

Abstract
Chronic hepatitis C is characterized by metabolic disorders and a microenvironment in the liver dominated by oxidative stress, inflammation and regeneration processes that lead in the long term to hepatocellular carcinoma. Many lines of evidence suggest that mitochondrial dysfunctions, including modification of metabolic fluxes, generation and elimination of oxidative stress, Ca2+ signaling and apoptosis, play a central role in these processes. However, how these dysfunctions are induced by the virus and whether they play a role in disease progression and neoplastic transformation remains to be determined. Most in vitro studies performed so far have shown that several of the hepatitis C virus (HCV) proteins localize to mitochondria, but the consequences of these interactions on mitochondrial functions remain contradictory, probably due to the use of artificial expression and replication systems. In vivo studies are hampered by the fact that innate and adaptive immune responses will overlay mitochondrial dysfunctions induced directly in the hepatocyte by HCV. Thus, the molecular aspects underlying HCV-induced mitochondrial dysfunctions and their roles in viral replication and the associated pathology need yet to be confirmed in the context of productively replicating virus and physiologically relevant in vitro and in vivo model systems.
Keywords: hepatitis C virus, pathology, hepatocarcinogenesis, mitochondria, oxidative stress, metabolism, calcium signaling, apoptosis
1. Introduction
Hepatitis C virus (HCV) is a major cause of chronic viral hepatitis with 150–170 million chronic carriers worldwide. Chronic hepatitis C frequently exhibits an insidious course of disease marked by progressive liver injuries that progress, often over several decades, from fibrosis to cirrhosis and, ultimately, hepatocellular carcinoma (HCC) [1]. Spontaneous resolution of infection is rare, and it is estimated that in 80% of cases, HCV establishes a chronic infection that is associated with complex clinical manifestations, including inflammation, insulin resistance (IR) and steatosis. Disease progression and response to therapy are greatly influenced by viral and host genetic factors, including viral genotype and IL28B polymorphisms [2]. Furthermore, risks factors, like alcohol consumption, or metabolic predispositions/diseases, including diabetes, insulin resistance, obesity and steatosis, can aggravate the course and progression of disease [3,4]. The fact that metabolic disturbances and the risk of hepatocarcinogenesis are known to drop back to baseline with successful antiviral response [3] support the notion that metabolic alterations induced by HCV play an important role in viral propagation, as well as the associated pathogenesis.
HCV is a 9.6 kb-long positive-sense, single-stranded RNA virus of the Flaviviridae family. Its genome encodes a polyprotein that is post-translationally cleaved to produce three structural proteins comprising the capsid protein, Core, and the two glycoproteins, E1 and E2, the p7 protein, as well as six non-structural proteins, including the protease, NS2, the helicase and protease, NS3, and its cofactor, NS4A, NS4B, NS5A and RNA-dependent RNA-polymerase, NS5B [5]. Some of the viral proteins have already been demonstrated to interact with and alter cellular signaling pathways [6]. Overall, it has been shown that HCV proteins strongly interact with endoplasmic reticulum (ER)-derived membranes, the site where the viral proteins are synthesized and matured, and induce the formation of “membranous web” structures in which viral replication is thought to occur [7]. HCV proteins have also been localized to the Golgi apparatus and the outer mitochondrial membrane (OMM), and notably, the Core protein, here referred to as Core, is known to be present at the surface of lipid droplets. Particularly, the formation of membranous webs at the ER and the close vicinity of Core to juxtaposed lipid droplets are thought to be important for the HCV assembly process, which is likely to occur at the ER/lipid droplet interface [8] and which remains a much investigated topic.
Mitochondria play a central role in the regulation of metabolic fluxes and the energy status of the cell. They sense and can counteract cellular stress and are the central switch that decides between cell growth, proliferation, differentiation and apoptosis. HCV has been shown not only to associate with, but also to alter, mitochondrial functions and signaling with important consequences for viral replication and the pathogenesis associated with chronic hepatitis C. The interactions between HCV and mitochondria within the hepatocyte and their potential pathological consequences are the topic of this review and are discussed in detail below.
2. Mitochondrial Structure and Function
Mitochondria are frequently referred to as "cellular power plants", because they generate most of the cell's supply of adenosine triphosphate (ATP). As sensors of the cells energy status, they decide over cell death, growth, proliferation and differentiation, but are also involved in a range of other processes [9]. Mitochondria have been implicated in a significant number of human diseases, such as cardiovascular disorders, including atherosclerosis, ischemic heart disease, ischemia-reperfusion injury and cardiac failure, as well as neurodegenerative disorders related to mitochondria-derived oxidative stress, such as Huntington’s disease, Parkinson’s disease and Alzheimer’s disease. Finally, mitochondrial functions have been shown to play a role in the aging process and diabetes [10].
Mitochondria comprise an OMM, which encloses the entire organelle and has a protein-to-phospholipid ratio similar to that of the eukaryotic plasma membrane (Figure 1). The OMM contains porins and translocase complexes that allow diffusion of small proteins or translocation of factors displaying a specific signaling sequence at their N-terminus, respectively. The OMM can associate with the ER membrane via protein tethering complexes, in a structure called mitochondria-associated ER-membrane or MAM [11]. MAMs form discrete junctions where the OMM and inner mitochondrial membrane (IMM) meet, in order to allow an exchange between ER and mitochondria. Indeed, MAMs are responsive to ER stress [12] and vital for transfer of Ca2+ and lipid from the ER to mitochondria. MAM fractions are enriched in a number of Ca2+ transfer proteins, like sarco/endoplasmic reticulum calcium ATPase (SERCA), which replenishes ER Ca2+ stores and inositol triphosphate receptor (IP3R), which together with the voltage-dependent anion channel (VDAC), cyclophilin D and other factors, forms part of the mitochondrial (mt) permeability transition pore (mPTP) and mediates Ca2+ transfer between the ER and mitochondria [13]. Furthermore, MAMs appear to be an intermediate destination between the rough ER and the Golgi in the pathway that leads to very-low-density lipoprotein (VLDL) assembly and secretion [14]. The intermembrane space is the space between the outer and the inner membranes. Because the OMM is freely permeable to small molecules, the concentrations of small molecules, such as ions and sugars in the intermembrane space, are the same as in the cytosol. However, because transport of large proteins depends on the activity of translocase complexes in the OMM, the protein composition of this space is different from the protein composition of the cytosol. One protein that is localized to the intermembrane space in this way is, e.g., cytochrome c. The IMM is compartmentalized into numerous cristae, which expand the surface area of the IMM and contains proteins that catalyze oxidative phosphorylation, generate ATP in the matrix and regulate metabolite passage and protein trafficking into and out of the matrix, as well as mt fusion and fission. In addition, the IMM is highly impermeable, and almost all ions and molecules require special membrane transporters to enter or exit. The impermeability of the IMM allows the buildup of a membrane potential across the inner membrane (mtΔΨ), which is formed by the action of the enzymes of the electron transport chain (ETC) and which is required for ATP synthesis. The matrix, the space enclosed by the inner membrane, contains a highly-concentrated mixture of hundreds of enzymes, whose major functions include oxidation of fatty acids and the tricarboxylic acid cycle (TCA) cycle.
Figure 1.
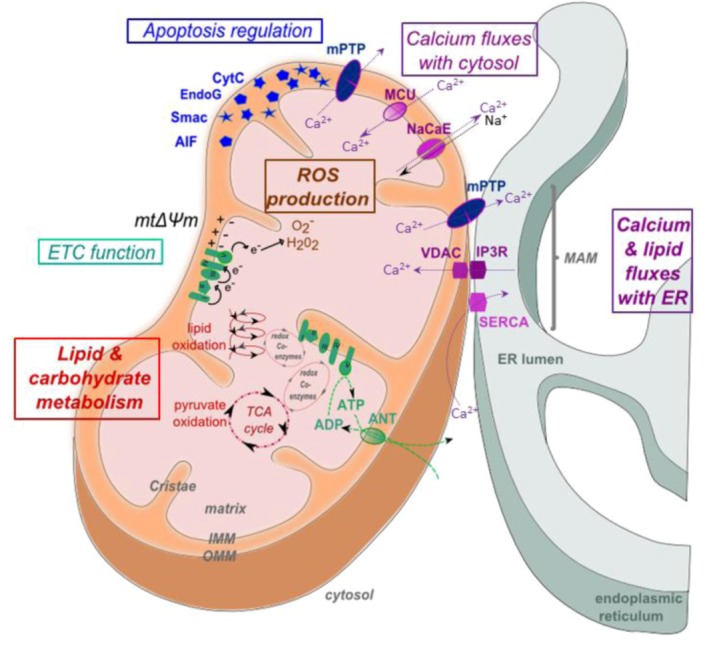
Structure and functions of mitochondria. Mitochondrial membranes and compartments comprise the matrix, inner mitochondrial membrane (IMM), outer mitochondrial membrane (OMM), cristae and mitochondria-associated membrane (MAM). MAMs are points of contact between mitochondria and smooth endoplasmic reticulum (ER), enriched in certain factors, including the inositol triphosphate receptor (IP3R), voltage-dependent anion channel (VDAC) and sarco/endoplasmic reticulum calcium ATPase (SERCA). The main functions of mitochondria are represented in color codes. In red: fatty acids and acetyl coenzyme A (acetyl-CoA) are oxidized by β-oxidation (also called Lynen helix) and the tricarboxylic acid cycle (TCA), respectively, and energy is transferred onto redox coenzymes. In green: the electron transport chain (ETC) is composed of five respiratory complexes (numbered I to V) and uses the electrons of the coenzymes generated by β-oxidation and the TCA cycle as substrates to generate a proton gradient (mtΔΨ) across the inner membrane. The flux of protons from the intermembrane space through complex V (ATP synthase) back into the mitochondrial matrix finally leads to ATP generation. ATP is exported to cytosol via adenine nucleotide translocase (ANT). In brown: electrons that leak from the ETC react with O2, forming superoxide anion O2•- and hydrogen peroxide H2O2, also referred to as reactive oxygen species (ROS). In blue: a number of caspase activators are retained in the intermitochondrial membrane space, including, e.g., cytochrome c, endonuclease G and apoptosis-inducing factor (AIF). These are released into the cytosol by pro-apoptotic signaling, which leads to opening of the mitochondrial permeability transition pore (mPTP). In violet: mitochondria constantly exchange calcium with the cytosol via the mitochondrial calcium uniporter (MCU) and Na+Ca2+ exchanger (NaCaE) and with the ER via the mPTP, which comprises, e.g., IP3R and VDAC.
Mitochondrial energy production is modulated by cell energetic demand, which induces a Ca2+ influx into the mt matrix mediated by the Ca2+ uniporter (MCU) and Na+Ca2+ exchanger (NaCaE), which regulate the TCA cycle rate [15]. Indeed, TCA dehydrogenase enzymes and adenosine nucleotide translocase are Ca2+ sensitive [16,17,18]. Ca2+ fluxes into mitochondria have a major impact not only on metabolism, but also on stress and survival. Indeed, mitochondria can serve as Ca2+ buffers, taking up substantial amounts of Ca2+ from the cytosol or the ER at the expense of loss of mtΔΨ in order to shape and buffer cellular Ca2+ signals. Mitochondrial Ca2+ overload, in turn, triggers the opening of the mPTP and induces apoptosis. The fact that mPTP opening is sensitive to antioxidants forms an important link between Ca2+ and reactive oxygen species (ROS) [19,20].
