiovascular changes by long-term inhibition of the renin–angiotensin system in aging. Hypertension. 1995;25:437–442. doi: 10.1161/01.HYP.25.3.437. [DOI] [PubMed] [Google Scholar]
- Inserra F, Romano LA, de Cavanagh EM, Ercole L, Ferder LF, Gomez RA. Renal interstitial sclerosis in aging: effects of enalapril and nifedipine. J Am Soc Nephrol. 1996;7:676–680. doi: 10.1681/ASN.V75676. [DOI] [PubMed] [Google Scholar]
- Iyer SN, Chappell MC, Averill DB, Diz DI, Ferrario CM. Vasodepressor actions of angiotensin-(1-7) unmasked during combined treatment with lisinopril and losartan. Hypertension. 1998;31:699–705. doi: 10.1161/01.HYP.31.2.699. [DOI] [PubMed] [Google Scholar]
- Jornayvaz FR, Shulman GI. Regulation of mitochondrial biogenesis. Essays Biochem. 2010;47:69–84. doi: 10.1042/bse0470069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Judge S, Jang YM, Smith A, Hagen T, Leeuwenburgh C. Age-associated increases in oxidative stress and antioxidant enzyme activities in cardiac interfibrillar mitochondria: implications for the mitochondrial theory of aging. FASEB J. 2005;19:419–421. doi: 10.1096/fj.04-2622fje. [DOI] [PubMed] [Google Scholar]
- Judge S, Jang YM, Smith A, Selman C, Phillips T, Speakman JR, et al. Exercise by lifelong voluntary wheel running reduces subsarcolemmal and interfibrillar mitochondrial hydrogen peroxide production in the heart. Am J Physiol Regul Integr Comp Physiol. 2005;289:R1564–R1572. doi: 10.1152/ajpregu.00396.2005. [DOI] [PubMed] [Google Scholar]
- Kim JA, Wei Y, Sowers JR. Role of mitochondrial dysfunction in insulin resistance. Circ Res. 2008;102:401–414. doi: 10.1161/CIRCRESAHA.107.165472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobzik L, Reid MB, Bredt DS, Stamler JS. Nitric oxide in skeletal muscle. Nature. 1994;372:546–548. doi: 10.1038/372546a0. [DOI] [PubMed] [Google Scholar]
- Kohara K, Tabuchi Y, Senanayake P, Brosnihan KB, Ferrario CM. Reassessment of plasma angiotensins measurement: effects of protease inhibitors and sample handling procedures. Peptides. 1991;12:1135–1141. doi: 10.1016/0196-9781(91)90070-6. [DOI] [PubMed] [Google Scholar]
- Lai Y, Thomas GD, Yue Y, Yang HT, Li D, Long C, et al. Dystrophins carrying spectrin-like repeats 16 and 17 anchor nNOS to the sarcolemma and enhance exercise performance in a mouse model of muscular dystrophy. J Clin Invest. 2009;119:624–635. doi: 10.1172/JCI36612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkin LM, Reynolds TH, Supiano MA, Kahn BB, Halter JB. Effect of aging and obesity on insulin responsiveness and glut-4 glucose transporter content in skeletal muscle of Fischer 344 × Brown Norway rats. J Gerontol A Biol Sci Med Sci. 2001;56:B486–B492. doi: 10.1093/gerona/56.11.B486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman J. Enzymes in sarcoidosis. Angiotensin-converting-enzyme (ACE) Clin Lab Med. 1989;9:745–755. [PubMed] [Google Scholar]
- Lipman RD, Chrisp CE, Hazzard DG, Bronson RT. Pathologic characterization of brown Norway, brown Norway × Fischer 344, and Fischer 344 × brown Norway rats with relation to age. J Gerontol A Biol Sci Med Sci. 1996;51:B54–B59. doi: 10.1093/gerona/51A.1.B54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lira VA, Brown DL, Lira AK, Kavazis AN, Soltow QA, Zeanah EH, et al. Nitric oxide and AMPK cooperatively regulate PGC-1 in skeletal muscle cells. J Physiol. 2010;588:3551–3566. doi: 10.1113/jphysiol.2010.194035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Martínez-Ruiz A, Cadenas S, Lamas S. Nitric oxide signaling: classical, less classical, and nonclassical mechanisms. Free Radic Biol Med. 2011;51:17–29. doi: 10.1016/j.freeradbiomed.2011.04.010. [DOI] [PubMed] [Google Scholar]
- Marzetti E, Groban L, Wohlgemuth SE, Lees HA, Lin M, Jobe H, et al. Effects of short-term GH supplementation and treadmill exercise training on physical performance and skeletal muscle apoptosis in old rats. Am J Physiol Regul Integr Comp Physiol. 2008;294:R558–R567. doi: 10.1152/ajpregu.00620.2007. [DOI] [PubMed] [Google Scholar]
