

ABSTRACT
The efficacy of current influenza vaccines requires a close antigenic match between circulating and vaccine strains. As such, timely identification of emerging influenza virus antigenic variants is central to the success of influenza vaccination programs. Empirical methods to determine influenza virus antigenic properties are time-consuming and mid-throughput and require live viruses. Here, we present a novel, experimentally validated, computational method for determining influenza virus antigenicity on the basis of hemagglutinin (HA) sequence. This method integrates a bootstrapped ridge regression with antigenic mapping to quantify antigenic distances by using influenza HA1 sequences. Our method was applied to H3N2 seasonal influenza viruses and identified the 13 previously recognized H3N2 antigenic clusters and the antigenic drift event of 2009 that led to a change of the H3N2 vaccine strain.
IMPORTANCE
This report supplies a novel method for quantifying antigenic distance and identifying antigenic variants using sequences alone. This method will be useful in influenza vaccine strain selection by significantly reducing the human labor efforts for serological characterization and will increase the likelihood of correct influenza vaccine candidate selection.
Introduction
Seasonal influenza causes approximately 24,000 deaths and 200,000 hospitalizations in the United States annually (1–3), and an influenza pandemic may kill millions of people in a short time. Vaccination is the primary option to reduce influenza outbreaks (4). Mutations in the influenza virus surface glycoproteins hemagglutinin (HA) and neuraminidase (NA) can cause antigenic drift, because these proteins, especially HA, are the primary targets for host immunity (5, 6). Because influenza viruses frequently undergo antigenic change in response to host immunity, circulating strains are continually monitored to optimize the antigenic matches between vaccine and predicted community strains, a process that is the key to a successful influenza vaccine program (7). However, identifying antigenic variants is not a trivial task, and current systems rely on empirical determination of antigenicity by using methods such as hemagglutination inhibition (HI) assays. Each year, thousands of scientists worldwide, including those from 5 World Health Organization (WHO) collaborative centers, more than 136 national influenza centers in 106 countries, more than 80 participating laboratories in the United States’ National Respiratory and Enteric Virus Surveillance System, and numerous vaccine companies, generate data to inform influenza vaccine strain selection (7). Vaccine mismatches can lead to vaccine failure, disease outbreaks, and huge economic burdens (8, 9). As such, developing additional rapid and robust methods to identify antigenic variants of influenza virus remains a major public health endeavor.
Because influenza virus sequence data can be collected rapidly and economically, sequence-based antigenic characterization can shorten influenza variant detection time and increase influenza surveillance coverage, thus facilitating influenza vaccine strain selection. A few attempts at predicting influenza virus antigenic variants based on the basis of genomic sequences have been previously reported. For instance, Lee and Chen developed a simple method to correlate HI titer with the number of mutations between test and reference viral HA sequences (10). Liao et al. applied multiple and logistic regression analyses to compare HA mutations to HI values (26). Huang et al. developed a decision tree algorithm in drift variant prediction by using information theory to derive association rules from HI data (12). Most recently, a naive Bayes classifier was developed to derive features solely on the basis of sequence comparison results, and these features were used to compare antigenic similarities between sequences (13). However, to the best of our knowledge, none of these methods was able to quantify influenza antigenic distance and to infer influenza antigenicity for antigenic variant identification.
Here, we developed and validated a novel computational method that integrates antigenic mapping and machine learning approaches to quantify antigenic distances by using influenza HA1 sequences (Fig. 1). This method, so-called antigenicity prediction via bootstrapped ridge selection (Antigen-Bridges), integrates a bootstrapped ridge regression with antigenic mapping to quantify antigenic distances by using influenza HA1 sequences. This method was used with H3N2 influenza A virus sequences and identified the genomic signatures associated with HA antigenicity and the mutations responsible for antigenic drift events in H3N2 seasonal influenza viruses. By using historical serologic data, the antigenic scoring function derived from this framework was validated to quantify the antigenic distances solely on the basis of HA1 sequences, with an accuracy of approximately 80%, while over 95% of historical vaccine strains were predicted as antigenic variants.
FIG 1 .

Simplified framework of Antigen-Bridges. (A) Sequence alignments and antigenic mapping to construct genetic and antigenic profiles; (B) ridge regression to identify antigenicity-associated sites and to detect mutations driving antigenic drift events; (C) antigenic distance prediction function to quantify antigenic distances and identify antigenic variants on the basis of their HA1 protein sequence.
RESULTS
Antigen-Bridges: sequence-based antigenic distance quantification by machine learning.
We developed a model using HA1 protein sequences to quantify influenza antigenic distances by integrating antigenic mapping and machine learning. In this model, serologic data (i.e., HI titers) are first transformed into pairwise antigenic distances between viruses by using matrix completion-multiple dimensional scaling (14) followed by transformation of the HA1 sequence alignment into a genetic distance matrix (Fig. 1). Because surface residues of HA1 are predominantly responsible for antigenicity (5, 6), the genetic information of the sites on the protein surface was the only information used to construct the genetic distance matrix. A novel machine learning method, antigenicity prediction via bootstrapped ridge selection (Antigen-Bridges), was selected to correlate the antigenic distance matrix with the genetic distance matrix.
The proposed computational method was developed on the basis of the hypothesis that only a few features (instead of the entire data set) are necessary to determine the overall data characteristics. The rationale of this hypothesis is that the sites affecting influenza antigenicity are mostly in the head structures of the HA protein, such as the antibody-binding sites Sa, Sb, Ca1, Ca2, and Cb in H1N1 (6) and sites A, B, C, D, and E in H3N2 viruses (5, 15). For H3N2 viruses, approximately 100 residues exist in these 5 antibody-binding sites, and only a few of these residues have frequently changed during antigenic drifts since 1968 (16–21). Furthermore, it is well documented that approximately 75% of epitopes have 15 to 25 residues (22), and only a few of these are responsible for the majority of antibody binding (23), which may be useful in predicting antigenic variants. The ridge regression selects the residues that are most likely to be involved in determining antigenicity by selecting those that minimize the difference between the genetic distance matrix and the antigenic distance matrix. After we trained and tested the model, it assigned each residue a weight indicating the influence of this residue on influenza antigenicity—the larger the weight, the greater the influence of the residue on influenza antigenicity. The number of antigenicity-associated residues was decided by performing cross-validation and bootstrapping, which aims to obtain the best match between the genetic distance matrix and the antigenic distance matrix. By integrating weights derived from ridge selection and the biophysical properties of the selected antigenicity-associated sites, a sequence-based antigenicity scoring function can be developed to quantify the antigenic distance between any two HA1 sequences. By using this function, the antigenic distance between a newly isolated influenza virus and a known virus can be quantified solely from its HA1 sequence and an antigenic map constructed using the distance matrix (14). By applying the computational model to two antigenic clusters, we could also identify the single and multiple sites that drive the antigenic drift.
Antigenic profiling derived from HA1 sequences by Antigen-Bridges matches the antigenic profile derived from serological data.
