

Abstract
p38 Mitogen-activated protein kinase (MAPK) is one of the most ancient signaling molecules and is involved in multiple cellular processes, including cell proliferation, cell growth, and cell death. In the heart, enhanced activation of p38 MAPK is associated with ischemia/reperfusion injury and the onset of heart failure. In the present study, we investigated the function of p38 MAPK in regulating cardiac contractility and its underlying mechanisms. In cultured adult rat cardiomyocytes, activation of p38 MAPK by adenoviral gene transfer of an activated mutant of its upstream kinase, MKK3bE, led to a significant reduction in baseline contractility, compared with uninfected cells or those infected with a control adenoviral vector (Adv-β-galactosidase). The inhibitory effect of MKK3bE on contractility was largely prevented by coexpressing a dominant-negative mutant of p38 MAPK or treating cells with a p38 MAPK inhibitor, SB203580. Conversely, inhibition of endogenous p38 MAPK activity by SB203580 rapidly and reversibly enhanced cell contractility in a dose-dependent manner, without altering L-type Ca2+ currents or Ca2+i transients. MKK3bE-induced p38 activation had no significant effect on pHi, whereas SB203580 had a minor effect to elevate pHi. Furthermore, activation of p38 MAPK was unable to increase troponin I phosphorylation. Thus, we conclude that the negative inotropic effect of p38 MAPK is mediated by decreasing myofilament response to Ca2+, rather than by altering Ca2+i homeostasis and that the reduced myofilament Ca2+ sensitivity is unlikely attributable to troponin I phosphorylation or alterations in pHi. These findings reveal a novel function of p38 MAPK and shed a new light on our understanding of the coincidence of p38 MAPK activation and the onset of heart failure.
Keywords: p38 mitogen-activated protein kinase, cardiac contractility, excitation-contraction coupling, troponin I, intracellular pH
Mitogen-activated protein kinase (MAPK) superfamily is one of the most important signal transduction systems conserved in all eukaryotes.1–4 There are three major subgroups identified, including the extracellular signal–regulated kinase (ERK1/2), p38 MAPK, and c-jun-NH2-terminal kinase (JNK). p38 MAPK is a subfamily of stress-activated MAPKs (SAPKs) known to be involved in a multitude of cellular events, such as inflammation, cell injury, cell proliferation or differentiation, and cell growth or death.5–7
In the heart, activation of p38 MAPK has been observed in pressure overload or ischemia/infarction-induced cardiac hypertrophy and heart failure in humans8,12,13 and animal models.14–19 In cultured cardiac myocytes, activation of p38 MAPK induces myocyte hypertrophy and apoptosis18,20 and is also implicated in the preconditioning process and ischemia/reperfusion injury.21–25 Our recent studies have shown that β-adrenergic stimulation is able to activate p38 MAPK via a protein kinase A (PKA)-dependent mechanism and that activation of p38 MAPK provides a negative feedback to PKA-mediated positive contractile response in intact cardiac myocytes.26 Furthermore, evidence from our recent in vivo studies in transgenic mice has demonstrated that cardiac-specific activation of p38 MAPK markedly attenuates cardiac contractility.27 However, the mechanism underlying the negative inotropic effect of p38 MAPK is not yet well understood.
The goal of the present investigation was to determine the possible molecular and cellular mechanisms underlying the negative inotropic effect of p38 MAPK signaling in cardiac myocytes. To avoid the potential complication caused by developmental compensation in transgenic animals, in the present study, we have investigated the effects of p38 MAPK activation on cardiomyocyte contractility under well-controlled experimental settings, ie, in isolated, cultured adult rat ventricular myocytes in conjunction with specific genetic and pharmacological manipulations of p38 MAPK activity. Specifically, p38 MAPK signaling is either enhanced by adenoviral gene transfer of a constitutively activated specific p38 MAPK activator, MKK3bE,28 or suppressed by a dominant-negative mutant of p38 or a synthetic inhibitor, SB203580.29 Using these approaches, we have examined the possible effects of p38 MAPK activation or inhibition on the main events of cardiac excitation-contraction (E-C) coupling, including L-type Ca2+ currents (ICa), Ca2+i transients, and cell contraction and phosphorylation of contractile proteins as well as pHi.
The present results have revealed that activation of p38 MAPK decreases cell contractility, whereas inhibition of the kinase markedly increases cell contraction amplitude without altering ICa and Ca2+i transient, suggesting that p38 MAPK depresses the myofilament response to Ca2+i. Moreover, the present results rule out the possibility that change of pHi or phosphorylation of troponin I (a key myofilament protein involved in the regulation of myofilament response to Ca2+i)30,31 plays an essential role in p38 MAPK–induced suppression of myofilament Ca2+ response.
Materials and Methods
Isolation, Culture, and Adenoviral Infection of Adult Rat Ventricular Myocytes
Single cardiac myocytes were isolated from the hearts of Sprague-Dawley rats (2 to 3 months old, weight 225 to 300 grams; Charles River Laboratories, Kingston, NY) using a standard enzymatic technique, as described previously.32 All experimental protocols were performed in accordance with the Guide for the Care and Use of Laboratory Animals (NIH) and approved by the Animal Care and Use Committee at the National Institute on Aging. The isolated myocytes were then cultured and infected with HA-tagged MKK3bE adenoviral vector (Adv-MKK3bE) at a multiplicity of infection (MOI) of 400 (≈20 plaque-forming units [pfu]/mL), with a method recently developed in this laboratory.33 In a subset of experiments, myocytes were coinfected by Adv-p38dn (a p38 MAPK dominant-negative mutant) or Adv-β-gal (a marker gene, β-galactosidase) as a negative control, all at MOI of 400. Before culture, myocytes were washed 3 times with medium 199 supplemented with L-carnitine (2 mmol/L), N-2-mercaptopropionyl glycine (5 mmol/L), taurine (5 mmol/L), insulin (0.1 μmol/L), 2% FBS, and penicillin-streptomycin (100 IU/mL) and then plated at ≈0.5 × 104/cm2 with the same medium in the culture dishes precoated with 10 μg/mL mouse laminin. Adenovirus-mediated gene transfer was implemented by adding a minimal volume of the FBS-free medium 199 containing gene-carrying adenoviruses for 2 hours. All experiments were performed after 24 hours of adenoviral infection.