- Marzetti E, Wohlgemuth SE, Lees HA, Chung HY, Giovannini S, Leeuwenburgh C. Age-related activation of mitochondrial caspase-independent apoptotic signaling in rat gastrocnemius muscle. Mech Ageing Dev. 2008;129:542–549. doi: 10.1016/j.mad.2008.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marzetti E, Carter CS, Wohlgemuth SE, Lees HA, Giovannini S, Anderson B, et al. Changes in IL-15 expression and death-receptor apoptotic signaling in rat gastrocnemius muscle with aging and life-long calorie restriction. Mech Ageing Dev. 2009;130:272–280. doi: 10.1016/j.mad.2008.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marzetti E, Privitera G, Simili V, Wohlgemuth SE, Aulisa L, Pahor M, et al. Multiple pathways to the same end: mechanisms of myonuclear apoptosis in sarcopenia of aging. Sci World J. 2010;10:340–349. doi: 10.1100/tsw.2010.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marzetti E, Calvani R, Bernabei R, Leeuwenburgh C. Apoptosis in skeletal myocytes: a potential target for interventions against sarcopenia and physical frailty—a mini-review. Gerontology. 2012;58:99–106. doi: 10.1159/000330064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marzetti E, Lees HA, Manini TM, Buford TW, Aranda JM, Jr, Calvani R, et al. Skeletal muscle apoptotic signaling predicts thigh muscle volume and gait speed in community-dwelling older persons: an exploratory study. PLoS One. 2012;7:e32829. doi: 10.1371/journal.pone.0032829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayhew DL, Kim JS, Cross JM, Ferrando AA, Bamman MM. Translational signaling responses preceding resistance training-mediated myofiber hypertrophy in young and old humans. J Appl Physiol. 2009;107:1655–1662. doi: 10.1152/japplphysiol.91234.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller MJ, Thompson JH, Liu X, Eloby-Childress S, Sadowska-Krowicka H, Zhang XJ, et al. Failure of L-NAME to cause inhibition of nitric oxide synthesis: role of inducible nitric oxide synthase. Inflamm Res. 1996;45:272–276. doi: 10.1007/BF02280990. [DOI] [PubMed] [Google Scholar]
- Mitsuishi M, Miyashita K, Muraki A, Itoh H. Angiotensin II reduces mitochondrial content in skeletal muscle and affects glycemic control. Diabetes. 2009;58:710–717. doi: 10.2337/db08-0949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nègre-Salvayre A, Hirtz C, Carrera G, Cazenave R, Troly M, Salvayre R, et al. A role for uncoupling protein-2 as a regulator of mitochondrial hydrogen peroxide generation. FASEB J. 1997;11:809–815. [PubMed] [Google Scholar]
- Nilakantan V, Halligan NL, Nguyen TK, Hilton G, Khanna AK, Roza AM, et al. Post-translational modification of manganese superoxide dismutase in acutely rejecting cardiac transplants: role of inducible nitric oxide synthase. J Heart Lung Transplant. 2005;24:1591–1599. doi: 10.1016/j.healun.2005.01.009. [DOI] [PubMed] [Google Scholar]
- Nisoli E, Clementi E, Paolucci C, Cozzi V, Tonello C, Sciorati C, et al. Mitochondrial biogenesis in mammals: the role of endogenous nitric oxide. Science. 2003;299:896–899. doi: 10.1126/science.1079368. [DOI] [PubMed] [Google Scholar]
- Nussler AK, Billiar TR. Inflammation, immunoregulation, and inducible nitric oxide synthase. J Leukoc Biol. 1993;54:171–178. [PubMed] [Google Scholar]
- Pasini E, Le Douairon LS, Flati V, Assanelli D, Corsetti G, Speca S, et al. Effects of treadmill exercise and training frequency on anabolic signaling pathways in the skeletal muscle of aged rats. Exp Gerontol. 2012;47:23–28. doi: 10.1016/j.exger.2011.10.003. [DOI] [PubMed] [Google Scholar]
- Perreault M, Marette A. Targeted disruption of inducible nitric oxide synthase protects against obesity-linked insulin resistance in muscle. Nat Med. 2001;7:1138–1143. doi: 10.1038/nm1001-1138. [DOI] [PubMed] [Google Scholar]