Using the proposed computational method to analyze H3N2 influenza virus datasets spanning from 1968 to 2007 identified 39 antigenicity-associated sites, including 35 sites in the 5 reported antibody-binding sites A to E (5): 9 in A (126, 131, 133, 135, 137, 140, 142, 144, and 145), 11 in B (155, 156, 158, 159, 160, 163, 188, 189, 193, 196, and 197), 4 in C (50, 53, 276, and 278), 6 in D (121, 172, 173, 214, 219, and 226), and 5 in E (57, 62, 63, 82, and 262) (see Fig. S1A and Table S1 in the supplemental material). Residues 25, 202, 222, and 225 were also identified as being antigenicity associated (see Table S1).
We attempted to use the identified antigenicity-associated sites to develop a scoring function (Fig. 1) to quantify antigenic distances solely on the basis of HA1 sequences. Using this scoring function, we constructed a sequence-based H3 antigenic map, which we compared with a map generated by using HI data. The Pearson’s correlation coefficient between the HI-derived antigenic map (see Fig. S1B) and the sequence-derived map (see Fig. S1C) was 0.9355, indicating a high level of congruence between the methods.
Mutations driving H3N2 influenza antigenic drift predicted by Antigen-Bridges were validated by bench experiments.
The H3N2 HI data of the 1968 to 2007 H3N2 viruses describe at least 13 major antigenic clusters: HK68, EN72, VI75, TX77, BK79, SI87, BE89, BE92, WU95, SY97, FU02, CA04, and BR07 (16, 24). We used our model to identify the mutations responsible for these antigenic transitions. The 12 antigenic drift events were caused by 3 single-residue mutations, 6 double mutations, and 3 multiple-residue mutations, with positions 135, 145, 156, and 193 being involved in at least 2 of the events (Table 1).
TABLE 1 .
Predominant mutations that drove H3N2 antigenic drifts from 1968 to 2007
| Antigenic drift | Mutation(s) |
|---|---|
| HK68 → EN72 | G144D |
| EN72 → VI75 | S145N-S193D |
| VI75 → TX77 | D53K-E82K |
| TX77 → BA79 | K156E |
| BK79 → SI87 | Y155H-K189R |
| SI87 → BE89 | G135E-N145K-N193S |
| BE89 → BE92 | E135K-K145N-E156K |
| BE92 → WU95 | N145K-G172D |
| WU95 → SY97 | K62E-K156Q-E158K |
| SY97 → FU02 | Q156H |
| FU02 → CA04 | K145N-Y159F |
| CA04 → BR07 | S193F-D225N |
The residues responsible for prior H3N2 antigenic drifts are at either single or multiple antibody-binding sites (Table 1) (http://sysbio.cvm.msstate.edu/H3N2MachineLearning/FIGW1.pdf), as reported previously (5). For example, substitutions at residues 189 and 155 that caused the antigenic drift from cluster BK79 to SI87 are in the B binding site; substitutions at residues 135, 145, and 156 that caused the antigenic drift from cluster BE89 to BE92 are in the neighboring binding sites A and B; and substitutions at residues 62, 156, and 158 that caused the antigenic drift from cluster WU95 to SY97 are in sites B and E, which are relatively far apart in H3 HA’s three-dimensional structure (http://sysbio.cvm.msstate.edu/H3N2MachineLearning/FIGW1.pdf).
The computationally identified mutations driving 3 antigenic drift events (BE92 to WU95, WU95 to SY97, and SY97 to FU02) were selected for experimental validation. Single and multiple mutants were generated by performing site-directed mutagenesis and reverse genetics with the following templates: A/Johannesburg/33/1994(H3N2) (JO/33; representing antigenic cluster BE92), A/Nanchang/933/1995(H3N2) (NA/933; representing cluster WU95), and A/Sydney/05/1997(H3N2) (SY/05; representing cluster SY97). Although 7 mutants were generated, NA933-E158K was not successfully rescued despite multiple attempts. These mutations were expected to relocate the virus from one antigenic cluster to the targeted antigenic cluster in the antigenic map. For example, the N145K and G172D mutations of JO/33 were expected to relocate JO/33 from antigenic cluster BE92 to antigenic cluster WU95. All of the introduced mutations led to at least a one-unit change in antigenic distance from the parental wild-type strains, corresponding to a 2-fold change in HI titer (see Table S2 in the supplemental material). Simultaneous mutations N145K and G172D and simultaneous mutations K62E, K156Q, and E158K were required to relocate the mutant from cluster BE92 to WU95 and from cluster WU95 to WY97, although N145K and K156Q dominated the changes leading to these 2 antigenic drift events. Q156H moved the mutant from cluster SY97 to FU02 (Fig. 2). The results of microneutralization (MN) assays confirmed those of HI assays (see Table S3).
FIG 2 .

Experimental validation of select predicted residues’ ability to drive antigenic drift events. In total, 17 single-, double-, or multiple-site mutants of the residues shown in Table 1 were generated (see Table S2 in the supplemental material for the list). HI experiments were performed with these mutants and their corresponding wild-type strains (JO/33, NA/933, and SY/05). The experimentally generated mutants are denoted by the suffix “EXP.” To facilitate the comparison, we used Antigen-Bridges to project the mutated HA (Fig. 1). The computationally simulated mutants (Table 1) are denoted by the suffix “SIM.” For example, 145EXP in Fig. 2A represents an experimentally derived N145K mutant of JO/33 wild-type virus, whereas 145SIM is a computer-simulated N145K mutant of JO/33s HA sequence. Here, 1 unit corresponds to a 2-fold change in HI value. The overall Pearson’s correlation coefficient r between the experimental and predicted antigenic distance matrices is 0.7148 (see Table S4 in the supplemental material). The two testing antigenic clusters are labeled in gray and blue, respectively. The viruses in white are not antigenically defined. The wild-type strains used in experiments are marked in yellow. The experimentally generated mutants are marked in pink if they are located in the antigenic cluster where their corresponding wild-type strain is located and in purple if in the expected antigenic cluster by mutation(s). The computationally generated mutants are marked in maroon if they are located in the antigenic cluster where their corresponding wild-type strain is located and in green if in the expected antigenic cluster by mutation(s).
To confirm our computational results, we generated 10 additional viruses having mutations outside the predominant sites previously examined: JO/33-rg-K135T, JO/33-rg-R197Q, JO/33-rg-N262S, JO/33-rg-S278N, NA/933-rg-G142R, NA/933-rg-V144I, NA/933-rg-V196A, SY/05-rg-L25I, SY/05-rg-H155T, and SY/05-rg-W222R. The results of serologic experiments showed that these mutants had antigenic profiles that differed from those of the parental wild-type strains (see Table S4 in the supplemental material). The Pearson correlation coefficient between antigenic distances estimated by using HI data and those estimated by using HA1 sequence data was 0.7148 (see Table S4), indicating that other residues outside the dominant epitope sites also contribute to antigenic drift.
Antigen-Bridges can predict the antigenic variants for the next influenza season(s).