p38 MAPK, ERK, or JNK Phosphorylation and p38 MAPK Activity Assay
Phosphorylation of p38 MAPK, ERK, and JNK was measured by Western blotting with phosphospecific (phospho-) p38 MAPK, phospho-ERK, or phospho-JNK antibodies, as described previously.26 The same membrane was stripped in strip buffer (62.5 mmol/L Tris, 100 mmol/L β-mercaptoethanol, and 2% SDS, pH 6.7) at 50°C for 30 minutes and then reprobed with a second primary antibody to determine the total protein abundance of p38 MAPK, ERK, and JNK using a similar procedure.
p38 MAPK activity was detected by using MAPK assay kits (New England Biolabs), as previously described.26 To detect the inhibitory effect of SB203580 on p38 activity, 1 mg total protein was immunoprecipitated overnight with phospho-p38 MAPK monoclonal antibody. The complex was washed and divided equally into several tubes, and the reaction was performed in kinase buffer containing 200 μmol/L ATP and 2 μg activating transcription factor (ATF)-2 fusion protein (a substrate of p38 MAPK), with different concentrations of SB203580 for 30 minutes at 30°C. Samples were then subjected to SDS-PAGE, and immunoblotting was performed with anti–phospho-ATF-2 antibody.
Phosphorylation of Recombinant Troponin I
Purified recombinant proteins, including cardiac troponin complex (cTn), GST-ATFs, GST-p38 (wild type), GST-p38-mut (GST-p38 kinase dead mutant), His-tagged MKK6bE (His-MKK6bE, an activator of p38 MAPK), and PKAc (protein kinase A catalytic subunit) were incubated in the presence of 32P-γ-ATP in kinase buffer at 30°C for 30 minutes. Phosphorylated proteins were separated by 4% to 12% SDS PAGE and the 32P-labeled proteins were visualized using a Phospho-Imager (Molecular Dynamics).
pHi Measurement
Intracellular pH was measured by a fluorescent indicator, SNARF-1 (carboxy-seminaphthorhodafluor-1), as described previously.34 The emission spectrum of SNARF-1, when excited at 530 nm, contains two well-separated peaks at ≈590 and 640 nm, corresponding to the acidic and basic forms of the indicator. The absolute values of cytosolic pH in individual myocytes, before and after SB203580 treatment, were obtained from a standard pH curve using in vitro calibration.
Cell Contraction Measurement
Cells were placed on the stage of an inverted microscope (Zeiss, model IM-35), perfused with the HEPES-buffered solution (in mmol/L: NaCl 137, KCl 5.4, MgCl2 1.2, NaH2PO4 1, CaCl2 1, glucose 20, and HEPES 20 [pH 7.4]), and electrically stimulated at 0.5 Hz at 23°C. Cell length was monitored by an optical edge-tracking method using a photodiode array (model 1024 SAQ; Reticon) at a time resolution of 3 ms. Cell contraction was measured by the percent shortening of cell length after electrical stimulation.
Simultaneous Recordings of Confocal Ca2+ Imaging, ICa, and Cell Contraction
Myocytes were placed on the stage of a Zeiss LSM-410 inverted confocal microscope (Carl Zeiss, Inc) and excited by the 488-nm line of an argon laser. Intracellular Ca2+ transients and cell shortening were measured using a Ca2+ indicator, fluo-4 AM, and line-scan imaging (the scan line oriented along the long axis of the myocyte), as described previously.35 The perforated patch-clamp technique was used to simultaneously record ICa, with an Axopatch 200B patch-clamp amplifier (Axon Instruments, Inc). Patch pipette (1.5 to 2.5 MΩ) was filled with a solution containing (in mmol/L) CsCl 120, NaCl2 10, MgCl 1, MgATP 5, HEPES 10, K5fluo-4 0.2, and β-escin 0.05. The seal resistance was ranged from 7 to 20 MΩ following 10 to 30 minutes after sealing. Patches that failed to fall in this range were discarded. After the establishment of voltage control of the cell, the cell perfusion solution was switched to a recording solution at room temperature (23°C) containing (in mmol/L) NaCl 137, CsCl 5.4, MgCl2 1.2, NaH2PO4 1, CaCl2 2, glucose 20, HEPES 20, and tetrodotoxin 0.02 (pH 7.4) in the presence or absence of SB203580. ICa was elicited by a 200-ms depolarization pulse from a holding potential of −50 mV to a test potential of 0 mV at 0.1Hz. The magnitude of ICa was indexed by the difference between the peak inward current and the current at the end of the 200-ms pulse.