The generation of ROS is largely due to the activities of the ETC and mitochondrial dehydrogenases [21] (Figure 2). Under normal conditions, only a small percentage of electrons does not complete the whole ETC and, instead, directly leaks to O2, resulting in the formation of free-radical superoxide, O2•-, for around 0.1% of the O2 consumed [22,23]. It has also been shown that mitochondrial Ca2+ uptake can lead to radical production, but the underlying mechanism in not understood [24]. Finally, extramitochondrial sources of ROS exist, such as xanthine oxidase or NADPH oxidases [20]. The increase of ROS is interpreted by the cell as an encounter of stress, and ROS are therefore usually immediately eliminated. Indeed, ROS can oxidize proteins, lipids and DNA and, thus, directly contribute to cellular transformation. However, an increase in ROS can also generate numerous physiological responses, spanning from the induction of inflammatory mediators, such as the signal transducer and activator of transcription (STAT) and NFκB pathways, to induced cell death [25]. The detoxification of ROS is an important function of the cellular redox homeostasis system. Cells rapidly convert O2•- into H2O2 with the help of superoxide dismutases (SOD). H2O2 can then be degraded by a number of enzymes of the cellular redox system, including catalase (CAT), glutathione peroxidases (GPx), thioredoxins (Trx) and peroxiredoxins (PRX). Thus, under normal physiological conditions, ROS are present at low concentrations, which are maintained by a fine balance between Ca2+ uptake, mitochondrial energy production, ROS generation, ROS detoxification and redox signaling (Figure 3).
Figure 2.

ROS generation and scavenging in HCV infected cells. The main source of mt reactive oxygen species (ROS) is the electron transport chain (ETC); electrons leak from complexes I and IV and produce superoxide anion (O2•-) and hydrogen peroxide (H2O2), which is membrane permeable. Non-mt sources of O2•- and H2O2 are cytosolic xanthine oxidase (XO) and plasma membrane NAD(P)H oxidases. The cellular detoxification system scavenges ROS and converts it into H2O and O2: Mitochondrial (Mn) or cytosolic (Cu/Zn) superoxide dismutases (SOD) degrade O2•- into H2O2, which is then converted into H2O and O2 by either catalase (CAT), glutathione-peroxidases (GPx) or peroxiredoxin (PRX), which oxidize glutathione (GSH) in the process. Recycling of oxidized glutathione (GSSG) into GSH requires NADPH coenzyme. In addition to GSH, metallothionein (MT) and thioredoxins (Trx) are also used to scavenge ROS peroxidation products. If ROS are considerably augmented, hydroxyl radicals (HO•), the most damaging form of ROS, can be produced by the Fenton reaction. ROS producing or scavenging factors whose action is modified by HCV are indicated by red stars and include: 1) oxidation of the glutathione pool [40,60]; 2) decrease in NADPH content [61]; 3) increased ROS production from ETC complex I [40,61]; 4) induction of mitochondrial SOD expression [60]; 5) catalase activation [60]; 6) induction of glutathione peroxidase [49,62]; 7) oxidation of the thioredoxin pool [60]; 8) lipid peroxidation [49,63]; 9) induction of metallothionein [49,64]; and 10) cytoplasmic ROS production by NADPH oxidase activation [65,66].
Figure 3.

Metabolism in HCV infected cells. The major metabolic pathways that use glucose (Glc) to either drive nucleotide synthesis and NADPH production via the pentose phosphate shunt (PPP) or to drive the TCA cycle and respiration in mitochondria are depicted. TCA reactions catalyze the reduction of NAD+ and FAD into NADH and FADH2, which in turn feed the electron transport chain (ETC). The ETC maintains the mitochondrial membrane potential, mtΔΨ, required for ATP synthesis. Electrons that leak from complex I and IV of the ETC form reactive oxygen species (ROS). ROS scavenging requires reduced glutathione (GSH), which is oxidized in the process. Recycling of the GSH pool requires NADPH, which is mainly produced by the PP shunt. Glucose-6-phosphate (G6P), pyruvate (Pyr), lactate (Lac), acetyl coenzyme A (AcCoA), pyruvate dehydrogenase complex (PDC), citrate synthase (CS), aconitase (ACO), isocitrate dehydrogenase (ICDH), α-ketoglutarate dehydrogenase (αKGDH), succinyl coenzyme A ligase (SuCLA), succinate dehydrogenase (SDH), fumarate hydratase (FH), malate dehydrogenase (MDH). Factors and events targeted by HCV are indicated by red stars and include: activation of glycolytic enzymes (1) with potential re-routing of the flux into the pentose phosphate shunt, (2) induction of TCA cycle (3) and lipogenic enzymes (4) and complex I of the ETC (5).
3. Mitochondrial Morphology in Productive HCV Infection
The number, morphology and organization of mitochondria within a cell are correlated with energy needs and health. Indeed, apoptotic cell death is characterized by alterations in mt size, shape and distribution [25]. Several viruses inducing chronic infections have already been shown to alter mt morphology. Clustering of mitochondria is induced by hepatitis B virus (HBV) protein X [26]. In the context of human immunodeficiency virus (HIV), Vpr protein has been shown to condense the mt matrix, shorten mitochondria, induce swollen cristae and disappearance of OMM and degradation of mitochondria-ER contact [27,28]. Human T-cell leukemia virus 1 p13 protein induces mitochondria swelling and fragmentation [29], and expression of the PB1-F2 protein of the influenza virus induces a punctuate pattern of mitochondria organization [30]. Overall, virus-induced changes to the mitochondria network are generally associated with mt dysfunctions [31], but the respective roles of these mt dysfunctions in viral replication and the associated pathologies remain unclear.
HCV infection is known to cause autophagy, ER and oxidative stress and alter Ca2+ signaling. It is therefore thought to alter mt structure and functions [32,33,34,35,36,37,38,39,40,41,42,43,44]. Infection of Huh7.5 cells with replicative HCV induces a disruption of the mtΔΨ, followed by mt swelling and cytochrome c release [45]. Huh7 cells harboring the full-length genomic, as well as subgenomic replicons, display a decreased number of mitochondria per cell, with a concomitant increase of size [46]. Core or Core-induced ROS were thought, at least in part, to be responsible for these modifications. NS4A protein expression also induces a redistribution of mitochondria in the perinuclear region in Huh7 cells [47].
4. Physical Interactions between HCV and Mitochondria
Several HCV proteins have been shown to directly associate with mitochondria (Figure 5). Particularly, Core has been shown to associate not only with lipid droplets (LD) [48], but also with mitochondria in Core expressing cells [49,50,51], Core transgenic mice [40], Huh7 cells harboring HCV replicons [47,50] or HCV biopsies [52]. Upon ectopic expression, Core has also been shown to be enriched in MAM fractions on the mitochondrial surface [51]. While initial reports showed that Core associates exclusively to the OMM via a C-terminal motif [40,51], recent electronic microscopy data suggest that Core can also be associated with the IMM [46]. While confocal studies using HCVcc infected Huh7.5 cells did so far not confirm a direct interaction of Core with mitochondria [53,54], biochemical evidence suggests that the interaction does also take place in the context of productively replicating HCVcc [55]. In Huh7 cells expressing p7, NS4A, NS3/4A or harboring a subgenomic RNA replicon, p7 and NS4A were found localized to mitochondria and ER by confocal microscopy, subcellular-fractionation and electron microscopic analysis [47,55,56,57,58,59]. NS5A has been known to be present in particular parts of the ER membrane, so-called “membranous web” structures, where replication is thought to occur [7]. In addition, NS5A is also found on mitochondria and, more precisely, in the matrix and on the IMM [46,47,55]. Similarly, NS5B was recently localized by electron microscopy to the IMM, as well as OMM and the mt matrix [46].
Figure 5.

Effect on mitochondria.
5. Mitochondrial Functions Altered by HCV Infection
Overall, many lines of evidence suggest that several HCV proteins interact directly with mitochondria in hepatocytes and profoundly alter their functions in metabolism, redox balance, ROS scavenging and apoptosis (Figure 5). However, the molecular mechanisms underlying these direct physical interactions, how these interactions translate into altered mt functions and what roles altered mt functions play in the viral lifecycle and the pathogenesis associated with chronic hepatitis C are issues that are not yet well understood.
5.1. Metabolism
Glucose is the major carbon source for the TCA cycle in hepatocytes. It is converted by glycolysis into pyruvate and actively transported across the IMM, converted to acetyl-CoA, which then enters the TCA cycle (Figure 3). The enzymes of the TCA cycle are located in the mt matrix, with the exception of succinate dehydrogenase, which is bound to the IMM as part of electron transfer complex II (see below). The TCA cycle oxidizes the acetyl-CoA to carbon dioxide and, in the process, produces reduced cofactors, NADH and FADH2, the substrates for the ETC. The redox energy from NADH and FADH2, which is produced not only by the TCA cycle, but also by fatty acid oxidation and amino acid oxidation, is stepwise transferred to oxygen by electron passage through the four complexes of the ETC and used to pump protons into the intermembrane space, thus creating an electrochemical proton gradient across the IMM: the mtΔΨ. Protons then return to the matrix through the ATP synthase complex, and their potential energy is used to synthesize ATP. Generally, only a small percentage of electrons in the ETC form prematurely reduced oxygen, forming reactive oxygen species, such as superoxide. Under physiological conditions, ROS are efficiently scavenged to avoid mitochondrial and cellular oxidative damage.
In the context of a large scale proteomic study using HCVcc-infected Huh7.5 cells, a number of glycolytic enzymes and enzymes driving the pentose phosphate shunt (PPS) have been shown to be upregulated [33]. However, albeit an increased flux through the glycolytic pathway and a supposed increase in pyruvate production, pyruvate seemed to be used rather for lactate production than for use in the TCA cycle [33]. Furthermore, upregulation of enzymes driving the PPS, such as transketolase and transaldolase, indicates that glycolytic intermediates are channeled into this pathway, which constitutes the major source of NADPH and controls nucleotide synthesis and homeostasis. Indeed, replication in HCV replicon-containing cells is known to depend on sufficient levels of intracellular uridine and cytidine triphosphate [67], and HCVcc-infected cells display lowered ATP levels, while the ATP/ADP and ATP/AMP ratios remained unchanged [68]. However, concomitant with a rechanneling of glycolytic intermediates into the PPS, Diamond et al. also observed a coordinated increase in the abundance of TCA cycle enzymes, including citrate synthase, isocitrate dehydrogenase 2 and 3A, fumarate hydratase, malate dehydrogenases 1 and 2 and components of the ETC [33]. These findings suggest that HCVcc-infected cells maintain TCA cycle activity to produce the metabolic precursors for fatty acid synthesis. Indeed, Diamond et al. observed the upregulation of lipogenic enzymes, but also an increased abundance of proteins associated with peroxisomal and mitochondrial fatty acid oxidation, suggesting both active lipid synthesis and turnover at the same time. In line with these data, dodecenoyl coenzyme A delta isomerase, a mitochondrial fatty acid oxidation enzyme, has been identified to be required for HCV replication [69]. However, others have reported that HCV decreased expression of the fatty acid-activated transcription factor peroxisome proliferator-activated receptor alpha, which controls β-oxidation and lipoprotein metabolism, as well as its target gene carnitine palmitoyl acyl-CoA transferase 1 gene, which shuttles long-chain fatty acids across the membrane [70,71,72]. Furthermore, addition of a hypolipidemic agent to HCVcc-infected cells that restored β-oxidation and lipogenesis back to normal inhibited replication of the virus, but the contrary has also been reported [69,72]. Thus, it is becoming clear that HCV alters metabolic fluxes through mitochondria; however, the underlying molecular mechanisms remain to be confirmed. Furthermore, the kick-off effects of these changes on the production of oxidative stress and the hepatic redox balance and their roles in viral replication remain unclear.
5.2. Redox System
HCV infection has been associated with oxidative stress and changes in the host’s redox balance, as well as with significant increases in peroxides and oxidative damage in vivo (Figure 2). Liver biopsies of HCV patients display elevated levels of 8-oxo-desoxihydroxyguanosine (8-OHdG), a DNA oxidation product, and the lipid peroxides, 4-hydroxy-2-nonenal (4-HNE) and malondialdehyde (MDA) [73,74,75,76,77,78]. Elevated MDA levels have also been detected in sera of chronic hepatitis C patients [79,80,81,82]. Furthermore, HCV-induced HCC is associated with more oxidative stress markers than HBV-induced HCC [75,83]. Generally, oxidative stress markers have been shown to correlate with severity of inflammation, grade of fibrosis and hepatic iron storage markers [75,81], raising the question whether oxidative stress is due to host immunity and iron overload [76,77,84,85,86] or whether direct HCV-host cell interactions also contribute to ROS production.