The key component of selecting influenza vaccine strains is comparing the antigenic properties of circulating influenza viruses with those of viruses from previous seasons. Thus, an effective sequence-based antigenic variant identification system would be expected to predict the antigenic profile of a virus on the basis of historical training data. We used historical training data (from 1968 to the year to be predicted) to test the prediction accuracies of our model for future seasons. The threshold value used to define an influenza antigenic variant was 2 units (25). The prediction accuracy measures the percentile of predicted antigenic variants matching the antigenic variants in the benchmark data. Our results had an average accuracy of 83% (n = 18 years, from 1990 to 2007) for predicting antigenic variants emerging in the coming year (see Table S5 in the supplemental material). The prediction accuracy decreased to approximately 70% when we used the algorithm to predict antigenic variants coming in the next 5 years.
Although no sequence-based quantitative algorithm to measure influenza antigenic distance existed prior to this study, a few groups reported finding residues to be associated with antigenic variations of H3N2 virus. For example, Liao et al. found 25 residues via multiple and logistic regression analyses of HA mutations and HI values (26), and Smith et al. identified 44 residues via simple sequence alignments (16). To compare the effectiveness of our proposed computational method in quantifying antigenic distance to that of published residue sets and to confirm the validity of the 39 identified sites, we performed comparative analyses using historical data spanning from 1968 to 2003. Our results demonstrated that combining our 39 predicted sites and the weight matrix derived from Antigen-Bridges yields the best prediction accuracy (see Table S5 in the supplemental material). Furthermore, using the weight matrix derived from Antigen-Bridges improved the prediction accuracy of the 25-residue set (26) and 44-residue set (16).
Large-scale sequence-based antigenic profiling suggested less-punctuated changes in antigenic evolution of H3N2 seasonal influenza viruses.
Using publicly available nonredundant HA1 sequences (n = 3,332), we generated an exhaustive H3 antigenic map. Phylogenetically, the H3 influenza A viruses isolated from 1968 to 2012 show a gradual change over time (Fig. 3A). The previously reported 13 antigenic clusters were on this map, although some viruses did connect the antigenic clusters (Fig. 3B). The results of further analyses showed that the multiple residues that we had previously identified as causing some of the antigenic drifts (Table 1) did not always appear simultaneously. For example, the mutations K62E, K156Q, and E158K caused the antigenic drift from cluster WU95 to SY97 (Table 1), with the predominant viruses in the WU95 and SY97 clusters having 62K-156K-158E and 62E-156Q-158K residues, respectively. H3N2 viruses with 62K-156K-158E residues were predominant in 1996, and those with 62E-156Q-158K residues were predominant after the autumn of 1997. In February and March 2007, a few intermediate viruses connected the WU95 and SY97 clusters, and these viruses had 62K-156K-158K, 62K-156Q-158K, or 62E-156K-158K residues (see Table S6 in the supplemental material and http://sysbio.cvm.msstate.edu/H3N2MachineLearning). Thus, antigenic changes in H3N2 viruses are more likely to occur gradually than simultaneously.
FIG 3 .
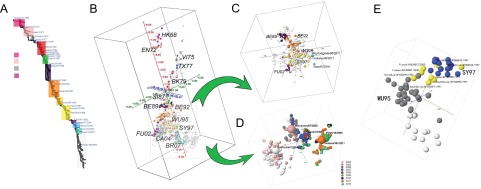
HA1 sequence-based H3N2 antigenic mapping. (A) Phylogenetic tree of H3N2 viruses (1968 to 2012). The selected vaccine strains recommended by the WHO are annotated in this tree. (B) The antigenic map of H3N2 influenza A virus based on nonredundant HA1 sequences (n = 3332). The scoring function was trained by using HI datasets of viruses from 1968 to 2007. The sequences with HI values are marked in color, and others are gray. (C) An antigenic submap of H3N2 viruses isolated from 1991 to 2001. The recently emerged swine origin H3N2v isolates (51) are marked in cyan. (D) An antigenic submap of H3N2 viruses isolated from 2004 to 2012. The viruses are color coded by year, with vaccine strains annotated in enlarged spheres. (E) The map of cluster WU95 and SY97, showing the gradual change. The viruses marked in yellow have 1 or more of the predominant mutations that drive antigenic drift from WU95 to SY97.
Detection of antigenic drifts by Antigen-Bridges.
A significant antigenic cluster emerged in 2009 and 2010 that corresponds to the change of the H3N2 vaccine candidate from A/Brisbane/10/2007(H3N2) to A/Perth/16/2009(H3N2) (27). The antigenic distance between the 2 viruses is about 2.39 units on our antigenic map, supporting the need for this change. In February 2012, the WHO suggested using A/Victoria/361/2011(H3N2)-like virus as the influenza vaccine candidate for the upcoming influenza season in the autumn of 2012. Antigen-Bridges indicates that the antigenic distance between A/Perth/16/2009(H3N2) and the 2012-2013 Northern Hemisphere vaccine candidate A/Victoria/361/2011(H3N2) is only 0.44 unit (Fig. 3D). Sequence comparison results show that 9 residues differ between A/Perth/16/2009(H3N2) and A/Victoria/361/2011(H3N2) (E1): S45N (antibody-binding site C), T48I (site C), K62E (site E), K144N (site A), A198S (site B), T212A (site D), S214I (site D), V223I, and N312S. Among these residues, K62E, K144N, and T212N were detected sporadically in the 2009-2010 season, more frequently in the 2010-2011 season, and predominantly in the 2011-2012 season.
Residues 62 and 144 are among the predominant antigenicity-associated sites identified by Antigen-Bridges. However, Antigen-Bridges also indicated a large extent of antigenic diversity among the epidemic H3N2 strains in the 2011-2012 season (Fig. 3). Besides those mutations in A/Victoria/361/2011(H3N2) (E1), sequence analyses showed that the common mutations D53N (antibody-binding site C) and N145S (site A) appeared in some strains but without clear temporal patterns. Both mutations are among our 39 predicted antigenicity-associated sites and could contribute to the large extent of antigenic diversity among the epidemic strains of the 2011-2012 season (Fig. 3D).
Estimated antigenicity of the variant H3N2 viruses using Antigen-Bridges.
In 2011 and 2012, more than 300 zoonotic infections with a variant H3N2 (H3N2v) virus were reported in people attending state fairs in the United States (28). In a proof-of-principle test of our model’s ability to estimate the antigenicity of an emerging virus, we produced a sequence-based antigenic map of the H3N2v viruses. This map suggested that the H3N2v isolates were antigenically related to the isolates in the BE92 and WU95 antigenic clusters (Fig. 3). This result was confirmed by HI data, which showed that the H3N2v viruses reacted to postinfection antisera raised against A/Ann Arbor/03/1993(H3N2) (AN/03), JO/33, or NA/933 (see Table S7 in the supplemental material). The percentages of sequence identity between H3N2v and either AN/03, JO/33, or NA/933 were 87.24% to 90.27%. On the basis of the public sequences, A/New York/571/1996(H3N2) has the smallest antigenic distance from these H3N2v viruses, varying from 0.44 to 0.98 units, with corresponding percentages of sequence identity from 89.36% to 89.97%.