Materials and Antibodies
FBS was purchased from Sigma. Laminin and penicillin-streptomycin were purchased from Gibco (Gaithersburg). SB203580 was purchased from Calbiochem (in the stock solution, SB203580 was dissolved in water at 1 mmol/L). Rabbit polyclonal antibodies against phospho-p38 and p38 were purchased from New England Biolabs. Anti-HA antibody was purchased from Boehringer Mannheim. Antibodies against phospho- and total SAPK/p38, ERK, and JNK were purchased from Cell Signal. Purified recombinant troponin complex includes cTnC (1–161) and cTnI (1–211), a kind gift from Dr Paul Rosevear (Washington University, St. Louis, Mo).
Statistical Evaluations
All data are presented as mean ± SE. Comparisons between control and treatments were performed using Student’s unpaired t test or ANOVA when appropriate. A value of P < 0.05 was considered to be statistically significant.
Results
Specific Activation of p38 MAPK in Cultured Rat Ventricular Myocytes by Adenovirus-Mediated Expression of MKK3bE
To study the functional role of p38 MAPK in regulating cardiomyocyte contractility, adult rat ventricular myocytes were infected with an adenovirus vector carrying HA-tagged MKK3bE (Adv-MKK3bE) at MOI 400 in culture for 24 hours. The efficiency of adenoviral infection was determined by infecting cells with an adenoviral vector carrying a marker gene, β-galactosidase (Adv-β-gal, MOI 400). Nearly 100% of myocytes were infected by Adv-β-gal, as indicated by β-gal–driven color reaction (Figure 1A). These infected myocytes still retained the rod-shaped morphology and functional integrity. Infection of myocytes with Adv-MKK3bE led to significant expression of MKK3bE protein, as detected by Western blotting using anti-HA antibodies (Figure 1B). As a result, the activity of p38 MAPK, an immediate substrate of MKK3bE, was also significantly increased, as manifested by 3.4-fold increase in its phosphorylation (Figure 1B). In contrast, the phosphorylation status of other members of the MAPK family, including JNK and ERK, was not affected in myocytes expressing MKK3bE (Figure 1B). These results suggest that adenovirus-mediated expression of MKK3bE in cultured adult rat ventricular myocytes specifically activates p38 MAPK.
Figure 1.
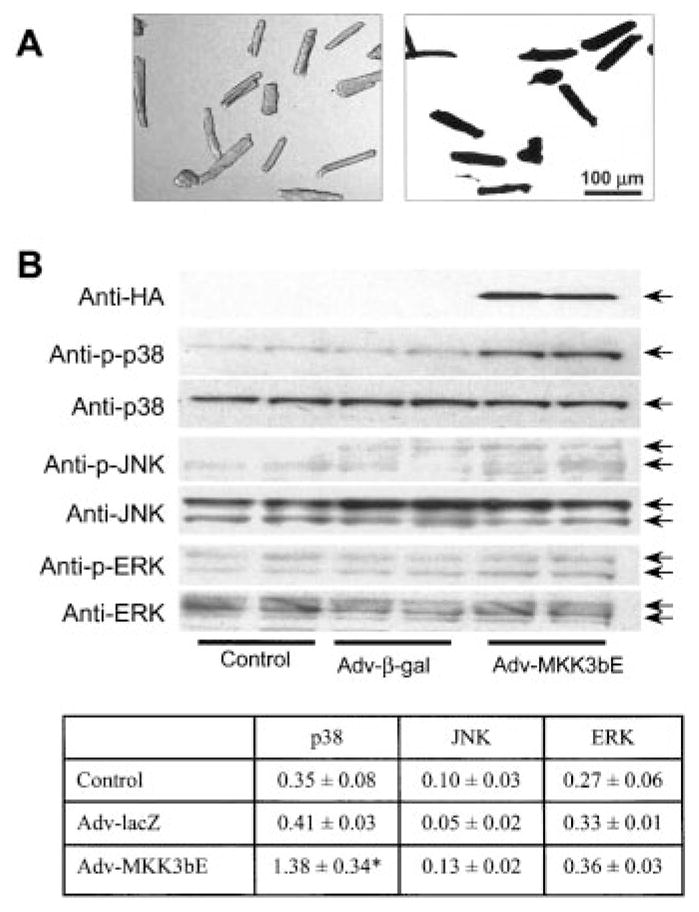
Specific activation of p38 MAPK in cultured adult rat ventricular myocytes by recombinant adenovirus-mediated gene transfer. A, Adult rat ventricular myocytes were cultured for 24 hours in the absence (left, transillumination image) or presence (right, β-galactosidase staining) of an adenoviral vector carrying a marker gene, β-galactosidase (Adv-β-gal) at MOI 400. Note that almost 100% of myocytes were infected by Adv-β-gal. B, Adenovirus-mediated expression of HA-tagged MKK3bE in cultured adult rat ventricular myocytes resulted in a specific increase in phosphorylation of p38 MAPK but not JNK or ERK. The inset shows the average data on phosphorylation of p38 MAPK, JNK, and ERK assayed by Western blot. Data are presented as mean ± SE of at least 3 independent experiments (*P < 0.01).
Activation of p38 MAPK Suppresses Contractility of Cardiac Myocytes
To determine the possible role of p38 MAPK activation in regulating cardiac contractility, the amplitude and kinetics of cell contraction were measured in myocytes infected with Adv-MKK3bE and in those infected by Adv-β-gal. As shown in Figure 2A, the contraction amplitude was significantly reduced by ≈45% in myocytes infected by Adv-MKK3bE compared with that in uninfected myocytes (3.9 ± 0.37% versus 7.6 ± 0.5% of resting cell length, respectively, P < 0.01) or those infected with Adv-β-gal (6.7 ± 0.4% of rest cell length, P < 0.01). Concomitantly, activation of p38 MAPK decreased maximal velocity of shortening (VS) and relaxation (VR) in myocytes expressing MKK3bE relative to uninfected controls (Figure 2B), without altering the time to peak or 50% relaxation time (data not shown). These results suggest that activation of p38 MAPK by MKK3bE overexpression markedly suppresses baseline contractility of intact cardiac myocytes. This is consistent with the fact that cardiac contractility was profoundly depressed in transgenic mice overexpressing MKK3bE in the heart.27
Figure 2.