Pointing to a direct role for HCV in ROS production, oxidative markers are found in patients with mild, moderate or no liver disease [87]. Furthermore, proteomic profiling has shown an upregulation of antioxidant enzymes at early (F1 to F3), but not late, stages of fibrosis [88]. One of the major sources for intracellular ROS production are mitochondria and particularly leaks in the ETC. Indeed, inhibition of the ETC has been observed to augment ROS generation [21]. In order to prevent oxidative damage and the induction of inflammatory processes, cells rapidly scavenge ROS. Thus, under normal physiological conditions, ROS are present at low concentrations. ROS also regulate the activity of target proteins through the reversible oxidation of critical protein thiols. Drastic increases in ROS can induce mPTP opening, leading to, e.g., cytochrome c or Ca2+ release and apoptosis [19,20]. Hepatocarcinogenesis is thought to be driven by permanent elevation of ROS that in turn induces chronic cytokine signaling and lipid, protein and DNA oxidation [6].
Several lines of evidence suggest that HCV can directly induce oxidative stress, including various cell lines overexpressing the HCV polyprotein or distinct viral proteins, as well as in cell lines containing replicons or HCVcc-infected Huh7.5 cells [40,49,60,65,89,90,91,92,93,94]. The ETC, in particular, has been identified as an important source of ROS in cells expressing various HCV proteins, as well as Core transgenic mice [32,38,49,61,63,91,95,96,97]. For example, expression of the HCV polyprotein inhibited complex I activity and depolarized the mt membrane, overall leading to an increase in mt ROS production [38]. Core protein expression has been shown to lead to ROS production in Huh7 or CHL cells, as well as in in HeLa and CHO cells using ROS sensitive fluorescent probes, such as DCFDA or DHE [49,55,60,92,94,98]. In addition, oxidative stress markers, like lipid peroxides and 8-OHdG, were increased in cells expressing Core [49,63]. This increase of ROS and peroxides was probably caused by mt dysfunctions for two reasons: it was accompanied by a loss of mtΔΨ and pharmacological inhibition of the ETC, which, however, also affects activity of NADPH oxidases and blocked the effect [49,63]. Mitochondria isolated from mice transgenic for Core, as well as E1 and E2 glycoproteins, also displayed a decrease in NADPH content and increased ROS production from complex I substrates accompanied by reduced activity of ETC complex I [40,61]. Besides production of mt ROS, Core is also involved in ROS production at the plasma membrane by activating NADPH oxidase 4 in human hepatoma cell lines [65,99], and productively replicating HCV has been shown to induce not only NADPH oxidase 4, but also isoform 1 [66]. Concomitantly with the induction of ROS, Core has been shown to alter the mitochondrial redox status. In mitochondria isolated from Core transgenic mice and Core expressing Huh7 cells, oxidation of the glutathione and thioredoxin pools was found to be increased [40,60,91]. Core expression has also been shown to induce the expression of mt, but not cytoplasmic SOD [60], suggesting that HCV generates ROS and, at the same time, strengthens the defense system against oxidative stress. Indeed, Core has been shown to induce metallothioneins and glutathione peroxidases [49,64]. Finally, data on Core-induced modulation of heme oxygenase-1 (HO-1) expression, an enzyme that generates redox active compounds, such as iron, remain contradictory [60,61,98,100,101].
Besides Core, the glycoproteins E1 and E2 are also associated with increased ROS production. E1- and E2-expressing hepatoma cells displayed increased levels of H2O2, and E1-, but not E2-, expressing cells had increased levels of superoxide anions, as well as lipid peroxides [63,98]. However, at least partially, these effects were mediated through activation of the Nrf2/ARE pathway, which induces the transcriptional activation of a battery of anti-oxidative stress response proteins and enzymes implicated in detoxification and glutathione generation, in ROS-dependent and -independent manners [98].
Ectopic expression of the non-structural proteins of HCV is also known to drive ROS production, to lead to the oxidation of thioredoxins and to activate catalase and, to a lesser extent, SOD and HO-1 in Huh7 cells [60]. NS3, for example, has been shown to induce superoxide anion, but not H2O2 production [63,98,99]. NS3 expression leads furthermore to an accumulation of lipid peroxides in Huh7 cells [63]. NS4B has been implicated in H2O2 production and activation of the Nrf2/ARE pathway in Huh7 cells, but the underlying mechanisms remain unknown [98]. NS5A expression has been shown to lead to ROS generation in CHL, HepG2 and Huh7 cells [90,96,98,102,103], but levels of lipid peroxides remained normal [63]. NS5A associates with ER membranes and induces Ca2+ fluxes from the ER to the mitochondria, which are the likely source of NS5A-induced ROS generation [96,103]. The fact that concomitantly with increased mt Ca2+ influx, NS5A decreases ATP generation, implies that infected cells cannot replenish ER Ca2+ stores via the SERCA pump [103]. Increased ROS generation and an increase of the total oxidized glutathione levels have been confirmed in the context of HCVcc-infected Huh7.5 cells [93,104].
The antioxidants, resveratrol, N-acetyl cysteine (NAC), and vitamins A, C and E were reported to improve replication in the OR6 reporter assay system for genome-length HCV RNA replication in hepatoma-derived HuH-7 cells [105,106,107]. Furthermore, lipid peroxides inhibit HCV replication in cells harboring replicons, and this effect can be reversed by vitamin E treatment [108]. In contrast, alcohol-induced increase of ROS has been reported to boost HCV replication [109,110]. The fact that HCV induces oxidative stress and that in vitro replication seems to be sensitive to the effect of antioxidants led to a number of experimental clinical trials, unfortunately, so far, with unclear results. Most clinical trials tested antioxidants in combination with the classical Peg-IFN/ribavirin combination. Frequently, a decrease in oxidative markers and histological inflammation was observed, but rarely a decrease in viral load [111,112,113,114,115]. Overall, the antioxidants seemed to improve liver condition and inflammation and, in some cases, also virological responses. However, it remains unclear whether HCV requires the changes to the cellular redox system for efficient viral replication [116].
5.3. Calcium Signaling
The close physical association between the ER and mitochondria mediated by MAMs results in Ca2+ micro domains at contact points that facilitate efficient Ca2+ transmission from the ER to the mitochondria [117]. Ca2+ transfer at MAMs requires tight interactions between IP3R, Grp75, VDAC and cyclophilin D, as well as the mtΔΨ-driven MCU at the IMM, which are part of the mPTP complex [15,117]. Regulating ER release of Ca2+ at the MAM is critical for cell homeostasis. Sufficient intra-organelle Ca2+ concentrations are required to stimulate metabolism by activating pyruvate dehydrogenase, isocitrate dehydrogenase and α-ketoglutarate dehydrogenase, all critical for maintenance of the TCA cycle [16,17]. Prolonged increases of Ca2+ can, in turn, interfere with the activity of these enzymes. Because the mtΔΨ depends on the activity of the ECT, which in turn is dependent on TCA cycle activity, interference with any of these processes will induce a substrate imbalance that will lead to the generation of ROS (Figure 3). Also, if Ca2+ signaling in the mitochondria passes a certain threshold, the mtΔΨ collapses, and the intrinsic pathway of apoptosis is triggered. The anti-apoptotic factor Bcl-2 has been shown to interact with IP3R to limit Ca2+ filling of the ER, leading to reduced efflux at the MAM and preventing collapse of the mtΔΨ post-apoptotic stimuli [118].
Figure 4.
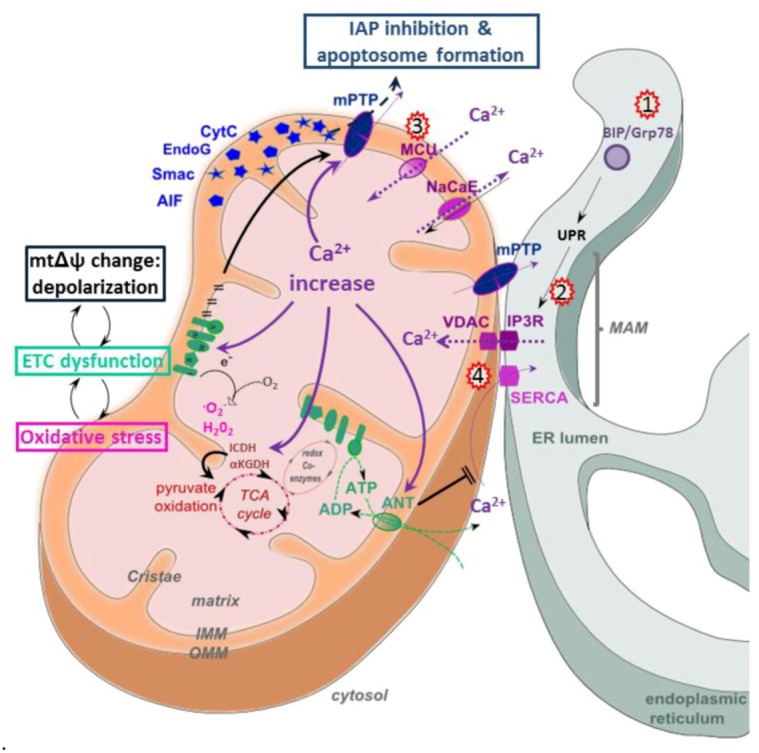
Calcium signaling in HCV infected cells. Mitochondria take up Ca2+ via the mtΔΨ-dependent calcium uniporter (MCU), the voltage-dependent anion channel (VDAC), the Na+/Ca2+ exchanger (NaCaE) and the mitochondrial permeability transition pore (mPTP). ER Ca2+ uptake is regulated by the sarco/endoplasmic reticulum calcium ATP-ase (SERCA). Ca2+ fluxes are represented by dotted violet arrows. Mt Ca2+ levels control energy metabolism by activating several enzymes of the TCA cycle, such as isocitrate dehydrogenase (ICDH) and α-ketoglutarate dehydrogenase (αKGDH), as well as adenosine nucleotide translocase (ANT), which is a component of the mPTP. Mt Ca2+ increase (violet arrows) inhibits ETC function by decreasing mtΔΨ, as well as complex V activity and induces ATP depletion by mPTP opening, leading to the release of pro-apoptotic factors. Kick off effects of Ca2+ increase are marked by black arrows and comprise changes in mtΔΨ, mPTP opening, amplification of ETC inhibition and concomitant ROS generation and ATP depletion. Factors and events targeted by HCV are indicated by red stars and include 1) increase of BIP/Grp78, an endoplasmic reticulum chaperone [35,119], 2) Ca2+ transfer from the ER into mitochondria, mediated by NS5A-induced ER stress [96] and by Core expression [55], 3) Core-induced increase of MCU activity [32] and 4) Core-induced SERCA expression [119].