DISCUSSION
The proposed computational framework integrates a machine learning approach with an antigenic mapping approach and quantifies influenza antigenic distance on the basis of HA1 sequence. In this study, this framework is applied and validated in H3N2 influenza A viruses. H3N2 is used as an example because antigenic drift occurred more frequently in H3N2 viruses than in any other subtypes of seasonal influenza viruses, including H1N1, 2009 H1N1, and influenza B viruses. Furthermore, the human vaccine strain used against the H3N2 virus was updated more frequently than those against other subtypes. For example, the vaccine strain against H3N2 virus has been updated at least 27 times since 1968 (20 times from 1977 to 2009) but only 9 times for H1N1 virus from 1977 to 2009 and 15 times for influenza B virus from 1972 to 2011. However, our method can be easily adapted to study other influenza subtypes. Because antibody binding sites may differ among subtypes of influenza A viruses, we will likely need to derive a subtype-specific quantification function. For example, the results of our recent study of H5N1 highly pathogenic avian influenza virus suggested that the antigenicity-associated sites of H5N1 viruses are not necessarily the same as those of H3N2 and H1N1 viruses (29). To use this method for other subtypes of influenza A viruses, we can use serologic data of the target subtype to train and build a subtype-specific scoring function.
Surveillance data show that new antigenic drift variants of epidemiological importance contain a mean of 13.2 HA amino acid substitutions, with more than half of them in antigenic sites (20). Our proposed computational model identified 39 antigenicity-associated sites, with the dominant sites determining the antigenic drift events from 1968 to 2007. Ndifon et al. (30) presented a competitive model to predict antibody escape, proposing that antigenic drift events would be associated with amino acid changes that occur in epitopes with high neutralization efficiencies (i.e., epitopes A, B, and D) rather than in those with low neutralization efficiencies (i.e., epitopes C and E). Supporting this prediction, the major residues our algorithm identified are in epitopes A and B. Among these predicted sites, 142, 156, 193, 219, and 225 have been linked to egg adaptation (31). More studies are needed to estimate the effect of these sites on our scoring function.
The proposed model quantifies antigenic distance based only on HA1 sequences. Although the NA gene of H3N2 also undergoes antigenic drift (32), HA’s effect on the antigenic profile is dominant over that of NA in both B- and T-cell priming because of HA-NA competition (33). Nevertheless, NA’s effect on influenza antigenicity will be explored to optimize the scoring function.
During influenza surveillance, tens of thousands of influenza viruses are isolated, and immunologic assays such as HI and MN are performed to detect antigenic variants. Although this is a robust system, issues such as the reduction seen in H3N2 virus binding to red blood cells (34, 35) can lead to problems performing and interpreting HI assays. Furthermore, because antigenic characterization is relatively labor-intensive, only a small portion (generally, fewer than 20%) of the influenza isolates sequenced will be antigenically characterized. In the absence of a reliable, cost-efficient, high-throughput assay, the sequence-based method proposed in this study can be used to substantially bolster the vaccine strain selection process. Our sequence-based method can significantly shorten influenza variant detection time and increase influenza surveillance coverage. Furthermore, this method can serve as an initial screen of antigenic variants and reduce current assay workloads in influenza surveillance, because few of the antigenic variants detected by using this sequence-based method are selected for experimental validation.
MATERIALS AND METHODS
Antigen-Bridges algorithm. (i) Generation of sequence-derived distance matrix for machine learning.
Two scoring functions, namely binary function and pattern-induced multisequence alignment (PIMA) function (36, 37), were used to generate the similarity matrix x. In the binary scoring function, the score of a pair of amino acids is 1 if they are different, and it is 0 otherwise. In the PIMA scoring function (37), the score of a pair of amino acids is shown in http://sysbio.cvm.msstate.edu/H3N2MachineLearning/FIGW2.pdf. A comparison shows that PIMA performs slightly better than binary function in accuracy; therefore, PIMA was used throughout this study (http://sysbio.cvm.msstate.edu/H3N2MachineLearning/TABLEW1.pdf).
(ii) Generation of the HI-derived antigenic distance matrix for machine learning.
As we did in our earlier study of antigenic maps, we sorted the viruses and antisera by temporal order (14). The resulting HI table had a banded structure: the entries in the diagonal zone of this matrix were composed of high reactors and missing values, whereas the entries in the rest of the matrix were either low reactors or missing values (http://sysbio.cvm.msstate.edu/H3N2MachineLearning/FIGW3.pdf). To minimize the effects of low reactors, the temporal model that was created by using the mathematical and computer modeling of the dynamic systems approach (14) with a window size of 12 years was adapted to generate the long-distance matrix Mlong (long function). The Mlong value was used to learn the weight matrix wlong.
We also recovered HI values without using a temporal model. The local function recovered only HI values spanning an interval of 12 years, and average values were used for entries having multiple learning processes because of the sliding window. The short-distance matrix Mshort (short function) was used to learn the weight matrix wshort.
(iii) Antigenicity determining feature selection using machine learning.
Ridge regression for feature selection is a machine learning strategy that is effective for selecting a small to moderate number of good features; in addition to Antigen-Bridges, Lasso (38) was applied to our data. We found that the proposed Antigen-Bridges method had a prediction accuracy that was slightly better than that of the more conventional Lasso method, with the added advantages of being more stable and more computationally efficient than Lasso.
Specifically, let x = [1,x′] and w = [w0, w1, w2, …, w329]. Then, ridge regression can be performed to obtain w by solving the following optimization equation:
where the regularization parameter λ can be tuned to optimize accuracy. The larger the weight value, or wj, of a residue j, the greater the influence the residue site will have on the antigenic distance quantification. To optimize the parameter λ in the ridge regression variable selection form, we set λ to be 0.01, 0.1, 1, 10, 100, 350, 400, 1,000, or 10,000. A comparison of the outcomes showed that a λ value of 350 yielded the highest accuracy, 0.8378, in predicting the antigenicity of viruses (see Table S8 in the supplemental material).
Ridge regression can assign a weight to each residue via learning. However, it is important to determine which residues predominantly drive influenza antigenicity, because not all residues in HA1 affect antigenicity. To determine the number of predominant residues, we plotted the root mean square error (RMSE) curve based on the variation of the number of residues (http://sysbio.cvm.msstate.edu/H3N2MachineLearning/FIGW4.pdf). The smaller the RMSE is, the better the learning results will be. Our results suggest that approximately 40 residues are enough to achieve the best performance.
To increase the feature selection stability and to minimize the likelihood of overfitting and false-positive errors, we randomly replaced 20% of the influenza viruses in each run of ridge regression learning and then selected 40 sites from each run for analysis. A total of 100 runs were performed, and the detection rates on each site were used as the confidence level for whether the site was an antigenicity-associated site. In these 100 runs, 60 individual residues were detected. The residues with a bootstrap value of at least 50 were deemed to be antigenicity-associated sites: 39 sites were thus identified (see Table S1 in the supplemental material).
(iv) Sequence-based antigenic distance-predicting function.
After determining the antigenicity-associated sites and their corresponding weights by using machine learning, we quantified the antigenic distance using the following function:
where the similarity x is derived by PIMA as described above, and w0 is a parameter optimized through cross-validation methods. To quantify the entries in the diagonal zone (http://sysbio.cvm.msstate.edu/H3N2MachineLearning/FIGW3.pdf), wshort was used; otherwise, wlong was used. The constant term w0 was 1.3958 for long distances and 0.5676 for short distances. Here, we adapted a linear function instead of using a nonlinear prediction function by reducing the computational burdens and increasing the scalability of the proposed method. However, we will explore using a nonlinear function in a future study. Note that the weight could be either positive or negative, because not all mutations will change influenza antigenicity in the same direction.