MKK3bE-induced reduction of baseline contractility of cardiac myocytes is mediated specifically by p38 MAPK activation. A, Cell contraction amplitude is markedly decreased in myocytes expressing MKK3bE compared with uninfected myocytes or those infected by Adv-β-gal. Coexpression of a p38 dominant-negative mutant (p38dn) reverses the MKK3bE-induced reduction of contraction amplitude. *P < 0.01 vs cells in the absence of Adv-MKK3bE or those coinfected by Adv-MKK3bE and Adv-p38dn. B, Activation of p38 MAPK significantly reduces the maximal shortening velocity (VS) and relaxation velocity (VR); this effect is also largely prevented by coexpression of the p38 dominant-negative mutant. *P < 0.01 vs the control group and the group coinfected by Adv-MKK3bE and Adv-p38dn. C, Inhibition of p38 MAPK by SB203580 (10 μmol/L) increases the contraction amplitude in myocytes infected by Adv-MKK3bE and control cells. D, Relative potentiating effect of SB203580 on augmenting cell contraction is significantly greater in Adv-MKK3bE–infected myocytes compared with the uninfected cells (the cell numbers are the same as in panel C). The number of cells for each group is indicated in the figure (5 hearts for each group).
To further demonstrate that the reduction of contractility in myocytes expressing MKK3bE is mediated specifically by p38 MAPK activation, a p38 dominant-negative mutant (p38dn) was coexpressed with MKK3bE in cultured cardiomyocytes. Whereas expression of the p38dn alone had no significant effect on contraction amplitude, coexpression of the p38dn mutant, but not β-gal, with MKK3bE fully restored the diminished baseline contractility (Figure 2A). Similarly, the reduction of the maximal shortening or relaxation velocity induced by MKK3bE overexpression was also largely prevented by coexpressing the p38dn mutant, but not β-gal (75.8 ± 11.6 and 69.3 ± 10.8 μm/ms for VS and VR, respectively, P > 0.05 versus MKK3bE-infected cells) (Figure 2B). In addition, inhibition of p38 MAPK with SB203580 (10 μmol/L) increased contraction amplitude in control cells and rescued the attenuated contractility in cells expressing MKK3bE (Figure 2C). The relative potency of SB203580 to augment the contraction amplitude was significantly greater in myocytes infected by Adv-MKK3bE compared with the uninfected cells (Figure 2D). Taken together, these results suggest that MKK3bE overexpression-induced contractile suppression is mediated by p38 MAPK signaling.
Inhibiting p38 MAPK Activity Enhances Contractility of Cardiac Myocytes
We investigated the possible response of cardiomyocyte contractility to p38 MAPK inhibition by the p38 MAPK inhibitor SB203580. Figure 3A shows a typical example of the inhibitory effect of SB203580 on endogenous p38 MAPK activity assayed by phosphorylation of its substrate protein ATF-2. SB203580 inhibited p38 MAPK activity in a dose-dependent manner. Treatment of adult rat cardiac myocytes with SB203580 (25 μmol/L) rapidly increased the cell contraction amplitude by nearly 2-fold; this effect was reversible on washout (Figure 3B). In accordance with its inhibitory effect on p38 MAPK activity, the positive inotropic effect of SB203580 in cardiomyocytes is dose dependent, with a threshold concentration at 1 μmol/L, and a maximal augmentation of 2.5-fold in contraction amplitude at 50 μmol/L (Figure 3C), when the endogenous p38 activity was almost completely inhibited (Figure 3A). These data indicate that the suppression of contractility is one of the primary consequences of p38 MAPK activation in cardiac myocytes.
Figure 3.

Inhibition of p38 MAPK activity by SB203580 induces a positive inotropic effect. A, Dose-dependent inhibitory effect of SB203580 (SB) on endogenous p38 MAPK activity. B, Rapid and reversible effect of SB203580 (SB, 25 μmol/L) to increase the contraction amplitude in a representative uninfected cardiomyocyte. Cell shortening is indicated by the upward deflection in the continuous chart recording (top). Individual contractile traces (bottom) (cell shortening indicated by the downward deflection) were obtained at the time points, as indicated (a, control; b, in the presence of SB; and c, after washout of SB), with a higher time resolution. C, Average dose response of contraction amplitude to SB203580. Data are presented as percent of control (n = 9 to 14 cells from 7 hearts for each data point).
Cellular Mechanism Underlying p38 MAPK–Mediated Modulation of E-C Coupling
To understand the cellular mechanism underlying the negative inotropic effect of p38 MAPK, we determined the possible effect of the p38 MAPK inhibitor SB203580 on the major events of E-C coupling, including sarcolemmal L-type Ca2+ current (ICa), Ca2+i transient, and cell contraction, simultaneously recorded using confocal imaging in conjunction with whole-cell patch-clamp techniques. Figure 4 shows a representative example of the effect of SB203580 (SB, 20 μmol/L), displaying, from top to bottom, line-scan images of Ca2+i, spatially averaged Ca2+ transients, cell contraction, whole-cell ICa, and membrane potential. SB203580 clearly increased the contraction amplitude, without augmenting the simultaneously recorded ICa or Ca2+i transient in the very same cell. In fact, SB203580 slightly reduced the amplitudes of ICa and Ca2+i transients, as illustrated in the superimposed traces. On average, SB203580 augmented the contraction amplitude to 127.2 ± 4.2% of control (n = 5, P < 0.02), without elevating either ICa (7.73 ± 0.28 and 7.08 ± 0.36 pA/pF, control versus SB, n = 5) or Ca2+i transients (4.7 ± 0.4 and 4.3 ± 0.4 F/F0, control versus SB, n = 5). The fact that SB203580 selectively enhanced cell contraction without reducing ICa or Ca2+i transients suggests that p38 MAPK reduces the contractility by desensitizing the response of contractile myofilaments to Ca2+i.