Pharmacological inhibition of ER-mitochondrial Ca2+ fluxes in a hepatoma cell line expressing the HCV polyprotein has been shown to normalize all aberrant effects induced by HCV: ETC complex I activity normalized, the loss of mtΔΨ was restored and ROS, as well as intramitochondrial Ca2+ concentrations returned to baseline. Time course and titrations of HCV polyprotein expression suggested furthermore that the uptake of Ca2+ into mitochondria is the earliest of these above events induced by HCV [38]. Thus, mitochondrial Ca2+ uptake may be the initial mt dysfunction induced by HCV and may trigger, in turn, complex I inhibition, loss of mtΔΨ and ROS generation. Another study confirming altered Ca2+ fluxes in HCV infection showed that the virus sensitizes Huh7.5 cells to t-butyl hydroperoxide-induced mtΔΨ loss and apoptosis. All these effects could be counteracted by intracellular Ca2+ chelation, again suggesting that a dysfunctioning of Ca2+ signaling is at their origin [55]. Whether increased mt Ca2+ levels in these studies were due to increased mt uptake or release from the ER remained unclear, but another independent study reported Core expressing hepatoma cells to display both increased mt Ca2+ entry and transfer of Ca2+ from the ER to mitochondria [32]. Finally, NS5A expression has been shown to activate ROS-mediated NFκB and STAT activation, and this process was sensitive to the effects of an MCU inhibitor, suggesting indirectly that alterations of Ca2+ fluxes precede ROS production [96,103]. In contrast to the above data, it has also been shown that Core triggers ER stress, including hyperexpression of the stress sensor Grp78/BiP, which in turn leads to mtΔΨ depolarization, cytochrome c release and apoptosis. These effects were due to SERCA impairment, followed by ER calcium depletion [119]. Using an ER-targeted aequorin calcium probe, the authors found that ER calcium depletion followed ER stress in Core-expressing cells, suggesting that ER stress was at the origin of these mt dysfunctions [119]. Grp78/BiP overexpression was also confirmed in HCV-infected SCID/Alb-uPA mice [35], but the causal effect on Ca2+ signaling was not investigated. Thus, overall, the details and the sequence of the molecular events by which HCV causes cellular stress and alters Ca2+ signaling remain unclear.
5.4. Apoptosis
Apoptosis or “programmed cell death” consists of a very well defined series of cellular events that lead to DNA condensation and fragmentation, followed by break down into small apoptotic bodies that are disposed by phagocytes. The intrinsic apoptotic pathway is characterized by mPTP-mediated release of cytochrome c or other caspase activating factors, such as Smac/Diablo and HtrA2/Omi from the mt intermembrane space into the cytoplasm. The release of these proteins occurs via the mPTP and is mediated by Bcl-2 family of proteins, whose members display both pro- (Bad, Bax, Bak or Bid) and anti- (Bcl-2, Bcl-xL, Mcl-1) apoptotic properties. Cytochrome c then forms a multi-protein complex, known as the ‘apoptosome’, and initiates activation of the caspase cascade through caspase 9. In contrast, the extrinsic apoptotic pathway is activated by ligand binding to death receptors on the plasma membrane, which in turn induces mt cytochrome c release or formation of the death-inducing signaling complex (DISC), which activates the caspase cascade through caspase 8 [120].
In chronic hepatitis C patients, apoptosis has been suggested to occur in an estimated 0.54% to 20.00% of all hepatocytes based on TUNEL assays and electron microscopy [121,122,123]. However, the strong presence of infiltrating lymphocytes [121,124] and the strong correlation with liver pathology [121,122] and fibrosis grade [125], but not with viral load or genotype, suggest that apoptosis is strongly immune-mediated. Furthermore, with only 10% of hepatocytes estimated to be infected [126,127], it remains unclear whether apoptotic hepatocytes are really HCV infected and whether apoptosis is induced to clear the virus. Thus, so far, the only line of evidence in vivo that suggests a direct viral-induced induction of apoptosis comes from SCID mice, in which HCV-infected hepatocytes have been shown to undergo apoptosis [35].
In vitro, a direct induction of apoptosis by HCV has been demonstrated in various models and cell lines. Actinomycin D-treated HCV subgenomic replicon harboring Huh7 cells were found to have a decreased Δψm and to undergo higher rates of apoptosis than naive cells. They showed, furthermore, no difference in their sensitivity to TNF-α-mediated apoptosis, suggesting that HCV replication specifically induces mitochondria-mediated cell death [47]. Infection of Huh7.5 cells with the chimeric J6/JFH1 strain led to increased caspase 3 activation and Poly ADP ribose polymerase (PARP) cleavage, accompanied by mitochondrial Bax accumulation, a decreased Δψm and increased cytoplasmic cytochrome c [45]. In chimeric SCID/Alb-uPA mice infected with strain H77c, increased rates of apoptosis correlated with increased Bax and decreased Bcl-xL levels [35]. Whether ER stress or the UPR are involved or amplify HCV-induced mitochondria-mediated cell death remains a controversial topic [35,45].
Studies focusing on the roles of the individual viral proteins in the induction of apoptosis have particularly identified Core and NS5A as key regulators of ligand-mediated apoptosis, while the role of other viral proteins is still unclear. Core has been demonstrated to possess pro- and anti-apoptotic effects. Core expression induces ligand-independent apoptosis in Jurkat and Huh7 cells by activating caspases 3 [128,129,130], 8 [128] and 9 [129], as well as PARP cleavage [130,131] in SK-HEP-1, 293T or Huh7 cells. In contrast, physiological Core expression in HepG2 cells inhibited caspase 3 activity [132]. Core-induced cytochrome c release [129,130] and a loss of Δψm [129] were recorded in 293T and Huh7 cells, strongly suggesting that Core induces apoptosis through the mitochondria-mediated pathway. Moreover, a Bcl-2 homology 3 (BH3) domain has been identified in the Core protein, that may confer pro-apoptotic properties by mediating interaction and inhibition of anti-apoptotic Mcl-1 and cytochrome c release into the cytoplasm [130]. Core was also shown to interact with 14-3-3 epsilon protein, leading to Bax release and mitochondrial-mediated apoptosis [129]. In contrast to these data, another study reports that Core only augments TRAIL-mediated apoptosis by enhancing Bid cleavage and activation of the mitochondria apoptosis signaling pathway [133]. Yet, another report shows that Core-induced apoptosis only enhanced death-receptor-mediated apoptosis [134]. Furthermore, the capacity of Core to induce apoptosis seemed to depend on the genotype. Core of genotype 1b was more efficient at inducing apoptosis than Core of genotype 2a, and this was linked to the hydrophobicity of residue 119 [130]. In E1-expressing hepatoma cells, apoptosis depended on the presence of the C-terminal transmembrane domain of E1, presumably altering membrane permeability of E1 [135,136]. E2 has been shown to inhibit death receptor-induced apoptosis in hepatoma cells, presumably through inhibition of mitochondrial cytochrome c release [129], and at the same time, it has been shown to induce mitochondria-related and caspase-dependent apoptosis in the same hepatoma cell line [137]. NS2 is known to bind and directly inhibit CIDE-B-induced apoptosis via the mitochondrial pathway [138]. NS3/4A prevents apoptosis by cleaving Cardif, an adaptor protein in the RIG-I antiviral pathway. Upon cleavage, Cardif translocates to the mitochondrial membrane [139], but its precise role in apoptosis induction remains unknown. In contrast, NS3 induces caspase-8 dependent apoptosis in hepatocytes [140] by mechanisms that remain unknown. NS4A expression in Huh7 cells induces cell death, decreases Δψm, induces cytochrome c release and caspase 3, but not caspase 8, activation, strongly suggesting that NS4A induces apoptosis through the mitochondria-mediated pathway [47]. NS4B was also found to induce apoptosis via the mitochondrial death pathway in Huh7 cells, where it induced cytochrome c release and Δψm decrease, coupled with caspase 3, 7 and 9 activation and PARP cleavage [141]; however, in U-2 OS cells, its action was caspase-independent [134]. NS5A has been shown to inhibit apoptosis by induction of survivin expression, an inhibitor of caspases [142], by blocking cytochrome c release [143], by the sequestration of p53 in the cytoplasm to prevent pro-apoptotic gene transcription [144] and by activation of the PKB/Akt and NFkB signaling pathways [96,145]. Bcl-2 homology (BH) domains 1, 2 and 3 were identified in NS5A and the BH2 domain, in particular, has been shown to mediate interaction with Bax, thereby blocking apoptosis [146].
6. Conclusions
Many viral proteins interact directly with and interfere with signaling pathways that converge in mitochondria. The mitochondrion is the key organelle that determines the cellular response to stress by putting into action stress relief responses, induction of cytokines or apoptosis. Thus, it is not surprising that mitochondrial dysfunctions have been implied in many different forms of diseases. HCV-induced alterations of mitochondrial functions are likely to have a major impact on fibrogenesis and disease progression towards liver cancer by creating a liver microenvironment dominated by oxidative stress and concomitant cytokine signaling and liver regeneration. Restoration of mitochondrial functions is thus an important factor to block and, ultimately, reverse disease progression in chronic hepatitis C, and recent in vitro data on the mPTP inhibitor, alisporivir, seem to confirm this notion [147]. However, the molecular details underlying HCV-induced mitochondrial dysfunctions remain confusing and even contradictory at this stage, probably due to the use of very heterologous and artificial in vitro expression and replication systems that are based on various different hepatic and non-hepatic cell lines. The lack of an infectious HCV tissue culture system based on physiologically and metabolically relevant cell systems is rendering the investigation of the impact of productively replicating HCV virus on mitochondrial functions in vitro very difficult. The possibility to replicate HCV in primary human and murine hepatocytes and the availability of immune-deficient mice with humanized livers will open up new avenues to revisit and validate our existing knowledge and may allow us to use HCV as a tool to better understand the role of mitochondria in other forms of liver disease.
Acknowledgments
This work was supported from grants from the ANRS, Region Rhone Alpes and the ANR.
Conflict of Interest
The authors declare no conflict of interest.