- Piotrkowski B, Fraga CG, de Cavanagh EM. Mitochondrial function and nitric oxide metabolism are modified by enalapril treatment in rat kidney. Am J Physiol Regul Integr Comp Physiol. 2007;292:R1494–R1501. doi: 10.1152/ajpregu.00540.2006. [DOI] [PubMed] [Google Scholar]
- Pullen AH. The distribution and relative sizes of three histochemical fibre types in the rat tibialis anterior muscle. J Anat. 1977;123:1–19. [PMC free article] [PubMed] [Google Scholar]
- Rafikov R, Fonseca FV, Kumar S, Pardo D, Darragh C, Elms S, et al. eNOS activation and NO function: structural motifs responsible for the posttranslational control of endothelial nitric oxide synthase activity. J Endocrinol. 2011;210:271–284. doi: 10.1530/JOE-11-0083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid JL. From kinetics to dynamics: are there differences between ACE inhibitors? Eur Heart J. 1997;18(Suppl E):E14–E18. doi: 10.1093/eurheartj/18.suppl_E.14. [DOI] [PubMed] [Google Scholar]
- Reid MB, Lannergren J, Westerblad H. Respiratory and limb muscle weakness induced by tumor necrosis factor-alpha: involvement of muscle myofilaments. Am J Respir Crit Care Med. 2002;166:479–484. doi: 10.1164/rccm.2202005. [DOI] [PubMed] [Google Scholar]
- Rennie MJ. Anabolic resistance: the effects of aging, sexual dimorphism, and immobilization on human muscle protein turnover. Appl Physiol Nutr Metab. 2009;34:377–381. doi: 10.1139/H09-012. [DOI] [PubMed] [Google Scholar]
- Reuter H, Adam C, Grönke S, Zobel C, Frank KF, Müller-Ehmsen J, et al. The increased angiotensin II (type 1) receptor density in myocardium of type 2 diabetic patients is prevented by blockade of the renin–angiotensin system. Diabetologia. 2006;49:3067–3074. doi: 10.1007/s00125-006-0444-8. [DOI] [PubMed] [Google Scholar]
- Rice KM, Linderman JK, Kinnard RS, Blough ER. The Fischer 344/NNiaHSd × Brown Norway/BiNia is a better model of sarcopenia than the Fischer 344/NNiaHSd: a comparative analysis of muscle mass and contractile properties in aging male rat models. Biogerontology. 2005;6:335–343. doi: 10.1007/s10522-005-4808-0. [DOI] [PubMed] [Google Scholar]
- Santos EL, de Picoli SK, da Silva ED, Batista EC, Martins PJ, D'Almeida V, et al. Long term treatment with ACE inhibitor enalapril decreases body weight gain and increases life span in rats. Biochem Pharmacol. 2009;78:951–958. doi: 10.1016/j.bcp.2009.06.018. [DOI] [PubMed] [Google Scholar]
- Schmeisser A, Soehnlein O, Illmer T, Lorenz HM, Eskafi S, Roerick O, et al. ACE inhibition lowers angiotensin II-induced chemokine expression by reduction of NF-kappaB activity and AT1 receptor expression. Biochem Biophys Res Commun. 2004;325:532–540. doi: 10.1016/j.bbrc.2004.10.059. [DOI] [PubMed] [Google Scholar]
- Servais S, Couturier K, Koubi H, Rouanet JL, Desplanches D, Sornay-Mayet MH, et al. Effect of voluntary exercise on H2O2 release by subsarcolemmal and intermyofibrillar mitochondria. Free Radic Biol Med. 2003;35:24–32. doi: 10.1016/S0891-5849(03)00177-1. [DOI] [PubMed] [Google Scholar]
- Shankar RR, Wu Y, Shen HQ, Zhu JS, Baron AD. Mice with gene disruption of both endothelial and neuronal nitric oxide synthase exhibit insulin resistance. Diabetes. 2000;49:684–687. doi: 10.2337/diabetes.49.5.684. [DOI] [PubMed] [Google Scholar]
- Song W, Kwak HB, Kim JH, Lawler JM. Exercise training modulates the nitric oxide synthase profile in skeletal muscle from old rats. J Gerontol A Biol Sci Med Sci. 2009;64:540–549. doi: 10.1093/gerona/glp021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spier SA, Delp MD, Meininger CJ, Donato AJ, Ramsey MW, Muller-Delp JM. Effects of ageing and exercise training on endothelium-dependent vasodilatation and structure of rat skeletal muscle arterioles. J Physiol. 2004;556:947–958. doi: 10.1113/jphysiol.2003.060301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamler JS, Meissner G. Physiology of nitric oxide in skeletal muscle. Physiol Rev. 2001;81:209–237. doi: 10.1152/physrev.2001.81.1.209. [DOI] [PubMed] [Google Scholar]