Figure 4.

Simultaneous recording of Ca2+i transients, cell contraction, and ICa in the presence or absence of the p38 MAPK inhibitor SB203580. From top to bottom, Confocal image of fluo-4 fluorescence, spatially averaged fluo-4 fluorescent transient (F/F0), cell contraction, whole-cell ICa, and the voltage-clamp protocol. Data were recorded immediately before and 5 minutes after application of SB203580 (SB, 20 μmol/L) in a typical rat ventricular myocyte. Similar results were obtained in 5 cells.
Response of pHi or Phosphorylation of Troponin I to p38 MAPK Activation
Phosphorylation of troponin I and pHi are the two major mechanisms that may affect myofilament Ca2+ response. As shown in Figure 5, top, activated p38 MAPK failed to directly phosphorylate purified troponin I in vitro (lanes 7 and 8), whereas it did markedly phosphorylate recombinant ATF-2 (lanes 3 and 4), its native substrate. As a positive control, activated PKA induced an evident increase in phosphorylation of troponin I (lanes 10 and 11), as previously documented.30,31 In addition, we examined the possible involvement of pHi using a fluorescent indicator, SNARF-1. Activation of p38 MAPK by expressing MKK3bE tended to decrease pHi compared with myocytes expressing β-gal, but the difference in pH between the two groups was not statistically significant (7.11 ± 0.02 and 7.09 ± 0.02 in cells infected by Adv-β-gal and Adv-MKK3bE, respectively; n = 16 cells for both groups, P > 0.05), whereas inhibition of p38 MAPK by SB203580 (20 μmol/L for 10 minutes) caused a minor yet significant increase in pHi (7.14 ± 0.01 and 7.17 ± 0.01 in the absence and presence of SB203580, respectively; n = 12 cells, P > 0.05) in freshly isolated myocytes (control cells). These results suggest that pHi does not play a major role in p38 MAPK–elicited negative inotropic effect. Taken together, p38 MAP-K–induced suppression of myofilament Ca2+ response is unlikely mediated by intracellular acidification or troponin I phosphorylation.
Figure 5.

Response of troponin I phosphorylation to p38 MAPK activation. p38 MAPK activation cannot directly phosphorylate cardiac troponin I in vitro. Lanes 3 and 4, p38 MAPK–induced phosphorylation of recombinant ATF-2, a native substrate for p38. Lanes 10 and 11, PKA-induced phosphorylation of troponin I, as a positive control. Lanes 7 and 8, Absence of troponin I phosphorylation by p38 MAPK under the same conditions. Purified recombinant proteins, including cardiac troponin complex (cTn), GST-ATFs, GST-p38 (wild type), GST-p38-mut (GST-p38 kinase dead mutant), His-tagged MKK6bE (His-MKK6bE, an activator of p38 MAPK), and PKAc (protein kinase A catalytic subunit) were incubated as indicated in the presence of 32P-γ-ATP at 30°C for 30 minutes (see Materials and Methods).
Discussion
Role of p38 MAPK in Regulating Cardiac Contractility
Intensive studies have focused on the involvement of p38 MAPK in cellular responses to various assaults or stresses (eg, UV light, osmotic stress, heat shock, and mechanical or chemical stress). In the present study, we focused on the functional role of p38 MAPK in regulating cardiac contractility. Our results provide several lines of evidence to indicate that increased p38 MAPK activation markedly inhibits the contractility of cardiac myocytes. First, specific activation of p38 MAPK by adenovirus-mediated gene transfer of MKK3bE results in a significant reduction in the contraction amplitude. This is in good agreement with the observation that in transgenic animals with targeted activation of p38 MAPK, in a cardiac-specific manner, exhibit severely attenuated cardiac contractility.27 Second, the suppression of contractility by MKK3bE expression is specifically prevented by coexpressing a p38 MAPK dominant-negative mutant and can be rescued by the p38 MAPK inhibitor SB203580. Furthermore, inhibition of endogenous p38 MAPK by SB203580 rapidly and reversibly increases cell contractility in a dose-dependent manner. SB203580 at 50 μmol/L, which fully inhibits the endogenous p38 MAKP activity (Figure 3A), enhances the contraction amplitude by 2.5-fold. Taken together, we conclude that p38 MAPK activation exerts a robust negative inotropic effect in intact cardiac myocytes. This finding not only reveals a heretofore unappreciated function of p38 MAPK but also unravels a novel mechanism for the regulation of cardiac contractility by stress-activated signals, which may have significant implications in the pathophysiology of heart failure.