References and Notes
- 1.Lemon S.M., Walker C.M., Alter M.J., Yi M.–K. Hepatitis c virus. In: Knipe D.M., Howley P.M., editors. Fields Virology. 5th. 2007. pp. 1253–1304. [Google Scholar]
- 2.Ge D., Fellay J., Thompson A.J., Simon J.S., Shianna K.V., Urban T.J., Heinzen E.L., Qiu P., Bertelsen A.H., Muir A.J., et al. Genetic variation in il28b predicts hepatitis c treatment–induced viral clearance. Nature. 2009;461:399–401. doi: 10.1038/nature08309. [DOI] [PubMed] [Google Scholar]
- 3.Alaei M., Negro F. Hepatitis c virus and glucose and lipid metabolism. Diabetes Metab. 2008;34:692–700. doi: 10.1016/S1262-3636(08)74606-8. [DOI] [PubMed] [Google Scholar]
- 4.Bartosch B. Hepatitis c virus and its complex interplay with hepatic glucose and lipid metabolism. J. Hepatol. 2009;50:845–847. doi: 10.1016/j.jhep.2009.02.009. [DOI] [PubMed] [Google Scholar]
- 5.Moradpour D., Penin F., Rice C.M. Replication of hepatitis c virus. Nat. Rev. Microbiol. 2007;5:453–463. doi: 10.1038/nrmicro1645. [DOI] [PubMed] [Google Scholar]
- 6.Bartosch B. Hepatitis b and c viruses and hepatocellular carcinoma. Viruses. 2010;2:1504–1509. doi: 10.3390/v2081504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Egger D., Wolk B., Gosert R., Bianchi L., Blum H.E., Moradpour D., Bienz K. Expression of hepatitis c virus proteins induces distinct membrane alterations including a candidate viral replication complex. J. Virol. 2002;76:5974–5984. doi: 10.1128/JVI.76.12.5974-5984.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Miyanari Y., Atsuzawa K., Usuda N., Watashi K., Hishiki T., Zayas M., Bartenschlager R., Wakita T., Hijikata M., Shimotohno K. The lipid droplet is an important organelle for hepatitis c virus production. Nat. Cell Biol. 2007;9:1089–1097. doi: 10.1038/ncb1631. [DOI] [PubMed] [Google Scholar]
- 9.Goldenthal M.J., Marin–Garcia J. Mitochondrial signaling pathways: A receiver/integrator organelle. Mol. Cell Biochem. 2004;262:1–16. doi: 10.1023/B:MCBI.0000038228.85494.3b. [DOI] [PubMed] [Google Scholar]
- 10.Duchen M.R. Roles of mitochondria in health and disease. Diabetes. 2004;53:96–102. doi: 10.2337/diabetes.53.2007.S96. [DOI] [PubMed] [Google Scholar]
- 11.Raturi A., Simmen T. Where the endoplasmic reticulum and the mitochondrion tie the knot: The mitochondria–associated membrane (mam) Biochim. Biophys. Acta. 2013;1833:213–224. doi: 10.1016/j.bbamcr.2012.04.013. [DOI] [PubMed] [Google Scholar]
- 12.Szabadkai G., Bianchi K., Varnai P., De Stefani D., Wieckowski M.R., Cavagna D., Nagy A.I., Balla T., Rizzuto R. Chaperone–mediated coupling of endoplasmic reticulum and mitochondrial ca2+ channels. J. Cell Biol. 2006;175:901–911. doi: 10.1083/jcb.200608073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Filippin L., Magalhaes P.J., Di Benedetto G., Colella M., Pozzan T. Stable interactions between mitochondria and endoplasmic reticulum allow rapid accumulation of calcium in a subpopulation of mitochondria. J. Biol. Chem. 2003;278:39224–39234. doi: 10.1074/jbc.M302301200. [DOI] [PubMed] [Google Scholar]
- 14.Rusinol A.E., Cui Z., Chen M.H., Vance J.E. A unique mitochondria–associated membrane fraction from rat liver has a high capacity for lipid synthesis and contains pre–golgi secretory proteins including nascent lipoproteins. J. Biol. Chem. 1994;269:27494–27502. [PubMed] [Google Scholar]
- 15.Rimessi A., Giorgi C., Pinton P., Rizzuto R. The versatility of mitochondrial calcium signals: From stimulation of cell metabolism to induction of cell death. Biochim. Biophys. Acta. 1777:808–816. doi: 10.1016/j.bbabio.2008.05.449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Balaban R.S. Cardiac energy metabolism homeostasis: Role of cytosolic calcium. J. Mol. Cell Cardiol. 2002;34:1259–1271. doi: 10.1006/jmcc.2002.2082. [DOI] [PubMed] [Google Scholar]
- 17.Denton R.M., McCormack J.G. Ca2+ as a second messenger within mitochondria of the heart and other tissues. Annu. Rev. Physiol. 1990;52:451–466. doi: 10.1146/annurev.ph.52.030190.002315. [DOI] [PubMed] [Google Scholar]
- 18.Liu T., O'Rourke B. Regulation of mitochondrial ca2+ and its effects on energetics and redox balance in normal and failing heart. J. Bioenerg. Biomembr. 2009;41:127–132. doi: 10.1007/s10863-009-9216-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Galley H.F. Bench–to–bedside review: Targeting antioxidants to mitochondria in sepsis. Crit. Care. 2010;14:230. doi: 10.1186/cc9098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Murphy M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009;417:1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rigoulet M., Yoboue E.D., Devin A. Mitochondrial ros generation and its regulation: Mechanisms involved in h(2)o(2) signaling. Antioxid Redox Signal. 2011;14:459–468. doi: 10.1089/ars.2010.3363. [DOI] [PubMed] [Google Scholar]
- 22.Hansford R.G., Hogue B.A., Mildaziene V. Dependence of h2o2 formation by rat heart mitochondria on substrate availability and donor age. J. Bioenerg. Biomembr. 1997;29:89–95. doi: 10.1023/A:1022420007908. [DOI] [PubMed] [Google Scholar]
- 23.Tahara E.B., Navarete F.D., Kowaltowski A.J. Tissue–, substrate–, and site–specific characteristics of mitochondrial reactive oxygen species generation. Free Radic Biol. Med. 2009;46:1283–1297. doi: 10.1016/j.freeradbiomed.2009.02.008. [DOI] [PubMed] [Google Scholar]
- 24.Feissner R.F., Skalska J., Gaum W.E., Sheu S.S. Crosstalk signaling between mitochondrial ca2+ and ros. Front Biosci. 2009;14:1197–1218. doi: 10.2741/3303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Karbowski M., Youle R.J. Dynamics of mitochondrial morphology in healthy cells and during apoptosis. Cell Death Differ. 2003;10:870–880. doi: 10.1038/sj.cdd.4401260. [DOI] [PubMed] [Google Scholar]
- 26.Kim S., Kim H.Y., Lee S., Kim S.W., Sohn S., Kim K., Cho H. Hepatitis b virus x protein induces perinuclear mitochondrial clustering in microtubule– and dynein–dependent manners. J. Virol. 2007;81:1714–1726. doi: 10.1128/JVI.01863-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huang C.Y., Chiang S.F., Lin T.Y., Chiou S.H., Chow K.C. Hiv–1 vpr triggers mitochondrial destruction by impairing mfn2–mediated er–mitochondria interaction. PLoS One. 2012;7:e33657. doi: 10.1371/journal.pone.0033657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Radovanovic J., Todorovic V., Boricic I., Jankovic–Hladni M., Korac A. Comparative ultrastructural studies on mitochondrial pathology in the liver of aids patients: Clusters of mitochondria, protuberances, "minimitochondria," vacuoles, and virus–like particles. Ultrastruct. Pathol. 1999;23:19–24. doi: 10.1080/019131299281798. [DOI] [PubMed] [Google Scholar]
- 29.D'Agostino D.M., Ranzato L., Arrigoni G., Cavallari I., Belleudi F., Torrisi M.R., Silic–Benussi M., Ferro T., Petronilli V., Marin O., et al. Mitochondrial alterations induced by the p13ii protein of human t–cell leukemia virus type 1. Critical role of arginine residues. J. Biol. Chem. 2002;277:34424–34433. doi: 10.1074/jbc.M203023200. [DOI] [PubMed] [Google Scholar]
- 30.Yamada H., Chounan R., Higashi Y., Kurihara N., Kido H. Mitochondrial targeting sequence of the influenza a virus pb1–f2 protein and its function in mitochondria. FEBS Lett. 2004;578:331–336. doi: 10.1016/j.febslet.2004.11.017. [DOI] [PubMed] [Google Scholar]
- 31.Boya P., Pauleau A.L., Poncet D., Gonzalez–Polo R.A., Zamzami N., Kroemer G. Viral proteins targeting mitochondria: Controlling cell death. Biochim. Biophys. Acta. 2004;1659:178–189. doi: 10.1016/j.bbabio.2004.08.007. [DOI] [PubMed] [Google Scholar]
- 32.Li Y., Boehning D.F., Qian T., Popov V.L., Weinman S.A. Hepatitis c virus core protein increases mitochondrial ros production by stimulation of ca2+ uniporter activity. FASEB J. 2007;21:2474–2485. doi: 10.1096/fj.06-7345com. [DOI] [PubMed] [Google Scholar]
- 33.Diamond D.L., Syder A.J., Jacobs J.M., Sorensen C.M., Walters K.A., Proll S.C., McDermott J.E., Gritsenko M.A., Zhang Q., Zhao R., et al. Temporal proteome and lipidome profiles reveal hepatitis c virus–associated reprogramming of hepatocellular metabolism and bioenergetics. PLoS Pathog. 2010;6:e1000719. doi: 10.1371/journal.ppat.1000719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sir D., Chen W.L., Choi J., Wakita T., Yen T.S., Ou J.H. Induction of incomplete autophagic response by hepatitis c virus via the unfolded protein response. Hepatology. 2008;48:1054–1061. doi: 10.1002/hep.22464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Joyce M.A., Walters K.A., Lamb S.E., Yeh M.M., Zhu L.F., Kneteman N., Doyle J.S., Katze M.G., Tyrrell D.L. Hcv induces oxidative and er stress, and sensitizes infected cells to apoptosis in scid/alb–upa mice. PLoS Pathog. 2009;5:e1000291. doi: 10.1371/journal.ppat.1000291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Merquiol E., Uzi D., Mueller T., Goldenberg D., Nahmias Y., Xavier R.J., Tirosh B., Shibolet O. Hcv causes chronic endoplasmic reticulum stress leading to adaptation and interference with the unfolded protein response. PLoS One. 2011;6:e24660. doi: 10.1371/journal.pone.0024660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tardif K.D., Waris G., Siddiqui A. Hepatitis c virus, er stress, and oxidative stress. Trends Microbiol. 2005;13:159–163. doi: 10.1016/j.tim.2005.02.004. [DOI] [PubMed] [Google Scholar]
- 38.Piccoli C., Scrima R., Quarato G., D'Aprile A., Ripoli M., Lecce L., Boffoli D., Moradpour D., Capitanio N. Hepatitis c virus protein expression causes calcium–mediated mitochondrial bioenergetic dysfunction and nitro–oxidative stress. Hepatology. 2007;46:58–65. doi: 10.1002/hep.21679. [DOI] [PubMed] [Google Scholar]
- 39.Wang T., Weinman S.A. Causes and consequences of mitochondrial reactive oxygen species generation in hepatitis c. J. Gastroenterol. Hepatol. 2006;21(Suppl 3):S34–S37. doi: 10.1111/j.1440-1746.2006.04591.x. [DOI] [PubMed] [Google Scholar]
- 40.Korenaga M., Wang T., Li Y., Showalter L.A., Chan T., Sun J., Weinman S.A. Hepatitis c virus core protein inhibits mitochondrial electron transport and increases reactive oxygen species (ros) production. J. Biol. Chem. 2005;280:37481–37488. doi: 10.1074/jbc.M506412200. [DOI] [PubMed] [Google Scholar]
- 41.Ait–Goughoulte M., Kanda T., Meyer K., Ryerse J.S., Ray R.B., Ray R. Hepatitis c virus genotype 1a growth and induction of autophagy. J. Virol. 2008;82:2241–2249. doi: 10.1128/JVI.02093-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sir D., Kuo C.F., Tian Y., Liu H.M., Huang E.J., Jung J.U., Machida K., Ou J.H. Replication of hepatitis c virus rna on autophagosomal membranes. J. Biol. Chem. 2012;287:18036–18043. doi: 10.1074/jbc.M111.320085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Quan W., Jung H.S., Lee M.S. Role of autophagy in the progression from obesity to diabetes and in the control of energy balance. Arch. Pharm. Res. 2013 doi: 10.1007/s12272-013-0024-7. [DOI] [PubMed] [Google Scholar]