(v) Performance assessment.
On the basis of the 39 single-mutation sites identified by the proposed computational model (see Table S1 in the supplemental material), the antigenic distances of viruses in year k ∈ [1990, 2003] were predicted from their HA protein sequences by using the viruses in some year span before k as training data. Five schemes (Pred1, Pred2, Pred3, Pred4, and Pred5) were applied. Pred1 predicts the pairwise distances of viruses in each pair of consecutive years k and k − 1 for k ∈ [1990, 2003] by using viruses in [1968, k − 1] as training data. Pred2 predicts the distance between viruses in year k − 2 and those in years k and k − 1 by using viruses in [1968, k − 2] as training data. Similar definitions hold for Pred3, Pred4, and Pred5 (see Table S5 in the supplemental material).
Antigenic distances larger than 4-fold (as measured by HI titer) were treated as significant changes in antigenicity (25). The 4-fold change (2 units of antigenic distance) was used as the threshold to partition each pair of antigens into 2 categories, nonvariant or variant. Antigenic distances larger than 2 units were treated as variant (i.e., positive). We tested the prediction accuracy using viruses isolated after 1990 because of the paucity of viruses isolated from 1968 to 1989. The prediction accuracy measures the percentiles of antigenic variants (i.e., true positive) and nonvariants (i.e., true negative) in the testing samples. The prediction was documented as being true positive if the antigenic distance predicted by Antigen-Bridges was indeed an antigenic variant when the pairwise antigenic distance measured by antigenic mapping using HI data was 2 units or more. Likewise, the prediction was documented as being true negative if the antigenic distance predicted by Antigen-Bridges was an antigenic nonvariant when the pairwise antigenic distance measured by antigenic mapping by using HI data was less than 2 units. A total of 6,909 pairs of antigenic distances were used in this training and testing.
The Prediction receiver operating characteristic (ROC) curve is a graphical plot of the sensitivity, or true-positive rate, against the false-positive rate, or 1 specificity, and yields a binary classification system with different decision thresholds. The ROC curves were constructed using different antigenic distance thresholds to partition each pair of antigens into two categories, nonvariant or variant. The threshold values for the classifier boundary range from 0.1 to 7 in increments of 0.1; thus, each ROC curve includes 70 points. The ROC curve shows a systematic profile of the predictive performance of Antigen-Bridges (see Fig. S2 in the supplemental material). The prediction accuracy can be evaluated on a ROC curve by using the threshold of 2 units of antigenic distance as the decision threshold. All curves show that all 5 prediction schema (Pred1 to Pred5) have a true-positive rate of 70%, with a false-positive rate of less than 20%. Pred1 outperformed other methods (see Fig. S2).
Besides ROC curves, we also assess the performance by comparing RMSE and Pearson correlation coefficient (CC) as we did before (14). Usually, a smaller RMSE and a higher CC indicate better performance.
(vi) Comparison with other reported sets of antigenicity-determined features.
To prove the effectiveness of the identified 39 residues in antigenic distance measurements, we compared the predictive accuracies of our 39-residue set with that of 2 reported antigenicity-associated residue sets: a 25-residue set proposed by Liao et al. (26) and a 44-residue set proposed by Smith et al. (16). The data and accuracy definition used here is the same as the one described in the “Sequence-based antigenic distance-predicting function” section. Because neither Liao et al. (26) nor Smith et al. (16) reported a quantitative function, we used 2 approaches to compare the prediction accuracy of their predominant residues to those of ours: (i) assign equal weights to each residue and (ii) assign weights by using our algorithm Antigen-Bridges.
Selection of the mutations that drove historical antigenic drift events.
In addition to knowing the number of residues determining influenza antigenicity, it is important to understand the mutations that drove historical antigenic drift events. Here, the machine learning model was applied subsequently to 2 adjacent virus clusters among the groups HK68, EN72, VI75, TX77, BK79, SI87, BE89, BE92, WU95, SY97, FU02, CA04, and BR07. Similar to the method of antigenicity-associated site selection, selection of these mutations was made according to the RMSE curve, and each residue was assigned a weight, which was used to generate the scoring function as an equation (3). To determine the effect of each mutation, we used this scoring function to identify the single-site or multiple-site mutations driving antigenic drifts. The criteria to be deemed a predominant mutation were that a given mutation would move an influenza virus from its parental wild-type strain position to the center of the subsequent antigenic cluster in a corresponding antigenic map. The minimum set of mutations needed for the 12 historical H3N2 antigenic drift events are listed in Table 1 (see also Fig. 2 and http://sysbio.cvm.msstate.edu/H3N2MachineLearning/FIGW5.pdf for simulation cartographies). The residues associated with these antigenic drifts were a subset of those identified in our earlier analyses as being antigenicity-associated sites (see Table S1).
Surface residue and glycosylation site identification.
GETAREA software (http://curie.utmb.edu/getarea.html) (39) was used to predict whether or not residues were on HA’s surface. H3N2’s three-dimensional HA structure (Protein Data Bank [PDB] identifier [ID] 2VIU) was used as the template. A total of 142 residues were predicted to be located at the HA protein’s surface (http://sysbio.cvm.msstate.edu/H3N2MachineLearning/TABLEW2.pdf).
NetNGlyc (http://www.cbs.dtu.dk/services/NetNGlyc/) was used to identify potential glycosylation sites, and no antigenicity-associated sites were identified as being glycosylation sites. The 13 potential glycosylation sites are at residues 8, 22, 38, 45, 63, 81, 122, 126, 133, 144, 165, 246, and 285 and occur in one or more sequences of the 512 test viruses. Among these sites, residues 63, 126, 133, and 144 are among those 39 residues predicted to predominantly affect antigenicity. However, residue 144 was not predicted to be a glycosylation site among sequences of viruses isolated from 1968 to 1972, but it is predicted to drive antigenic drift from cluster HK68 to EN72 (Table 1). Because no antigenicity-associated sites were identified as being glycosylation sites, the number of glycosylation sites in H3N2 virus is relatively small, and glycosylation status can be affected by factors other than protein sequence. Thus, our prediction function does not consider glycosylation.
Viruses, sera, and serologic and sequence datasets.
The H3N2 influenza viruses used were provided by the Centers for Disease Control and Prevention and BEI Resources (see Table S2 in the supplemental material). The ferret antisera were generated by using 6- to 8-week-old ferrets that had HI baseline titers less than 1:10 against A/Brisbane/10/2007(H3N2), A/Brisbane/59/2007(H3N2), and A/California/4/2009(H1N1). The H3N2 HI table used for training contains sequences of 512 viruses and 133 serum samples collected from 1968 to 2007 and was created by combining an HI table containing data from 1968 to 2003 (16) and a sub-HI table containing data from 2002 to 2007 (40). The selected viruses must have been tested against at least 5% of the 133 serum samples, and their full-length HA1 sequences must be available in public databases. The 512 H3N2 viruses were grouped into 13 clusters: HK68, EN72, VI75, TX77, BK79, SI87, BE89, BE92, WU95, SY97, FU02, CA04, and BR07 (16). The full HI data are in Table S2. The HI table and viral sequences used are available at http://sysbio.cvm.msstate.edu/H3N2MachineLearning.