Cellular Mechanisms of p38 MAPK–Induced Negative Inotropic Effect: Suppression of Myofilament Ca2+ Response
We investigated the possible mechanism underlying the negative inotropic effect of p38 MAPK by examining the response of ICa, Ca2+i transient, and cell contraction to p38 MAPK inhibition with SB203580. The p38 MAPK inhibitor significantly increases cell contraction amplitude but slightly decreases the simultaneously recorded ICa and Ca2+i transient (Figure 4). This suggests that the negative inotropic effect of p38 MAPK is largely mediated by reducing the responsiveness of myofilaments to Ca2+i. It is noteworthy that the contractile response to SB203580 is considerably greater in field-stimulated myocytes (Figures 3B and 3C) than in voltage-clamped cells (Figure 4). This suggests that SB203580 might enhance myocyte contractility, in part, by changing action potential configuration.
Multiple mechanisms might be involved in regulating the interaction between myofilaments and Ca2+, including alterations in phosphorylation status of myofilament proteins and pHi. In particular, phosphorylation of troponin I is shown to decrease the Ca2+ affinity of troponin C that results in a reduced ability of Ca2+ to activate myofilaments.30,31 However, our in vitro assay demonstrates that p38 MAPK is unable to directly phosphorylate troponin I, suggesting that troponin I is not a direct downstream target of p38 MAPK. To further dissect the mechanism of p38 MAPK–mediated reduction in myocyte contractility, we also examined the response of pHi. Activation of p38 MAPK by overexpressing MKK3bE tends to decrease pHi, but the effect is not significant, whereas inhibition of p38 MAPK by SB203580 slightly elevates pHi. These data suggest that even if the change in pHi were involved in p38 MAPK–mediated negative inotropic effect, its contribution might be minor. Thus, p38 MAPK–induced suppression of cardiac contractility is unlikely mediated by intracellular acidification or troponin I phosphorylation.
It has been demonstrated that heat shock proteins such as HSP27 and αB-crystallin are phosphorylation targets of p38 MAPK in cardiac myocytes, and upon phosphorylation, those p38 MAPK target proteins are able to associate with cytoskeletal and sarcomere structures.36–39 Further studies are merited to determine whether p38 MAPK–mediated phosphorylation of these heat shock proteins alters myofilament response to Ca2+i, thus contributing to the negative inotropic effect.
Physiological and Pathological Relevance of p38 MAPK Activation in the Heart
It has been shown that β-adrenergic receptor–mediated activation of p38 MAPK negatively regulates the concurrent PKA-induced positive contractile response in intact cardiac myocytes.26 The opposing effects of β-adrenergic stimulation and p38 MAPK activation on cardiac contractility may have important physiological and pathological relevance. Because increased p38 MAPK activation is associated with the onset of heart failure,40 ischemic or reperfusion injury, and in vivo pressure overload,14–19 the negative inotropic effect of p38 MAPK and its inhibitory effect on β-adrenergic receptor/PKA-mediated contractile response may contribute, at least in part, to the diminished cardiac contractility under those pathological circumstances. Indeed, a number of studies have demonstrated that inhibiting p38 MAPK signaling improves contractile function in ischemia/reperfusion-injured hearts.13,19 Further studies are urgently awaited to evaluate such a strategy in other heart failure models induced by either genetic or physiological manipulations.
In summary, we have presented the first cellular evidence that enhanced p38 MAPK activation negatively regulates cardiomyocyte contractility, whereas inhibition of p38 MAPK activity leads to a positive inotropic effect. These findings not only reveal a novel function of p38 MAPK but also provide new insights for better understanding of the implication of enhanced p38 MAPK signaling in cardiac dysfunction under certain pathophysiological conditions, such as cardiac ischemic/reperfusion injury or chronic heart failure.
Acknowledgments
This work is supported in part by NIH grant HL62311 (Y.W.), NIH intramural research grant (H.C. and R.-P.X.), and Major State Basic Research Development Program of China (H.C.). The authors would like to thank Dr Edward G. Lakatta for his critical comments and Bruce Ziman for his excellent technical support.
References
- 1.Marshall CJ. Specificity of receptor tyrosine kinase signaling: transient versus sustained extracellular signal-regulated kinase activation. Cell. 1995;80:179–185. doi: 10.1016/0092-8674(95)90401-8. [DOI] [PubMed] [Google Scholar]
- 2.Cobb MH, Goldsmith EJ. How MAP kinases are regulated. J Biol Chem. 1995;270:14843–14846. doi: 10.1074/jbc.270.25.14843. [DOI] [PubMed] [Google Scholar]
- 3.Minder A, Karin M. Regulation and function of the JNK subgroup of MAP kinases. Biochim Biophys Acta. 1997;1333:F85–F104. doi: 10.1016/s0304-419x(97)00018-8. [DOI] [PubMed] [Google Scholar]
- 4.Kyriakis JM, Avruch J. Sounding the alarm: protein kinase cascades activated by stress and inflammation. J Biol Chem. 1996;271:24313–24316. doi: 10.1074/jbc.271.40.24313. [DOI] [PubMed] [Google Scholar]
- 5.Ono K, Han J. The p38 signal transduction pathway: activation and function. Cell Signal. 2000;12:1–13. doi: 10.1016/s0898-6568(99)00071-6. [DOI] [PubMed] [Google Scholar]
- 6.English J, Pearson G, Wilsbacher J, Swantek J, Karandikar M, Xu S, Cobb MH. New insights into the control of MAP kinase pathways. Exp Cell Res. 1999;1253:255–270. doi: 10.1006/excr.1999.4687. [DOI] [PubMed] [Google Scholar]
- 7.Nebreda AR, Porras A. p38 MAP kinases: beyond the stress response. Trends Biochem Sci. 2000;25:257–260. doi: 10.1016/s0968-0004(00)01595-4. [DOI] [PubMed] [Google Scholar]
- 8.Tamura K, Sudo T, Senftleben U, Dadak AM, Johnson R, Karin M. Requirement for p38α in erythropoietin expression: a role for stress kinases in erythropoiesis. Cell. 2000;102:221–231. doi: 10.1016/s0092-8674(00)00027-1. [DOI] [PubMed] [Google Scholar]
- 9.Deleted in proof.