- 44.Barbaro G., Di Lorenzo G., Asti A., Ribersani M., Belloni G., Grisorio B., Filice G., Barbarini G. Hepatocellular mitochondrial alterations in patients with chronic hepatitis c: Ultrastructural and biochemical findings. Am. J. Gastroenterol. 1999;94:2198–2205. doi: 10.1111/j.1572-0241.1999.01294.x. [DOI] [PubMed] [Google Scholar]
- 45.Deng L., Adachi T., Kitayama K., Bungyoku Y., Kitazawa S., Ishido S., Shoji I., Hotta H. Hepatitis c virus infection induces apoptosis through a bax–triggered, mitochondrion–mediated, caspase 3–dependent pathway. J. Virol. 2008;82:10375–10385. doi: 10.1128/JVI.00395-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chu V.C., Bhattacharya S., Nomoto A., Lin J., Zaidi S.K., Oberley T.D., Weinman S.A., Azhar S., Huang T.T. Persistent expression of hepatitis c virus non–structural proteins leads to increased autophagy and mitochondrial injury in human hepatoma cells. PLoS One. 2011;6:e28551. doi: 10.1371/journal.pone.0028551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nomura–Takigawa Y., Nagano–Fujii M., Deng L., Kitazawa S., Ishido S., Sada K., Hotta H. Non–structural protein 4a of hepatitis c virus accumulates on mitochondria and renders the cells prone to undergoing mitochondria–mediated apoptosis. J. Gen. Virol. 2006;87:1935–1945. doi: 10.1099/vir.0.81701-0. [DOI] [PubMed] [Google Scholar]
- 48.Barba G., Harper F., Harada T., Kohara M., Goulinet S., Matsuura Y., Eder G., Schaff Z., Chapman M.J., Miyamura T., et al. Hepatitis c virus core protein shows a cytoplasmic localization and associates to cellular lipid storage droplets. Proc. Natl. Acad. Sci. U.S.A. 1997;94:1200–1205. doi: 10.1073/pnas.94.4.1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Okuda M., Li K., Beard M.R., Showalter L.A., Scholle F., Lemon S.M., Weinman S.A. Mitochondrial injury, oxidative stress, and antioxidant gene expression are induced by hepatitis c virus core protein. Gastroenterology. 2002;122:366–375. doi: 10.1053/gast.2002.30983. [DOI] [PubMed] [Google Scholar]
- 50.Suzuki R., Sakamoto S., Tsutsumi T., Rikimaru A., Tanaka K., Shimoike T., Moriishi K., Iwasaki T., Mizumoto K., Matsuura Y., et al. Molecular determinants for subcellular localization of hepatitis c virus core protein. J. Virol. 2005;79:1271–1281. doi: 10.1128/JVI.79.2.1271-1281.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schwer B., Ren S., Pietschmann T., Kartenbeck J., Kaehlcke K., Bartenschlager R., Yen T.S., Ott M. Targeting of hepatitis c virus core protein to mitochondria through a novel c–terminal localization motif. J. Virol. 2004;78:7958–7968. doi: 10.1128/JVI.78.15.7958-7968.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kasprzak A., Seidel J., Biczysko W., Wysocki J., Spachacz R., Zabel M. Intracellular localization of ns3 and c proteins in chronic hepatitis c. Liver Int. 2005;25:896–903. doi: 10.1111/j.1478-3231.2005.01109.x. [DOI] [PubMed] [Google Scholar]
- 53.Rouille Y., Helle F., Delgrange D., Roingeard P., Voisset C., Blanchard E., Belouzard S., McKeating J., Patel A.H., Maertens G., et al. Subcellular localization of hepatitis c virus structural proteins in a cell culture system that efficiently replicates the virus. J. Virol. 2006;80:2832–2841. doi: 10.1128/JVI.80.6.2832-2841.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lai C.K., Jeng K.S., Machida K., Lai M.M. Hepatitis c virus egress and release depend on endosomal trafficking of core protein. J. Virol. 2010;84:11590–11598. doi: 10.1128/JVI.00587-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang T., Campbell R.V., Yi M.K., Lemon S.M., Weinman S.A. Role of hepatitis c virus core protein in viral–induced mitochondrial dysfunction. J. Viral Hepat. 2010;17:784–793. doi: 10.1111/j.1365-2893.2009.01238.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Horner S.M., Liu H.M., Park H.S., Briley J., Gale M., Jr. Mitochondrial–associated endoplasmic reticulum membranes (mam) form innate immune synapses and are targeted by hepatitis c virus. Proc. Natl. Acad. Sci. U.S.A. 2011;108:14590–14595. doi: 10.1073/pnas.1110133108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mottola G., Cardinali G., Ceccacci A., Trozzi C., Bartholomew L., Torrisi M.R., Pedrazzini E., Bonatti S., Migliaccio G. Hepatitis c virus nonstructural proteins are localized in a modified endoplasmic reticulum of cells expressing viral subgenomic replicons. Virology. 2002;293:31–43. doi: 10.1006/viro.2001.1229. [DOI] [PubMed] [Google Scholar]
- 58.Wolk B., Sansonno D., Krausslich H.G., Dammacco F., Rice C.M., Blum H.E., Moradpour D. Subcellular localization, stability, and trans–cleavage competence of the hepatitis c virus ns3–ns4a complex expressed in tetracycline–regulated cell lines. J. Virol. 2000;74:2293–2304. doi: 10.1128/JVI.74.5.2293-2304.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Griffin S., Clarke D., McCormick C., Rowlands D., Harris M. Signal peptide cleavage and internal targeting signals direct the hepatitis c virus p7 protein to distinct intracellular membranes. J. Virol. 2005;79:15525–15536. doi: 10.1128/JVI.79.24.15525-15536.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Abdalla M.Y., Ahmad I.M., Spitz D.R., Schmidt W.N., Britigan B.E. Hepatitis c virus–core and non structural proteins lead to different effects on cellular antioxidant defenses. J. Med. Virol. 2005;76:489–497. doi: 10.1002/jmv.20388. [DOI] [PubMed] [Google Scholar]
- 61.Fujinaga H., Tsutsumi T., Yotsuyanagi H., Moriya K., Koike K. Hepatocarcinogenesis in hepatitis c: Hcv shrewdly exacerbates oxidative stress by modulating both production and scavenging of reactive oxygen species. Oncology. 2011;81(Suppl 1):11–17. doi: 10.1159/000333253. [DOI] [PubMed] [Google Scholar]
- 62.Morbitzer M., Herget T. Expression of gastrointestinal glutathione peroxidase is inversely correlated to the presence of hepatitis c virus subgenomic rna in human liver cells. J. Biol. Chem. 2005;280:8831–8841. doi: 10.1074/jbc.M413730200. [DOI] [PubMed] [Google Scholar]
- 63.Machida K., Cheng K.T., Lai C.K., Jeng K.S., Sung V.M., Lai M.M. Hepatitis c virus triggers mitochondrial permeability transition with production of reactive oxygen species, leading to DNA damage and stat3 activation. J. Virol. 2006;80:7199–7207. doi: 10.1128/JVI.00321-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Li K., Prow T., Lemon S.M., Beard M.R. Cellular response to conditional expression of hepatitis c virus core protein in huh7 cultured human hepatoma cells. Hepatology. 2002;35:1237–1246. doi: 10.1053/jhep.2002.32968. [DOI] [PubMed] [Google Scholar]
- 65.Boudreau H.E., Emerson S.U., Korzeniowska A., Jendrysik M.A., Leto T.L. Hepatitis c virus (hcv) proteins induce nadph oxidase 4 expression in a transforming growth factor beta–dependent manner: A new contributor to hcv–induced oxidative stress. J. Virol. 2009;83:12934–12946. doi: 10.1128/JVI.01059-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.de Mochel N.S., Seronello S., Wang S.H., Ito C., Zheng J.X., Liang T.J., Lambeth J.D., Choi J. Hepatocyte nad(p)h oxidases as an endogenous source of reactive oxygen species during hepatitis c virus infection. Hepatology. 2010;52:47–59. doi: 10.1002/hep.23671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Stuyver L.J., Whitaker T., McBrayer T.R., Hernandez–Santiago B.I., Lostia S., Tharnish P.M., Ramesh M., Chu C.K., Jordan R., Shi J., et al. Ribonucleoside analogue that blocks replication of bovine viral diarrhea and hepatitis c viruses in culture. Antimicrob. Agents Chemother. 2003;47:244–254. doi: 10.1128/AAC.47.1.244-254.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ando T., Imamura H., Suzuki R., Aizaki H., Watanabe T., Wakita T., Suzuki T. Visualization and measurement of atp levels in living cells replicating hepatitis c virus genome rna. PLoS Pathog. 2012;8:e1002561. doi: 10.1371/journal.ppat.1002561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rasmussen A.L., Diamond D.L., McDermott J.E., Gao X., Metz T.O., Matzke M.M., Carter V.S., Belisle S.E., Korth M.J., Waters K.M., et al. Systems virology identifies a mitochondrial fatty acid oxidation enzyme, dodecenoyl coenzyme a delta isomerase, required for hepatitis c virus replication and likely pathogenesis. J. Virol. 2011;85:11646–11654. doi: 10.1128/JVI.05605-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dharancy S., Malapel M., Perlemuter G., Roskams T., Cheng Y., Dubuquoy L., Podevin P., Conti F., Canva V., Philippe D., et al. Impaired expression of the peroxisome proliferator-activated receptor alpha during hepatitis c virus infection. Gastroenterology. 2005;128:334–342. doi: 10.1053/j.gastro.2004.11.016. [DOI] [PubMed] [Google Scholar]
- 71.Cheng Y., Dharancy S., Malapel M., Desreumaux P. Hepatitis c virus infection down–regulates the expression of peroxisome proliferator–activated receptor alpha and carnitine palmitoyl acyl–coa transferase 1a. World J. Gastroenterol. 2005;11:7591–7596. doi: 10.3748/wjg.v11.i48.7591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Syed G.H., Siddiqui A. Effects of hypolipidemic agent nordihydroguaiaretic acid on lipid droplets and hepatitis c virus. Hepatology. 2011;54:1936–1946. doi: 10.1002/hep.24619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Shimoda R., Nagashima M., Sakamoto M., Yamaguchi N., Hirohashi S., Yokota J., Kasai H. Increased formation of oxidative DNA damage, 8–hydroxydeoxyguanosine, in human livers with chronic hepatitis. Cancer Res. 1994;54:3171–3172. [PubMed] [Google Scholar]
- 74.Mahmood S., Kawanaka M., Kamei A., Izumi A., Nakata K., Niiyama G., Ikeda H., Hanano S., Suehiro M., Togawa K., et al. Immunohistochemical evaluation of oxidative stress markers in chronic hepatitis c. Antioxid. Redox Signal. 2004;6:19–24. doi: 10.1089/152308604771978318. [DOI] [PubMed] [Google Scholar]
- 75.Fujita N., Sugimoto R., Ma N., Tanaka H., Iwasa M., Kobayashi Y., Kawanishi S., Watanabe S., Kaito M., Takei Y. Comparison of hepatic oxidative DNA damage in patients with chronic hepatitis b and c. J. Viral Hepat. 2008;15:498–507. doi: 10.1111/j.1365-2893.2008.00972.x. [DOI] [PubMed] [Google Scholar]
- 76.Farinati F., Cardin R., De Maria N., Della Libera G., Marafin C., Lecis E., Burra P., Floreani A., Cecchetto A., Naccarato R. Iron storage, lipid peroxidation and glutathione turnover in chronic anti–hcv positive hepatitis. J. Hepatol. 1995;22:449–456. doi: 10.1016/0168-8278(95)80108-1. [DOI] [PubMed] [Google Scholar]