The HA1 sequences of 5,878 viruses isolated from 1968 to 2012 (including 9 swine origin viruses) and 26 vaccine strains were downloaded from the influenza virus resources (41). With 512 sequences from the benchmark data, there are 6,390 sequences in total. After removing redundant sequences, 3,332 sequences, including those of 5 H3N2v viruses (42), remained for the following analyses. These sequences were aligned by using MUSCLE (43) and reordered by isolation year to form the alignment file (http://sysbio.cvm.msstate.edu/H3N2MachineLearning/)
Antigenic map construction and phylogenetic tree construction.
The serologic data-based antigenic maps were constructed directly from HI data by using AntigenMap3D (14, 44). First, the antigenic distance matrices were measured by using the sequence-based antigenic distance predictive function, and then classical multidimensional scaling (45) was used to generate the antigenic map, which was visualized by using Jmol (46).
Phylogenetic trees were generated by using GARLI version 0.95 (47) to perform the maximum likelihood estimation method, and the bootstrap values were generated by using PAUP* version 4.0 beta (48) to implement neighbor-joining methods.
Experimental validation.
The mutagenesis, reverse genetics, and serologic assays were conducted as described (29). Briefly, the HAs of A/Johannesburg/33/1994(H3N2), A/Nanchang/933/1995(H3N2), and A/Sydney/05/1997(H3N2) were used as the templates for mutagenesis with the QuikChange II site-directed mutagenesis kit. All mutations were confirmed by sequencing. Viruses were generated by using pHW2000 clones of the mutated HAs and appropriate NAs, with the 6 remaining genes belonging to A/Puerto Rico/8/34(H1N1) (49). The mutagenesis targeted 2 types of residues: (i) those with mutations predicted to play a dominant role in driving antigenic drift from cluster BE92 to WU95, from cluster WU95 to SY97, and from cluster SY97 to FU02 (Table 1) and (ii) those with mutations predicted to play a dominant role in driving antigenic drift from cluster BE92 to WU95, cluster WU95 to SY97, or cluster SY97 to FU02 that were also predicted to be antigenicity-associated sites (see Table S1 in the supplemental material). A total of 19 mutants (17 single-site mutants and 2 multiple-site mutants) were rescued (see Table S2). Despite four trials, the single mutant JO/33-E158K was not rescued.
HI titer assays were performed as described (29) by using 0.5% turkey red blood cells. The MN assay was adapted from the protocol of the Centers for Disease Control and Prevention (50).
SUPPLEMENTAL MATERIAL
Mutations predicted by Antigen-Bridges to have caused the antigenic drifts reported from 1968 to 2007. (A) Ribbon diagram of the trimetric HA molecule (PDB: 2VIU), with the 39 predicted antigenicity-associated sites shown in sphere balls. The known antibody-binding sites A to E are marked in color. Residues not belonging to reported antibody-binding sites are shown in white. (B) Antigenic map of 512 H3N2 viruses isolated from 1968 to 2007. The viruses were grouped into 13 antigenic clusters: HK68, EN72, VI75, TX77, BA79, SI87, BE89, BE92, WU95, SY97, FU02, CA04, and BR07. The antigenic distance (mean ± standard deviation [SD]) between any 2 neighboring antigenic clusters is 2.37 ± 0.66 units, with 1 unit corresponding to a 2-fold change in HI experiments. (C) The HA1 sequence-derived antigenic map of the same set of H3N2 viruses used in http://sysbio.cvm.msstate.edu/H3N2MachineLearning/FIGW1.pdf. The Pearson correlation coefficient r of the antigenic distance matrices generating the 2 maps is 0.9355. Download
Prediction receiver operating characteristic (ROC) curves based on the 39 antigenicity-associated sites identified via sparse learning. The antigenic distances between newly isolated viruses were predicted from their HA protein sequences by using historical data as the training set. Download
Antigenicity-associated sites of H3N2 influenza A viruses identified via sparse learning methods for H3N2 influenza A viruses.
Antigenicity of recombinant viruses and parental wild-type viruses in HI assay.
Antigenicity of recombinant viruses and their parental wild-type viruses in microneutralization assay.
In vitro verification of antigenic distances predictions.
Utility of predominant residues predicted in various studies.
Mutation patterns of influenza viruses connecting antigenic clusters WU95 and SY07.
Serologic assays of H3N2v activity against seasonal H3N2 influenza A viruses.
Parameter optimization of ridge regression variable selection.
ACKNOWLEDGMENT
We thank Zhu Guo and Jianqiang Ye for their comments, Zhipeng Cai for technical assistance, and Xiyan Xu for providing H3N2 influenza A viruses.
This study was supported by RC1AI086830 from the U.S. National Institutes of Health.
Footnotes
Citation Sun H, Yang J, Zhang T, Long L-P, Jia K, Yang G, Webby RJ, Wan X-F. 2013. Using sequence data to infer the antigenicity of influenza virus. mBio 4(4):e00230-13. doi:10.1128/mBio.00230-13.
