

ABSTRACT
Bruton’s tyrosine kinase (Btk) is a signaling molecule that plays important roles in B-1 B cell development and innate myeloid cell functions and has recently been identified as a target for therapy of B cell lymphomas. We examined the contribution of B-1 B cells to resistance to Cryptococcus neoformans infection by utilizing X-linked immunodeficient (XID) mice (CBA-CaHN-XID), which possess a mutation in Btk. XID mice had significantly higher brain fungal burdens than the controls 6 weeks after infection with C. neoformans strain 52D (CN52D); however, consistent with the propensity for greater virulence of C. neoformans strain H99 (CNH99), CNH99-infected XID mice had higher lung and brain fungal burdens than the controls 3 weeks after infection. Further studies in a chronic CN52D model revealed markedly lower levels of total and C. neoformans-specific serum IgM in XID mice than in the control mice 1 and 6 weeks after infection. Alveolar macrophage phagocytosis was markedly impaired in CN52D-infected XID mice compared to the controls, with XID mice exhibiting a disorganized lung inflammatory pattern in which Gomori silver staining revealed significantly more enlarged, extracellular C. neoformans cells than the controls. Adoptive transfer of B-1 B cells to XID mice restored peritoneal B-1 B cells but did not restore IgM levels to those of the controls and had no effect on the brain fungal burden at 6 weeks. Taken together, our data support the hypothesis that IgM promotes fungal containment in the lungs by enhancing C. neoformans phagocytosis and restricting C. neoformans enlargement. However, peritoneal B-1 B cells are insufficient to reconstitute a protective effect in the lungs.
IMPORTANCE
Cryptococcus neoformans is a fungal pathogen that causes an estimated 600,000 deaths per year. Most infections occur in individuals who are immunocompromised, with the majority of cases occurring in those with HIV/AIDS, but healthy individuals also develop disease. Immunoglobulin M (IgM) has been linked to resistance to disease in humans and mice. In this article, we found that X-linked immunodeficient (XID) mice, which have markedly reduced levels of IgM, were unable to contain Cryptococcus in the lungs. This was associated with reduced yeast uptake by macrophages, an aberrant tissue inflammatory response, an enlargement of the yeast cells in the lungs, and fungal dissemination to the brain. Since XID mice have a mutation in the Bruton’s tyrosine kinase (Btk) gene, our data suggest that treatments aimed at blocking the function of Btk could pose a higher risk for cryptococcosis.
Introduction
The humoral immune response is critical for resistance to cryptococcosis, the disease caused by the fungal pathogen Cryptococcus neoformans. C. neoformans-specific antibodies exhibit many functions that benefit the host, such as promoting macrophage phagocytosis of C. neoformans, directly eliciting transcriptional responses in yeast upon binding, inhibiting fungal replication, and modulating the host inflammatory response (1–7). During infection, the polysaccharide capsule of C. neoformans, a type 2 T cell-independent (TI-2) antigen, elicits a largely IgM antigen-specific response (8). Defined C. neoformans capsule-specific mouse and human monoclonal IgM antibodies protect against C. neoformans in healthy mice (9, 10), and mice lacking secreted IgM (sIgM−/−) are more susceptible to C. neoformans (6). IgM deficiency is also associated with disease resistance in humans, as it was shown that HIV-associated reduction of antigen-specific IgM and IgM+ memory B cell levels were predictive of cryptococcosis, independent of CD4 T cell status (11).
In mice, B-1 B cells are an innate-like population of B cells that contribute to IgM production. B-1 B cells reside in the peritoneal and thoracic cavities and in the spleen and bone marrow; however, they can migrate to sites of inflammation (12–14). In the steady state, B-1 B cells produce IgM that is of low affinity, germ line encoded, and broadly reactive against conserved, repeating carbohydrate motifs shared by host cells and pathogens (15, 16). During infection, B-1 B cells secrete antigen-specific IgM and IgG in response to TI-2 antigens (13, 17–20). These cells exhibit functional differences from conventional B-2 B cells, which include the ability to secrete antibody in the absence of T cell help, produce antibody outside germinal centers, and phagocytose and present small particulate antigens (21).