- 77.Kageyama F., Kobayashi Y., Kawasaki T., Toyokuni S., Uchida K., Nakamura H. Successful interferon therapy reverses enhanced hepatic iron accumulation and lipid peroxidation in chronic hepatitis c. Am. J. Gastroenterol. 2000;95:1041–1050. doi: 10.1111/j.1572-0241.2000.01979.x. [DOI] [PubMed] [Google Scholar]
- 78.Paradis V., Mathurin P., Kollinger M., Imbert–Bismut F., Charlotte F., Piton A., Opolon P., Holstege A., Poynard T., Bedossa P. In situ detection of lipid peroxidation in chronic hepatitis c: Correlation with pathological features. J Clin Pathol. 1997;50:401–406. doi: 10.1136/jcp.50.5.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.De Maria N., Colantoni A., Fagiuoli S., Liu G.J., Rogers B.K., Farinati F., Van Thiel D.H., Floyd R.A. Association between reactive oxygen species and disease activity in chronic hepatitis c. Free Radic. Biol. Med. 1996;21:291–295. doi: 10.1016/0891-5849(96)00044-5. [DOI] [PubMed] [Google Scholar]
- 80.Romero F.J., Bosch–Morell F., Romero M.J., Jareno E.J., Romero B., Marin N., Roma J. Lipid peroxidation products and antioxidants in human disease. Environ. Health Perspect. 1998;106(Suppl 5):1229–1234. doi: 10.1289/ehp.98106s51229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Yadav D., Hertan H.I., Schweitzer P., Norkus E.P., Pitchumoni C.S. Serum and liver micronutrient antioxidants and serum oxidative stress in patients with chronic hepatitis c. Am. J. Gastroenterol. 2002;97:2634–2639. doi: 10.1111/j.1572-0241.2002.06041.x. [DOI] [PubMed] [Google Scholar]
- 82.Ko W.S., Guo C.H., Yeh M.S., Lin L.Y., Hsu G.S., Chen P.C., Luo M.C., Lin C.Y. Blood micronutrient, oxidative stress, and viral load in patients with chronic hepatitis c. World J. Gastroenterol. 2005;11:4697–4702. doi: 10.3748/wjg.v11.i30.4697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Nakashima T., Sumida Y., Yoh T., Kakisaka Y., Nakajima Y., Ishikawa H., Mitsuyoshi H., Kashima K., Nakamura H., Yodoi J. Thioredoxin levels in the sera of untreated viral hepatitis patients and those treated with glycyrrhizin or ursodeoxycholic acid. Antioxid Redox Signal. 2000;2:687–694. doi: 10.1089/ars.2000.2.4-687. [DOI] [PubMed] [Google Scholar]
- 84.Thorburn D., Curry G., Spooner R., Spence E., Oien K., Halls D., Fox R., McCruden E.A., MacSween R.N., Mills P.R. The role of iron and haemochromatosis gene mutations in the progression of liver disease in chronic hepatitis c. Gut. 2002;50:248–252. doi: 10.1136/gut.50.2.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Izumi N., Enomoto N., Uchihara M., Murakami T., Ono K., Noguchi O., Miyake S., Nouchi T., Fujisawa K., Marumo F., et al. Hepatic iron contents and response to interferon-alpha in patients with chronic hepatitis c. Relationship to genotypes of hepatitis c virus. Dig. Dis. Sci. 1996;41:989–994. doi: 10.1007/BF02091542. [DOI] [PubMed] [Google Scholar]
- 86.Hezode C., Cazeneuve C., Coue O., Roudot–Thoraval F., Lonjon I., Bastie A., Duvoux C., Pawlotsky J.M., Zafrani E.S., Amselem S., et al. Liver iron accumulation in patients with chronic active hepatitis c: Prevalence and role of hemochromatosis gene mutations and relationship with hepatic histological lesions. J. Hepatol. 1999;31:979–984. doi: 10.1016/S0168-8278(99)80308-0. [DOI] [PubMed] [Google Scholar]
- 87.Vendemiale G., Grattagliano I., Portincasa P., Serviddio G., Palasciamo G., Altomare E. Oxidative stress in symptom–free hcv carriers: Relation with alt flare–up. Eur. J. Clin. Invest. 2001;31:54–63. doi: 10.1046/j.1365-2362.2001.00747.x. [DOI] [PubMed] [Google Scholar]
- 88.Diamond D.L., Jacobs J.M., Paeper B., Proll S.C., Gritsenko M.A., Carithers R.L., Jr., Larson A.M., Yeh M.M., Camp D.G., 2nd, Smith R.D., et al. Proteomic profiling of human liver biopsies: Hepatitis c virus–induced fibrosis and mitochondrial dysfunction. Hepatology. 2007;46:649–657. doi: 10.1002/hep.21751. [DOI] [PubMed] [Google Scholar]
- 89.Chan S.W., Egan P.A. Hepatitis c virus envelope proteins regulate chop via induction of the unfolded protein response. FASEB J. 2005;19:1510–1512. doi: 10.1096/fj.04-3455fje. [DOI] [PubMed] [Google Scholar]
- 90.Qadri I., Iwahashi M., Capasso J.M., Hopken M.W., Flores S., Schaack J., Simon F.R. Induced oxidative stress and activated expression of manganese superoxide dismutase during hepatitis c virus replication: Role of jnk, p38 mapk and ap–1. Biochem J. 2004;378:919–928. doi: 10.1042/BJ20031587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Moriya K., Nakagawa K., Santa T., Shintani Y., Fujie H., Miyoshi H., Tsutsumi T., Miyazawa T., Ishibashi K., Horie T., et al. Oxidative stress in the absence of inflammation in a mouse model for hepatitis c virus–associated hepatocarcinogenesis. Cancer Res. 2001;61:4365–4370. [PubMed] [Google Scholar]
- 92.Lin W., Tsai W.L., Shao R.X., Wu G., Peng L.F., Barlow L.L., Chung W.J., Zhang L., Zhao H., Jang J.Y., et al. Hepatitis c virus regulates transforming growth factor beta1 production through the generation of reactive oxygen species in a nuclear factor kappab–dependent manner. Gastroenterology. 2010;138:2509–2518. doi: 10.1053/j.gastro.2010.03.008. 2518 e2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Polyak S.J., Morishima C., Lohmann V., Pal S., Lee D.Y., Liu Y., Graf T.N., Oberlies N.H. Identification of hepatoprotective flavonolignans from silymarin. Proc. Natl. Acad. Sci. U.S.A. 2010;107:5995–5999. doi: 10.1073/pnas.0914009107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Dionisio N., Garcia–Mediavilla M.V., Sanchez–Campos S., Majano P.L., Benedicto I., Rosado J.A., Salido G.M., Gonzalez–Gallego J. Hepatitis c virus ns5a and core proteins induce oxidative stress–mediated calcium signalling alterations in hepatocytes. J. Hepatol. 2009;50:872–882. doi: 10.1016/j.jhep.2008.12.026. [DOI] [PubMed] [Google Scholar]
- 95.Korenaga M., Okuda M., Otani K., Wang T., Li Y., Weinman S.A. Mitochondrial dysfunction in hepatitis c. J. Clin. Gastroenterol. 2005;39:S162–S166. doi: 10.1097/01.mcg.0000155517.02468.46. [DOI] [PubMed] [Google Scholar]
- 96.Gong G., Waris G., Tanveer R., Siddiqui A. Human hepatitis c virus ns5a protein alters intracellular calcium levels, induces oxidative stress, and activates stat–3 and nf–kappa b. Proc. Natl. Acad. Sci. U.S.A. 2001;98:9599–9604. doi: 10.1073/pnas.171311298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Hara Y., Hino K., Okuda M., Furutani T., Hidaka I., Yamaguchi Y., Korenaga M., Li K., Weinman S.A., Lemon S.M., et al. Hepatitis c virus core protein inhibits deoxycholic acid–mediated apoptosis despite generating mitochondrial reactive oxygen species. J. Gastroenterol. 2006;41:257–268. doi: 10.1007/s00535-005-1738-1. [DOI] [PubMed] [Google Scholar]
- 98.Ivanov A.V., Smirnova O.A., Ivanova O.N., Masalova O.V., Kochetkov S.N., Isaguliants M.G. Hepatitis c virus proteins activate nrf2/are pathway by distinct ros–dependent and independent mechanisms in huh7 cells. PLoS One. 2011;6:e24957. doi: 10.1371/journal.pone.0024957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Machida K., Cheng K.T., Sung V.M., Lee K.J., Levine A.M., Lai M.M. Hepatitis c virus infection activates the immunologic (type ii) isoform of nitric oxide synthase and thereby enhances DNA damage and mutations of cellular genes. J. Virol. 2004;78:8835–8843. doi: 10.1128/JVI.78.16.8835-8843.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ghaziani T., Shan Y., Lambrecht R.W., Donohue S.E., Pietschmann T., Bartenschlager R., Bonkovsky H.L. Hcv proteins increase expression of heme oxygenase–1 (ho–1) and decrease expression of bach1 in human hepatoma cells. J. Hepatol. 2006;45:5–12. doi: 10.1016/j.jhep.2005.12.020. [DOI] [PubMed] [Google Scholar]
- 101.Abdalla M.Y., Britigan B.E., Wen F., Icardi M., McCormick M.L., LaBrecque D.R., Voigt M., Brown K.E., Schmidt W.N. Down–regulation of heme oxygenase–1 by hepatitis c virus infection in vivo and by the in vitro expression of hepatitis c core protein. J. Infect. Dis. 2004;190:1109–1118. doi: 10.1086/423488. [DOI] [PubMed] [Google Scholar]
- 102.Garcia–Mediavilla M.V., Sanchez–Campos S., Gonzalez–Perez P., Gomez–Gonzalo M., Majano P.L., Lopez–Cabrera M., Clemente G., Garcia–Monzon C., Gonzalez–Gallego J. Differential contribution of hepatitis c virus ns5a and core proteins to the induction of oxidative and nitrosative stress in human hepatocyte–derived cells. J. Hepatol. 2005;43:606–613. doi: 10.1016/j.jhep.2005.04.019. [DOI] [PubMed] [Google Scholar]
- 103.Robinson L.C., Marchant J.S. Enhanced ca2+ leak from er ca2+ stores induced by hepatitis c ns5a protein. Biochem. Biophys. Res. Commun. 2008;368:593–599. doi: 10.1016/j.bbrc.2008.01.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Roe B., Kensicki E., Mohney R., Hall W.W. Metabolomic profile of hepatitis c virus–infected hepatocytes. PLoS One. 2011;6:e23641. doi: 10.1371/journal.pone.0023641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Yano M., Ikeda M., Abe K., Dansako H., Ohkoshi S., Aoyagi Y., Kato N. Comprehensive analysis of the effects of ordinary nutrients on hepatitis c virus rna replication in cell culture. Antimicrob. Agents Chemother. 2007;51:2016–2027. doi: 10.1128/AAC.01426-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Nakamura M., Saito H., Ikeda M., Hokari R., Kato N., Hibi T., Miura S. An antioxidant resveratrol significantly enhanced replication of hepatitis c virus. World J. Gastroenterol. 2010;16:184–192. doi: 10.3748/wjg.v16.i2.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kuroki M., Ariumi Y., Ikeda M., Dansako H., Wakita T., Kato N. Arsenic trioxide inhibits hepatitis c virus rna replication through modulation of the glutathione redox system and oxidative stress. J. Virol. 2009;83:2338–2348. doi: 10.1128/JVI.01840-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Huang H., Chen Y., Ye J. Inhibition of hepatitis c virus replication by peroxidation of arachidonate and restoration by vitamin e. Proc. Natl. Acad. Sci. U.S.A. 2007;104:18666–18670. doi: 10.1073/pnas.0708423104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Trujillo–Murillo K., Alvarez–Martinez O., Garza–Rodriguez L., Martinez–Rodriguez H., Bosques–Padilla F., Ramos–Jimenez J., Barrera–Saldana H., Rincon–Sanchez A.R., Rivas–Estilla A.M. Additive effect of ethanol and hcv subgenomic replicon expression on cox–2 protein levels and activity. J. Viral Hepat. 2007;14:608–617. doi: 10.1111/j.1365-2893.2006.00837.x. [DOI] [PubMed] [Google Scholar]
- 110.McCartney E.M., Semendric L., Helbig K.J., Hinze S., Jones B., Weinman S.A., Beard M.R. Alcohol metabolism increases the replication of hepatitis c virus and attenuates the antiviral action of interferon. J. Infect. Dis. 2008;198:1766–1775. doi: 10.1086/593216. [DOI] [PubMed] [Google Scholar]