REFERENCES
- 1. CDC 2010. Estimates of deaths associated with seasonal influenza—United States, 1976–2007. MMWR Morb. Mortal. Wkly. Rep. 59:1057–1062 [PubMed] [Google Scholar]
- 2. Thompson WW, Shay DK, Weintraub E, Brammer L, Cox N, Anderson LJ, Fukuda K. 2003. Mortality associated with influenza and respiratory syncytial virus in the United States. JAMA 289:179–186 [DOI] [PubMed] [Google Scholar]
- 3. Thompson WW, Shay DK, Weintraub E, Brammer L, Bridges CB, Cox NJ, Fukuda K. 2004. Influenza-associated hospitalizations in the United States. JAMA 292:1333–1340 [DOI] [PubMed] [Google Scholar]
- 4. Harper SA, Fukuda K, Uyeki TM, Cox NJ, Bridges CB, Centers for Disease Control and Prevention. CDC. Advisory Committee on Immunization Practices 2004. Prevention and control of influenza: recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm. Rep 53:1–40 [PubMed] [Google Scholar]
- 5. Wilson IA, Cox NJ. 1990. Structural basis of immune recognition of influenza virus hemagglutinin. Annu. Rev. Immunol. 8:737–771 [DOI] [PubMed] [Google Scholar]
- 6. Caton AJ, Brownlee GG, Yewdell JW, Gerhard W. 1982. The antigenic structure of the influenza virus A/PR/8/34 hemagglutinin (H1 subtype). Cell 31:417–427 [DOI] [PubMed] [Google Scholar]
- 7. WHO Writing Group. Ampofo WK, Baylor N, Cobey S, Cox NJ, Daves S, Edwards S, Ferguson N, Grohmann G, Hay A, Katz J, Kullabutr K, Lambert L, Levandowski R, Mishra AC, Monto A, Siqueira M, Tashiro M, Waddell AL, Wairagkar N, Wood J, Zambon M, Zhang W, Zhang W. 2012. Improving influenza vaccine virus selection: report of a WHO informal consultation held at WHO headquarters, Geneva, Switzerland, 14–16 June 2010. Influenza Other Respi. Viruses 6:142–152 21819547 [Google Scholar]
- 8. Kilbourne ED, Smith C, Brett I, Pokorny BA, Johansson B, Cox N. 2002. The total influenza vaccine failure of 1947 revisited: major intrasubtypic antigenic change can explain failure of vaccine in a post-World War II epidemic. Proc. Natl. Acad. Sci. U. S. A. 99:10748–10752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. de Jong JC, Beyer WE, Palache AM, Rimmelzwaan GF, Osterhaus AD. 2000. Mismatch between the 1997/1998 influenza vaccine and the major epidemic A(H3N2) virus strain as the cause of an inadequate vaccine-induced antibody response to this strain in the elderly. J. Med. Virol. 61:94–99 [PubMed] [Google Scholar]
- 10. Lee MS, Chen JS. 2004. Predicting antigenic variants of influenza A/H3N2 viruses. Emerg. Infect. Dis. 10(8):1385–1390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. CDC 2012. Update: influenza A (H3N2)v transmission and guidelines—five states, 2011. MMWR Morb. Mortal. Wkly. Rep. 60:1741–1744 [PubMed] [Google Scholar]
- 12. Huang JW, King CC, Yang JM. 2009. Co-evolution positions and rules for antigenic variants of human influenza A/H3N2 viruses. BMC Bioinformatics 10(Suppl 1):S41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mansfield KG. 2007. Viral tropism and the pathogenesis of influenza in the mammalian host. Am. J. Pathol. 171:1089–1092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cai Z, Zhang T, Wan XF. 2010. A computational framework for influenza antigenic cartography. PLoS Comput. Biol. 6:e1000949. 10.1371/journal.pcbi.1000949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Xu R, Ekiert DC, Krause JC, Hai R, Crowe JE, Jr, Wilson IA. 2010. Structural basis of preexisting immunity to the 2009 H1N1 pandemic influenza virus. Science 328:357–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Smith DJ, Lapedes AS, de Jong JC, Bestebroer TM, Rimmelzwaan GF, Osterhaus AD, Fouchier RA. 2004. Mapping the antigenic and genetic evolution of influenza virus. Science 305:371–376 [DOI] [PubMed] [Google Scholar]
- 17. Shih AC, Hsiao TC, Ho MS, Li WH. 2007. Simultaneous amino acid substitutions at antigenic sites drive influenza A hemagglutinin evolution. Proc. Natl. Acad. Sci. U. S. A. 104:6283–6288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jin H, Zhou H, Liu H, Chan W, Adhikary L, Mahmood K, Lee MS, Kemble G. 2005. Two residues in the hemagglutinin of A/Fujian/411/02-like influenza viruses are responsible for antigenic drift from A/Panama/2007/99. Virology 336:113–119 [DOI] [PubMed] [Google Scholar]
- 19. Zhou R, Das P, Royyuru AK. 2008. Single mutation induced H3N2 hemagglutinin antibody neutralization: a free energy perturbation study. J. Phys. Chem. B 112:15813–15820 [DOI] [PubMed] [Google Scholar]
- 20. Ansaldi F, Icardi G, Gasparini R, Campello C, Puzelli S, Bella A, Donatelli I, Salmaso S, Crovari P. 2005. New A/H3N2 influenza variant: a small genetic evolution but a heavy burden on the Italian population during the 2004–2005 season. J. Clin. Microbiol. 43:3027–3029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bush RM, Bender CA, Subbarao K, Cox NJ, Fitch WM. 1999. Predicting the evolution of human influenza A. Science 286:1921–1925 [DOI] [PubMed] [Google Scholar]
- 22. Rubinstein ND, Mayrose I, Halperin D, Yekutieli D, Gershoni JM, Pupko T. 2008. Computational characterization of B-cell epitopes. Mol. Immunol. 45:3477–3489 [DOI] [PubMed] [Google Scholar]
- 23. Air GM, Laver WG, Webster RG. 1990. Mechanism of antigenic variation in an individual epitope on influenza virus N9 neuraminidase. J. Virol. 64:5797–5803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Shu B, Garten R, Emery S, Balish A, Cooper L, Sessions W, Deyde V, Smith C, Berman L, Klimov A, Lindstrom S, Xu X. 2012. Genetic analysis and antigenic characterization of swine origin influenza viruses isolated from humans in the United States, 1990–2010. Virology 422:151–160 [DOI] [PubMed] [Google Scholar]
- 25. Smith DJ, Forrest S, Ackley DH, Perelson AS. 1999. Variable efficacy of repeated annual influenza vaccination. Proc. Natl. Acad. Sci. U. S. A. 96:14001–14006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Liao YC, Lee MS, Ko CY, Hsiung CA. 2008. Bioinformatics models for predicting antigenic variants of influenza A/H3N2 virus. Bioinformatics 24:505–512 [DOI] [PubMed] [Google Scholar]
- 27. Fiore AE, Uyeki TM, Broder K, Finelli L, Euler GL, Singleton JA, Iskander JK, Wortley PM, Shay DK, Bresee JS, Cox NJ, Centers for Disease Control and Prevention. CDC 2010. Prevention and control of influenza with vaccines: recommendations of the Advisory Committee on Immunization Practices (ACIP), 2010. MMWR Recomm. Rep. 59:1–62 [PubMed] [Google Scholar]
- 28. CDC 2012. Evaluation of rapid influenza diagnostic tests for influenza A (H3N2)v virus and updated case count—United States, 2012. MMWR Morb. Mortal. Wkly. Rep. 61:619–621 [PubMed] [Google Scholar]