- Thomas MM, Khan W, Betik AC, Wright KJ, Hepple RT. Initiating exercise training in late middle age minimally protects muscle contractile function and increases myocyte oxidative damage in senescent rats. Exp Gerontol. 2010;45:856–867. doi: 10.1016/j.exger.2010.07.003. [DOI] [PubMed] [Google Scholar]
- Torres SH, De Sanctis JB, de Briceño M, Hernández N, Finol HJ. Inflammation and nitric oxide production in skeletal muscle of type 2 diabetic patients. J Endocrinol. 2004;181:419–427. doi: 10.1677/joe.0.1810419. [DOI] [PubMed] [Google Scholar]
- Van Antwerpen P, Legssyer I, Zouaoui Boudjeltia K, Babar S, Moreau P, Moguilevsky N, et al. Captopril inhibits the oxidative modification of apolipoprotein B-100 caused by myeloperoxydase in a comparative in vitro assay of angiotensin converting enzyme inhibitors. Eur J Pharmacol. 2006;537:31–36. doi: 10.1016/j.ejphar.2006.03.022. [DOI] [PubMed] [Google Scholar]
- van de Wal RM, Plokker HW, Lok DJ, Boomsma F, van der Horst FA, van Veldhuisen DJ, et al. Determinants of increased angiotensin II levels in severe chronic heart failure patients despite ACE inhibition. Int J Cardiol. 2006;106:367–372. doi: 10.1016/j.ijcard.2005.02.016. [DOI] [PubMed] [Google Scholar]
- Villanueva C, Giulivi C. Subcellular and cellular locations of nitric oxide synthase isoforms as determinants of health and disease. Free Radic Biol Med. 2010;49:307–316. doi: 10.1016/j.freeradbiomed.2010.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang MX, Murrell DF, Szabo C, Warren RF, Sarris M, Murrell GA. Nitric oxide in skeletal muscle: inhibition of nitric oxide synthase inhibits walking speed in rats. Nitric Oxide. 2001;5:219–232. doi: 10.1006/niox.2001.0348. [DOI] [PubMed] [Google Scholar]
- Wei Y, Sowers JR, Clark SE, Li W, Ferrario CM, Stump CS. Angiotensin II-induced skeletal muscle insulin resistance mediated by NF-kappaB activation via NADPH oxidase. Am J Physiol Endocrinol Metab. 2008;294:E345–E351. doi: 10.1152/ajpendo.00456.2007. [DOI] [PubMed] [Google Scholar]
- Weisinger RS, Stanley TK, Begg DP, Weisinger HS, Spark KJ, Jois M. Angiotensin converting enzyme inhibition lowers body weight and improves glucose tolerance in C57BL/6J mice maintained on a high fat diet. Physiol Behav. 2009;98:192–197. doi: 10.1016/j.physbeh.2009.05.009. [DOI] [PubMed] [Google Scholar]
- Weissman BA, Gross SS (2001) Measurement of NO and NO synthase. Curr Protoc Neurosci 7.13.1–7.13.22 [DOI] [PubMed]
- Wilson KM, Magargal W, Berecek KH. Long-term captopril treatment. Angiotensin II receptors and responses. Hypertension. 1988;11:I148–I152. doi: 10.1161/01.hyp.11.2_pt_2.i148. [DOI] [PubMed] [Google Scholar]
- Wu JN, Edwards D, Berecek KH. Changes in renal angiotensin II receptors in spontaneously hypertensive rats by early treatment with the angiotensin-converting enzyme inhibitor captopril. Hypertension. 1994;23:819–822. doi: 10.1161/01.HYP.23.6.819. [DOI] [PubMed] [Google Scholar]
- Wu M, Katta A, Gadde MK, Liu H, Kakarla SK, Fannin J. Aging-associated dysfunction of Akt/protein kinase B: S-nitrosylation and acetaminophen intervention. PLoS One. 2009;4:e6430. doi: 10.1371/journal.pone.0006430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Hwang JC, Lees HA, Wohlgemuth SE, Knutson MD, Judge AR, et al. Long-term perturbation of muscle iron homeostasis following hindlimb suspension in old rats is associated with high levels of oxidative stress and impaired recovery from atrophy. Exp Gerontol. 2012;47:100–108. doi: 10.1016/j.exger.2011.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L, Zhu DY. Neuronal nitric oxide synthase: structure, subcellular localization, regulation, and clinical implications. Nitric Oxide. 2009;20:223–230. doi: 10.1016/j.niox.2009.03.001. [DOI] [PubMed] [Google Scholar]
- Zoll J, Monassier L, Garnier A, N'Guessan B, Mettauer B, Veksler V, et al. ACE inhibition prevents myocardial infarction-induced skeletal muscle mitochondrial dysfunction. J Appl Physiol. 2006;101:385–391. doi: 10.1152/japplphysiol.01486.2005. [DOI] [PubMed] [Google Scholar]