- 10.Deleted in proof.
- 11.Deleted in proof.
- 12.Cook SA, Sugden PH, Clerk A. Activation of c-Jun N-terminal kinases and p38-mitogen-activated protein kinases in human heart failure secondary to ischaemic heart disease. J Mol Cell Cardiol. 1999;31:1429–1434. doi: 10.1006/jmcc.1999.0979. [DOI] [PubMed] [Google Scholar]
- 13.Cain BS, Meldrum DR, Meng X, Dinarello CA, Shames BD, Banerjee A, Harken AH. p38-MAPK inhibition decreases TNF-α production and enhances postischemic human myocardial function. J Surg Res. 1999;83:7–12. doi: 10.1006/jsre.1998.5548. [DOI] [PubMed] [Google Scholar]
- 14.Bogoyevitch MA, Gillespie BJ, Ketterman AJ, Fuller SJ, Ben LR, Ashworth A, Marshall CJ, Sugden PH. Stimulation of the stress-activated mitogen-activated protein kinase subfamilies in perfused heart: p38/RK mitogen-activated protein kinases and c-Jun N-terminal kinases are activated by ischemia/reperfusion. Circ Res. 1996;79:162–173. doi: 10.1161/01.res.79.2.162. [DOI] [PubMed] [Google Scholar]
- 15.Yin T, Sandhu G, Wolfgang CD, Burrier A, Webb RL, Rigel DF, Hai T, Whelan J. Tissue-specific pattern of stress kinase activation in ischemic/reperfused heart and kidney. J Biol Chem. 1997;272:19943–19950. doi: 10.1074/jbc.272.32.19943. [DOI] [PubMed] [Google Scholar]
- 16.Clerk A, Fuller SJ, Michael A, Sugden PH. Stimulation of “stress-regulated” mitogen-activated protein kinases (stress-activated protein kinases/c-Jun N-terminal kinases and p38-mitogen-activated protein kinases) in perfused rat hearts by oxidative and other stresses. J Biol Chem. 1998;273:7228–7234. doi: 10.1074/jbc.273.13.7228. [DOI] [PubMed] [Google Scholar]
- 17.Izumi Y, Kim S, Murakami T, Yamanaka S, Iwao H. Cardiac mitogen-activated protein kinase activities are chronically increased in stroke-prone hypertensive rats. Hypertension. 1998;31:50–56. doi: 10.1161/01.hyp.31.1.50. [DOI] [PubMed] [Google Scholar]
- 18.Wang Y, Huang S, Sah VP, Ross J, Jr, Brown JH, Han J, Chien KR. Cardiac muscle cell hypertrophy and apoptosis induced by distinct members of the p38 mitogen-activated protein kinase family. J Biol Chem. 1998;273:2161–2168. doi: 10.1074/jbc.273.4.2161. [DOI] [PubMed] [Google Scholar]
- 19.Ma XL, Kumar S, Gao F, Louden CS, Lopez BL, Christopher TA, Wang C, Lee JC, Feuerstein GZ, Yue TL. Inhibition of p38 mitogen-activated protein kinase decreases cardiomyocyte apoptosis and improves cardiac function after myocardial ischemia and reperfusion. Circulation. 1999;99:1685–1691. doi: 10.1161/01.cir.99.13.1685. [DOI] [PubMed] [Google Scholar]
- 20.Zechner D, Thuerauf DJ, Hanford DS, McDonough PM, Glembotski CC. A role for the p38 mitogen-activated protein kinase pathway in myocardial cell growth, sarcomeric organization, and cardiac-specific gene expression. J Cell Biol. 1997;139:115–127. doi: 10.1083/jcb.139.1.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yue TL, Wang C, Gu JL, Ma XL, Kumar S, Lee JC, Feuerstein GZ, Thomas H, Maleeff B, Ohlstein EH. Inhibition of extracellular signal-–regulated kinase enhances ischemia/reoxygenation-induced apoptosis in cultured cardiac myocytes and exaggerates reperfusion injury in isolated perfused heart. Circ Res. 2000;86:692–699. doi: 10.1161/01.res.86.6.692. [DOI] [PubMed] [Google Scholar]
- 22.Marber MS. Ischemic preconditioning in isolated cells. Circ Res. 2000;86:926–931. doi: 10.1161/01.res.86.9.926. [DOI] [PubMed] [Google Scholar]
- 23.Ping P, Murphy E. Role of p38 mitogen-activated protein kinases in preconditioning: a detrimental factor or a protective kinase? Circ Res. 2000;86:921–922. doi: 10.1161/01.res.86.9.921. [DOI] [PubMed] [Google Scholar]
- 24.Mackay K, Mochly-Rosen D. An inhibitor of p38 mitogen-activated protein kinase protects neonatal cardiac myocytes from ischemia. J Biol Chem. 1999;274:6272–6279. doi: 10.1074/jbc.274.10.6272. [DOI] [PubMed] [Google Scholar]
- 25.Barancik M, Htun P, Strohm C, Kilian S, Schaper W. Inhibition of the cardiac p38-MAPK pathway by SB203580 delays ischemic cell death. J Cardiovasc Pharmacol. 2000;35:474–483. doi: 10.1097/00005344-200003000-00019. [DOI] [PubMed] [Google Scholar]