- 29. Cai Z, Ducatez MF, Yang J, Zhang T, Long LP, Boon AC, Webby RJ, Wan XF. 2012. Identifying antigenicity-associated sites in highly pathogenic H5N1 influenza virus hemagglutinin by using sparse learning. J. Mol. Biol. 422:145–155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ndifon W, Wingreen NS, Levin SA. 2009. Differential neutralization efficiency of hemagglutinin epitopes, antibody interference, and the design of influenza vaccines. Proc. Natl. Acad. Sci. U. S. A. 106:8701–8706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Stevens J, Chen LM, Carney PJ, Garten R, Foust A, Le J, Pokorny BA, Manojkumar R, Silverman J, Devis R, Rhea K, Xu X, Bucher DJ, Paulson JC, Cox NJ, Klimov A, Donis RO. 2010. Receptor specificity of influenza A H3N2 viruses isolated in mammalian cells and embryonated chicken eggs. J. Virol. 84:8287–8299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Westgeest KB, de Graaf M, Fourment M, Bestebroer TM, van Beek R, Spronken MI, de Jong JC, Rimmelzwaan GF, Russell CA, Osterhaus AD, Smith GJ, Smith DJ, Fouchier RA. 2012. Genetic evolution of the neuraminidase of influenza A (H3N2) viruses from 1968 to 2009 and its correspondence to haemagglutinin evolution. J. Gen. Virol. 93:1996–2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Johansson BE, Moran TM, Kilbourne ED. 1987. Antigen-presenting B cells and helper T cells cooperatively mediate intravirionic antigenic competition between influenza A virus surface glycoproteins. Proc. Natl. Acad. Sci. U. S. A. 84:6869–6873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Morishita T, Nobusawa E, Nakajima K, Nakajima S. 1996. Studies on the molecular basis for loss of the ability of recent influenza A (H1N1) virus strains to agglutinate chicken erythrocytes. J. Gen. Virol. 77:2499–2506 [DOI] [PubMed] [Google Scholar]
- 35. Nobusawa E, Ishihara H, Morishita T, Sato K, Nakajima K. 2000. Change in receptor-binding specificity of recent human influenza A viruses (H3N2): a single amino acid change in hemagglutinin altered its recognition of sialyloligosaccharides. Virology 278:587–596 [DOI] [PubMed] [Google Scholar]
- 36. Valdar WS. 2002. Scoring residue conservation. Proteins 48:227–241 [DOI] [PubMed] [Google Scholar]
- 37. Smith RF, Smith TF. 1992. Pattern-induced multi-sequence alignment (PIMA) algorithm employing secondary structure-dependent gap penalties for use in comparative protein modelling. Protein Eng. 5:35–41 [DOI] [PubMed] [Google Scholar]
- 38. Tibshirani R. 1996. Regression shrinkage and selection via the LASSO. J. R. Stat. Soc. B Stat. Methodol. 58:267–288 [Google Scholar]
- 39. Fraczkiewicz R, Braun W. 1998. Exact and efficient analytical calculation of the accessible surface areas and their gradients for macromolecules. J. Comput. Chem. 19:319–333 [Google Scholar]
- 40. Russell CA, Jones TC, Barr IG, Cox NJ, Garten RJ, Gregory V, Gust ID, Hampson AW, Hay AJ, Hurt AC, de Jong JC, Kelso A, Klimov AI, Kageyama T, Komadina N, Lapedes AS, Lin YP, Mosterin A, Obuchi M, Odagiri T, Osterhaus AD, Rimmelzwaan GF, Shaw MW, Skepner E, Stohr K, Tashiro M, Fouchier RA, Smith DJ. 2008. Influenza vaccine strain selection and recent studies on the global migration of seasonal influenza viruses. Vaccine 26(Suppl 4):D31–D34 [DOI] [PubMed] [Google Scholar]
- 41. Bao Y, Bolotov P, Dernovoy D, Kiryutin B, Zaslavsky L, Tatusova T, Ostell J, Lipman D. 2008. The influenza virus resource at the National Center for Biotechnology Information. J. Virol. 82:596–601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Garnier JL, Merino R, Kimoto M, Izui S. 1988. Resistance to tolerance induction to human gamma globulin (HGG) in autoimmune BXSB/MpJ mice: functional analysis of antigen-presenting cells and HGG-specific T helper cells. Clin. Exp. Immunol. 73:283–288 [PMC free article] [PubMed] [Google Scholar]
- 43. Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32:1792–1797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Barnett JL, Yang J, Cai Z, Zhang T, Wan XF. 2012. AntigenMap 3D: an online antigenic cartography resource. Bioinformatics 28:1292–1293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Triana-Baltzer GB, Gubareva LV, Klimov AI, Wurtman DF, Moss RB, Hedlund M, Larson JL, Belshe RB, Fang F. 2009. Inhibition of neuraminidase inhibitor-resistant influenza virus by DAS181, a novel sialidase fusion protein. PLoS One 4:e7838. 10.1371/journal.pone.0007838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Chan RW, Chan MC, Wong AC, Karamanska R, Dell A, Haslam SM, Sihoe AD, Chui WH, Triana-Baltzer G, Li Q, Peiris JS, Fang F, Nicholls JM. 2009. DAS181 inhibits H5N1 influenza virus infection of human lung tissues. Antimicrob. Agents Chemother. 53:3935–3941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zwickl DJ. 2006. Genetic algorithm approaches for the phylogenetic analysis of large biological sequence datasets under the maximum likelihood criterion. University of Texas at Austin, Austin, TX [Google Scholar]
- 48. Swofford DL. 1998. PAUP*: phylogenic analysis using parsimony. Sinauer, Sunderland, MA [Google Scholar]
- 49. Ducatez MF, Cai Z, Peiris M, Guan Y, Ye Z, Wan XF, Webby RJ. 2011. Extent of antigenic cross-reactivity among highly pathogenic H5N1 influenza viruses. J. Clin. Microbiol. 49:3531–3536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Rowe T, Abernathy RA, Hu-Primmer J, Thompson WW, Lu X, Lim W, Fukuda K, Cox NJ, Katz JM. 1999. Detection of antibody to avian influenza A (H5N1) virus in human serum by using a combination of serologic assays. J. Clin. Microbiol. 37:937–943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lindstrom S, Garten R, Balish A, Shu B, Emery S, Berman L, Barnes N, Sleeman K, Gubareva L, Villanueva J, Klimov A. 2012. Human infections with novel reassortant influenza A(H3N2)v viruses, United States, 2011. Emerg. Infect. Dis. 18:834–837 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Mutations predicted by Antigen-Bridges to have caused the antigenic drifts reported from 1968 to 2007. (A) Ribbon diagram of the trimetric HA molecule (PDB: 2VIU), with the 39 predicted antigenicity-associated sites shown in sphere balls. The known antibody-binding sites A to E are marked in color. Residues not belonging to reported antibody-binding sites are shown in white. (B) Antigenic map of 512 H3N2 viruses isolated from 1968 to 2007. The viruses were grouped into 13 antigenic clusters: HK68, EN72, VI75, TX77, BA79, SI87, BE89, BE92, WU95, SY97, FU02, CA04, and BR07. The antigenic distance (mean ± standard deviation [SD]) between any 2 neighboring antigenic clusters is 2.37 ± 0.66 units, with 1 unit corresponding to a 2-fold change in HI experiments. (C) The HA1 sequence-derived antigenic map of the same set of H3N2 viruses used in http://sysbio.cvm.msstate.edu/H3N2MachineLearning/FIGW1.pdf. The Pearson correlation coefficient r of the antigenic distance matrices generating the 2 maps is 0.9355. Download
Prediction receiver operating characteristic (ROC) curves based on the 39 antigenicity-associated sites identified via sparse learning. The antigenic distances between newly isolated viruses were predicted from their HA protein sequences by using historical data as the training set. Download
Antigenicity-associated sites of H3N2 influenza A viruses identified via sparse learning methods for H3N2 influenza A viruses.
Antigenicity of recombinant viruses and parental wild-type viruses in HI assay.
Antigenicity of recombinant viruses and their parental wild-type viruses in microneutralization assay.
In vitro verification of antigenic distances predictions.
Utility of predominant residues predicted in various studies.
Mutation patterns of influenza viruses connecting antigenic clusters WU95 and SY07.
Serologic assays of H3N2v activity against seasonal H3N2 influenza A viruses.
Parameter optimization of ridge regression variable selection.