B-1 B cells are subdivided into two populations based on expression of CD5, CD5+ B-1a B cells and CD5− B-1b B cells (22), which are derived from different progenitors (23). The contribution of each population to immunity appears to be pathogen specific. Whereas antigen-specific IgM from B-1a B cells protected against Francisella tularensis (19, 20, 24), B-1b B cells were required for Streptococcus pneumoniae immunization to elicit antigen-specific antibodies and to promote resolution of borreliosis (18, 25). In C. neoformans-infected C57BL/6 mice, the peritoneal B-1a B cell subset exhibited greater binding to C. neoformans ex vivo than peritoneal B-1b and B-2 B cells and were shown to mediate a reduction in the lung and brain fungal burden in the early innate immune response (26). B-1 B cells, primarily of the B-1b B cell subset, are capable of migrating to sites of inflammation and differentiating into macrophage-like cells (27) and in vitro-generated B-1-derived macrophages internalize and kill C. neoformans in a nitric oxide-dependent manner (14). However, the role that B-1 B cells play in control of chronic cryptococcal infection has not been examined and that of B-1 B cell-derived IgM in disease resistance has not been established.
In this study, we used X-linked immunodeficient (XID) mice (CBA-CaHN-XID) to examine the role of B-1 B cells in control of chronic pulmonary cryptococcosis. XID mice, which carry a mutation in the Bruton’s tyrosine kinase (Btk) gene that is expressed in B cells and myeloid cells (28, 29), lack B-1a B cells and have reduced levels of natural IgM (29–31). Six weeks after pulmonary infection, C. neoformans strain 52D (CN52D)-infected XID mice had higher brain fungal burdens, a reduced ability to phagocytize C. neoformans, and lower levels of C. neoformans-specific IgM than CBA/CaJ controls. However, restoration of peritoneal cavity B-1 B cells in XID mice did not restore IgM to control levels or reduce brain fungal burdens, suggesting that a threshold amount of IgM and/or other B cell subsets are required to prevent dissemination of chronic pulmonary C. neoformans infection to the brain in mice.
RESULTS
Fungal burden and survival of XID mice infected with C. neoformans strain 52D.
As a previous study demonstrated that XID mice are more susceptible to intravenous infection with a serotype D strain (ATCC 13690) than CBA/Ca control mice (32), we examined whether XID mice are also more susceptible to pulmonary infection via the intranasal route. On day 3 after infection with C. neoformans strain 52D (CN52D), fungal burdens (shown as log10 mean CFU ± standard error of the mean [SEM]) were similar (P > 0.05; n = 5) in lungs (5.71 ± 0.1 for XID mice and 5.92 ± 0.05 for CBA/CaJ mice) and brains (1.81 ± 0.48 for XID mice and 1.67 ± 0.43 for CBA/CaJ mice). One week after infection, lung and brain fungal burden remained comparable (Fig. 1A). However, 6 weeks after infection, XID mice had brain fungal burdens that were 100-fold higher than those of CBA/CaJ controls (Fig. 1A). Only 30% of infected XID mice survived 154 days, while 75% of CBA/CaJ controls lived this long; however, this difference was not statistically significant (P = 0.11) (Fig. 1B).
FIG 1 .

Brain fungal burden is increased in XID mice. (A) Lung and brain fungal burden in XID mice and CBA/CaJ control mice after 1 and 6 weeks of infection with C. neoformans strain 52D (5 mice on day 7 [D7] and 15 mice on day 42 [D42]). Each symbol represents the value for an individual mouse, with the mean of one experiment (day 7) and 3 independent experiments (day 42) indicated by the horizontal black bars. The values for the brain fungal burden in XID mice and CBA/CaJ mice on day 42 are significantly different (P < 0.001) as indicated by the three asterisks. (B) Survival of infected XID mice and control mice (n = 10). Time (in days) postinfection (PI) is shown on the x axis. The graph shows the results of one experiment with 10 mice per group.
To determine whether the increased fungal burdens in the brains of XID mice resulted from an increase in yeast escape from lungs or an increase in brain invasion, we infected mice intravenously. Brain fungal burdens (log10 mean CFU ± SEM) in XID (6.83 ± 0.13) and CBA/CaJ (6.95 ± 0.03) mice were similar 2 weeks after infection, but lung (5.96 ± 0.05 in XID mice and 5.13 ± 0.19 in CBA/CaJ mice) and liver (5.04 ± 0.06 in XID mice and 4.66 ± 0.03 in CBA/CaJ mice) fungal burdens were higher (P < 0.05; n = 4 or 5) in XID mice than in CBA/CaJ controls. This suggests that XID mice were unable to control fungal growth in infected organs.