- 26.Zheng M, Zhang SJ, Zhu WZ, Ziman B, Kobilka B, Xiao RP. β2-Adrenergic receptor-induced p38-MAPK activation is mediated by protein kinase A rather than by Gi or Gβγ (in adult mouse cardiomyocytes. J Biol Chem. 2000;275:40635–40640. doi: 10.1074/jbc.M006325200. [DOI] [PubMed] [Google Scholar]
- 27.Liao P, Georgakopoulos D, Kovacs A, Zheng M, Lerner D, Pu H, Saffitz J, Chien K, Xiao RP, Kass DA, Wang Y. The in vivo role of p38 MAP kinases in cardiac remodeling and restrictive cardiomyopathy. Proc Natl Acad Sci U S A. 2001;98:12283–12288. doi: 10.1073/pnas.211086598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Han J, Wang X, Jiang Y, Ulevitch RJ, Lin S. Identification and characterization of a predominant isoform of human MKK3. FEBS Lett. 1997;403:19–22. doi: 10.1016/s0014-5793(97)00021-5. [DOI] [PubMed] [Google Scholar]
- 29.Barancik M, Bohacova VV, Kvackajova J, Hudecova S, Krizanova O, Breier A. SB203580, a specific inhibitor of p38-MAPK pathway, is a new reversal agent of P-glycoprotein-mediated multidrug resistance. Eur J Pharm Sci. 2001;14:29–36. doi: 10.1016/s0928-0987(01)00139-7. [DOI] [PubMed] [Google Scholar]
- 30.Li G, Martin AF, Solaro RJ. Localization of regions of troponin I important in deactivation of cardiac myofilaments by acidic pH. J Mol Cell Cardiol. 2001;33:1309–1320. doi: 10.1006/jmcc.2000.1392. [DOI] [PubMed] [Google Scholar]
- 31.Solaro RJ. Troponin I, stunning, hypertrophy, and failure of the heart. Circ Res. 1999;84:122–124. doi: 10.1161/01.res.84.1.122. [DOI] [PubMed] [Google Scholar]
- 32.Spurgeon HA, Stern MD, Baartz G, Raffaeli S, Hansford RG, Talo A, Lakatta EG, Capogrossi MC. Simultaneous measurement of Ca2+, contraction, and potential in cardiac myocytes. Am J Physiol. 1990;258:H574–H586. doi: 10.1152/ajpheart.1990.258.2.H574. [DOI] [PubMed] [Google Scholar]
- 33.Zhou YY, Wang SQ, Zhu WZ, Chruscinski A, Kobilka BK, Ziman B, Wang S, Lakatta EG, Cheng H, Xiao RP. Culture and adenoviral infection of adult mouse cardiac myocytes: methods for cellular genetic physiology. Am J Physiol. 2000;279:H429–H436. doi: 10.1152/ajpheart.2000.279.1.H429. [DOI] [PubMed] [Google Scholar]
- 34.Blank PS, Silverman HS, Chung OY, Hogue BA, Stern MD, Hansford RG, Lakatta EG, Capogrossi MC. Cytosolic pH measurements in single cardiac myocytes using carboxy-seminaphthorhodafluor-1. Am J Physiol. 1992;263:H276–H284. doi: 10.1152/ajpheart.1992.263.1.H276. [DOI] [PubMed] [Google Scholar]
- 35.Cheng H, Lederer WJ, Cannell MB. Calcium sparks: elementary events underlying excitation-contraction coupling in heart muscle. Science. 1993;262:740–744. doi: 10.1126/science.8235594. [DOI] [PubMed] [Google Scholar]
- 36.Dana A, Skarli M, Papakrivopoulou J, Yellon DM. Adenosine A1 receptor induced delayed preconditioning in rabbits: induction of p38 mitogen-activated protein kinase activation and Hsp27 phosphorylation via a tyrosine kinase– and protein kinase C–dependent mechanism. Circ Res. 2000;86:989–997. doi: 10.1161/01.res.86.9.989. [DOI] [PubMed] [Google Scholar]
- 37.Armstrong SC, Delacey M, Ganote CE. Phosphorylation state of hsp27 and p38-MAPK during preconditioning and protein phosphatase inhibitor protection of rabbit cardiomyocytes. J Mol Cell Cardiol. 1999;31:555–567. doi: 10.1006/jmcc.1998.0891. [DOI] [PubMed] [Google Scholar]
- 38.Hoover HE, Thuerauf DJ, Martindale JJ, Glembotski CC. αB-Crystallin gene induction and phosphorylation by MKK6-activated p38: a potential role for αB-crystallin as a target of the p38 branch of the cardiac stress response. J Biol Chem. 2000;275:23825–23833. doi: 10.1074/jbc.M003864200. [DOI] [PubMed] [Google Scholar]
- 39.Sakamoto K, Urushidani T, Nagao T. Translocation of HSP27 to sarcomere induced by ischemic preconditioning in isolated rat hearts. Biochem Biophys Res Commun. 2000;269:137–142. doi: 10.1006/bbrc.2000.2233. [DOI] [PubMed] [Google Scholar]
- 40.Adams JW, Sakata Y, Davis MG, Sah VP, Wang Y, Liggett SB, Chien KR, Brown JH, Dorn GW., 2nd Enhanced Gαq signaling: a common pathway mediates cardiac hypertrophy and apoptotic heart failure. Proc Natl Acad Sci U S A. 1998;95:10140–10145. doi: 10.1073/pnas.95.17.10140. [DOI] [PMC free article] [PubMed] [Google Scholar]