- 111.von Herbay A., Stahl W., Niederau C., Sies H. Vitamin e improves the aminotransferase status of patients suffering from viral hepatitis c: A randomized, double–blind, placebo–controlled study. Free Radical Res. 1997;27:599–605. doi: 10.3109/10715769709097863. [DOI] [PubMed] [Google Scholar]
- 112.Par A., Roth E., Miseta A., Hegedus G., Par G., Hunyady B., Vincze A. Effects of supplementation with the antioxidant flavonoid, silymarin, in chronic hepatitis c patients treated with peg–interferon + ribavirin. A placebo–controlled double blind study. Orv Hetil. 2009;150:73–79. doi: 10.1556/OH.2009.28517. [DOI] [PubMed] [Google Scholar]
- 113.Farias M.S., Budni P., Ribeiro C.M., Parisotto E.B., Santos C.E., Dias J.F., Dalmarco E.M., Frode T.S., Pedrosa R.C., Wilhelm Filho D. Antioxidant supplementation attenuates oxidative stress in chronic hepatitis c patients. Gastroenterol. Hepatol. 2012;35:386–394. doi: 10.1016/j.gastrohep.2012.03.004. [DOI] [PubMed] [Google Scholar]
- 114.Melhem A., Stern M., Shibolet O., Israeli E., Ackerman Z., Pappo O., Hemed N., Rowe M., Ohana H., Zabrecky G., et al. Treatment of chronic hepatitis c virus infection via antioxidants: Results of a phase i clinical trial. J. Clin. Gastroenterol. 2005;39:737–742. doi: 10.1097/01.mcg.0000174023.73472.29. [DOI] [PubMed] [Google Scholar]
- 115.Gane E.J., Weilert F., Orr D.W., Keogh G.F., Gibson M., Lockhart M.M., Frampton C.M., Taylor K.M., Smith R.A., Murphy M.P. The mitochondria–targeted anti–oxidant mitoquinone decreases liver damage in a phase ii study of hepatitis c patients. Liver Int. 2010;30:1019–1026. doi: 10.1111/j.1478-3231.2010.02250.x. [DOI] [PubMed] [Google Scholar]
- 116.Choi J., Lee K.J., Zheng Y., Yamaga A.K., Lai M.M., Ou J.H. Reactive oxygen species suppress hepatitis c virus rna replication in human hepatoma cells. Hepatology. 2004;39:81–89. doi: 10.1002/hep.20001. [DOI] [PubMed] [Google Scholar]
- 117.Rieusset J., Fauconnier J., Paillard M., Belaidi E., Tubbs E., Chauvin M.A., Durand A., Bravard A., Teixeira G., Bartosch B., et al. Disruption of cyclophilin d–mediated calcium transfer from the er to mitochondria contributes to hepatic er stress and insulin resistance. Hepatology. 2012 [Google Scholar]
- 118.Distelhorst C.W., Bootman M.D. Bcl–2 interaction with the inositol 1,4,5–trisphosphate receptor: Role in ca(2+) signaling and disease. Cell Calcium. 2011;50:234–241. doi: 10.1016/j.ceca.2011.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Benali–Furet N.L., Chami M., Houel L., De Giorgi F., Vernejoul F., Lagorce D., Buscail L., Bartenschlager R., Ichas F., Rizzuto R., et al. Hepatitis c virus core triggers apoptosis in liver cells by inducing er stress and er calcium depletion. Oncogene. 2005;24:4921–4933. doi: 10.1038/sj.onc.1208673. [DOI] [PubMed] [Google Scholar]
- 120.Elmore S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007;35:495–516. doi: 10.1080/01926230701320337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Calabrese F., Pontisso P., Pettenazzo E., Benvegnu L., Vario A., Chemello L., Alberti A., Valente M. Liver cell apoptosis in chronic hepatitis c correlates with histological but not biochemical activity or serum hcv–rna levels. Hepatology. 2000;31:1153–1159. doi: 10.1053/he.2000.7123. [DOI] [PubMed] [Google Scholar]
- 122.Bantel H., Lugering A., Heidemann J., Volkmann X., Poremba C., Strassburg C.P., Manns M.P., Schulze–Osthoff K. Detection of apoptotic caspase activation in sera from patients with chronic hcv infection is associated with fibrotic liver injury. Hepatology. 2004;40:1078–1087. doi: 10.1002/hep.20411. [DOI] [PubMed] [Google Scholar]
- 123.Kronenberger B., Ruster B., Lee J.H., Sarrazin C., Roth W.K., Herrmann G., Zeuzem S. Hepatocellular proliferation in patients with chronic hepatitis c and persistently normal or abnormal aminotransferase levels. J. Hepatol. 2000;33:640–647. doi: 10.1016/S0168-8278(00)80017-3. [DOI] [PubMed] [Google Scholar]
- 124.Lau J.Y., Xie X., Lai M.M., Wu P.C. Apoptosis and viral hepatitis. Semin. Liver Dis. 1998;18:169–176. doi: 10.1055/s-2007-1007152. [DOI] [PubMed] [Google Scholar]
- 125.Canbay A., Friedman S., Gores G.J. Apoptosis: The nexus of liver injury and fibrosis. Hepatology. 2004;39:273–278. doi: 10.1002/hep.20051. [DOI] [PubMed] [Google Scholar]
- 126.Hiramatsu N., Hayashi N., Haruna Y., Kasahara A., Fusamoto H., Mori C., Fuke I., Okayama H., Kamada T. Immunohistochemical detection of hepatitis c virus–infected hepatocytes in chronic liver disease with monoclonal antibodies to core, envelope and ns3 regions of the hepatitis c virus genome. Hepatology. 1992;16:306–311. doi: 10.1002/hep.1840160205. [DOI] [PubMed] [Google Scholar]
- 127.Liang Y., Shilagard T., Xiao S.Y., Snyder N., Lau D., Cicalese L., Weiss H., Vargas G., Lemon S.M. Visualizing hepatitis c virus infections in human liver by two–photon microscopy. Gastroenterology. 2009;137:1448–1458. doi: 10.1053/j.gastro.2009.07.050. [DOI] [PubMed] [Google Scholar]
- 128.Moorman J.P., Prayther D., McVay D., Hahn Y.S., Hahn C.S. The c–terminal region of hepatitis c core protein is required for fas–ligand independent apoptosis in jurkat cells by facilitating fas oligomerization. Virology. 2003;312:320–329. doi: 10.1016/S0042-6822(03)00208-3. [DOI] [PubMed] [Google Scholar]
- 129.Lee S.K., Park S.O., Joe C.O., Kim Y.S. Interaction of hcv core protein with 14–3–3epsilon protein releases bax to activate apoptosis. Biochem. Biophys. Res. Commun. 2007;352:756–762. doi: 10.1016/j.bbrc.2006.11.098. [DOI] [PubMed] [Google Scholar]
- 130.Mohd–Ismail N.K., Deng L., Sukumaran S.K., Yu V.C., Hotta H., Tan Y.J. The hepatitis c virus core protein contains a bh3 domain that regulates apoptosis through specific interaction with human mcl–1. J. Virol. 2009;83:9993–10006. doi: 10.1128/JVI.00509-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Goh P.Y., Tan Y.J., Lim S.P., Lim S.G., Tan Y.H., Hong W.J. The hepatitis c virus core protein interacts with ns5a and activates its caspase–mediated proteolytic cleavage. Virology. 2001;290:224–236. doi: 10.1006/viro.2001.1195. [DOI] [PubMed] [Google Scholar]
- 132.Sacco R., Tsutsumi T., Suzuki R., Otsuka M., Aizaki H., Sakamoto S., Matsuda M., Seki N., Matsuura Y., Miyamura T., et al. Antiapoptotic regulation by hepatitis c virus core protein through up–regulation of inhibitor of caspase–activated dnase. Virology. 2003;317:24–35. doi: 10.1016/j.virol.2003.08.028. [DOI] [PubMed] [Google Scholar]
- 133.Chou A.H., Tsai H.F., Wu Y.Y., Hu C.Y., Hwang L.H., Hsu P.I., Hsu P.N. Hepatitis c virus core protein modulates trail–mediated apoptosis by enhancing bid cleavage and activation of mitochondria apoptosis signaling pathway. J. Immunol. 2005;174:2160–2166. doi: 10.4049/jimmunol.174.4.2160. [DOI] [PubMed] [Google Scholar]
- 134.Berg C.P., Schlosser S.F., Neukirchen D.K., Papadakis C., Gregor M., Wesselborg S., Stein G.M. Hepatitis c virus core protein induces apoptosis–like caspase independent cell death. Virol. J. 2009;6:213. doi: 10.1186/1743-422X-6-213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Ciccaglione A.R., Marcantonio C., Tritarelli E., Equestre M., Magurano F., Costantino A., Nicoletti L., Rapicetta M. The transmembrane domain of hepatitis c virus e1 glycoprotein induces cell death. Virus Res. 2004;104:1–9. doi: 10.1016/j.virusres.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 136.Ciccaglione A.R., Marcantonio C., Costantino A., Equestre M., Rapicetta M. Expression of hcv e1 protein in baculovirus–infected cells: Effects on cell viability and apoptosis induction. Intervirology. 2003;46:121–126. doi: 10.1159/000069747. [DOI] [PubMed] [Google Scholar]
- 137.Chiou H.L., Hsieh Y.S., Hsieh M.R., Chen T.Y. Hcv e2 may induce apoptosis of huh–7 cells via a mitochondrial–related caspase pathway. Biochem. Biophys. Res. Commun. 2006;345:453–458. doi: 10.1016/j.bbrc.2006.04.118. [DOI] [PubMed] [Google Scholar]
- 138.Erdtmann L., Franck N., Lerat H., Le Seyec J., Gilot D., Cannie I., Gripon P., Hibner U., Guguen–Guillouzo C. The hepatitis c virus ns2 protein is an inhibitor of cide–b–induced apoptosis. J. Biol. Chem. 2003;278:18256–18264. doi: 10.1074/jbc.M209732200. [DOI] [PubMed] [Google Scholar]
- 139.Meylan E., Curran J., Hofmann K., Moradpour D., Binder M., Bartenschlager R., Tschopp J. Cardif is an adaptor protein in the rig–i antiviral pathway and is targeted by hepatitis c virus. Nature. 2005;437:1167–1172. doi: 10.1038/nature04193. [DOI] [PubMed] [Google Scholar]
- 140.Prikhod'ko E.A., Prikhod'ko G.G., Siegel R.M., Thompson P., Major M.E., Cohen J.I. The ns3 protein of hepatitis c virus induces caspase–8–mediated apoptosis independent of its protease or helicase activities. Virology. 2004;329:53–67. doi: 10.1016/j.virol.2004.08.012. [DOI] [PubMed] [Google Scholar]
- 141.Zhao P., Han T., Guo J.J., Zhu S.L., Wang J., Ao F., Jing M.Z., She Y.L., Wu Z.H., Ye L.B. Hcv ns4b induces apoptosis through the mitochondrial death pathway. Virus Res. 2012;169:1–7. doi: 10.1016/j.virusres.2012.04.006. [DOI] [PubMed] [Google Scholar]
- 142.Zhou Y., Gong G.Z. Hepatitis c virus ns5a protein upregulates survivin gene expression. Zhonghua Gan Zang Bing Za Zhi. 2006;14:414–417. [PubMed] [Google Scholar]
- 143.Lin L.Y., Li S.C., Lu S.L. Hepatitis c virus nonstructural 5a protein inhibits tumor necrosis factor alpha mediated apoptosis of hepg2 cells. Zhonghua Nei Ke Za Zhi. 2003;42:392–395. [PubMed] [Google Scholar]
- 144.Lan K.H., Sheu M.L., Hwang S.J., Yen S.H., Chen S.Y., Wu J.C., Wang Y.J., Kato N., Omata M., Chang F.Y., et al. Hcv ns5a interacts with p53 and inhibits p53–mediated apoptosis. Oncogene. 2002;21:4801–4811. doi: 10.1038/sj.onc.1205589. [DOI] [PubMed] [Google Scholar]
- 145.Street A., Macdonald A., Crowder K., Harris M. The hepatitis c virus ns5a protein activates a phosphoinositide 3–kinase–dependent survival signaling cascade. J. Biol. Chem. 2004;279:12232–12241. doi: 10.1074/jbc.M312245200. [DOI] [PubMed] [Google Scholar]
- 146.Chung Y.L., Sheu M.L., Yen S.H. Hepatitis c virus ns5a as a potential viral bcl–2 homologue interacts with bax and inhibits apoptosis in hepatocellular carcinoma. Int. J. Cancer. 2003;107:65–73. doi: 10.1002/ijc.11303. [DOI] [PubMed] [Google Scholar]
- 147.Quarato G., D'Aprile A., Gavillet B., Vuagniaux G., Moradpour D., Capitanio N., Piccoli C. The cyclophilin inhibitor alisporivir prevents hepatitis c virus– mediated mitochondrial dysfunction. Hepatology. 2012;55:1333–1343. doi: 10.1002/hep.25514. [DOI] [PubMed] [Google Scholar]