Cellular infiltration and cytokine production in lungs.
XID mice are more susceptible to Mycobacterium bovis BCG infection, which was associated with changes in lung leukocyte composition and histological pattern (33). We performed fluorescence-activated cell sorting (FACS) analysis to examine lung leukocyte populations in XID and CBA/CaJ control strains after CN52D infection. First, we examined cellular recruitment to the lungs during the innate immune response. On day 3 after infection, XID mice had fewer lung B-2 B cells (values are shown as mean [×104] ± SEM [×104]) than CBA/CaJ controls (2.5 ± 0.6 [XID] and 7.6 ± 2.1 [CBA/CaJ]) (P < 0.05; n = 5), but the numbers of B-1a B cells (0.03 ± 0.01 [XID] and 0.1 ± 0.06 [CBA/CaJ]) and B-1b B cells (1.1 ± 0.3 [XID] and 1.1 ± 0.3 [CBA/CaJ]) were similar. There was no difference in the number of neutrophils in XID (8.0 ± 1.6) and CBA/CaJ (6.2 ± 0.7) mice or eosinophils (1.2 ± 0.2 [XID] and 1.7 ± 0.5 [CBA/CaJ]).
Next, we examined cellular infiltrates 6 weeks after infection. Compared to CBA/CaJ controls, the number of cells expressing cell surface markers of B-1a B cells was minimal in the lungs of XID mice (Fig. 2A), and there were fewer B-1b B cells (Fig. 2A). XID mice also exhibited reduced numbers of lung B-2 B cells (Fig. 2B) with more neutrophils (Fig. 2B) and eosinophils (Fig. 2B). Other leukocyte populations in XID and CBA/CaJ mice did not differ (Fig. 2B).
FIG 2 .
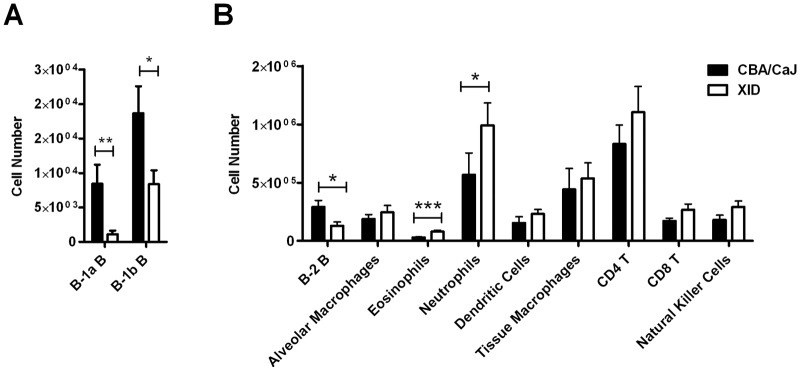
The reduction in B cells in XID lungs is accompanied by increased neutrophil and eosinophil recruitment. Lung B-1 B cells (A) and other lung cellular populations (B) were assessed by FACS at week 6 after infection with C. neoformans strain 52D. Values are means plus SEMs (error bars) of three independent experiments (n = 12 or 13). Values that are significantly different are indicated by bars and asterisks as follows: *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Neutrophil recruitment is induced by interleukin 17 (IL-17) (34), and IL-17 contributes to early control of C. neoformans infection (35, 36). However, IL-17 and other cytokine levels in the lungs of XID mice were comparable to CBA/CaJ controls 1 and 6 weeks after infection (Table 1), and brain cytokine levels were also similar (n = 4 or 5; data not shown).
TABLE 1 .
Lung cytokine production in mice infected with C. neoformans strain 52D at the indicated time postinfection
| Day PIa | Mouse strain | Cytokine level (pg/ml) (mean ± SEM) |
||||||
|---|---|---|---|---|---|---|---|---|
| IL-4 | IL-6 | IL-10 | IL-12 | IL-17 | IFN-γ | TGF-βb | ||
| 7 | CBA/CaJ | 781 ± 53 | 183 ± 43 | 257 ± 49 | 234 ± 25 | 489 ± 36 | 116 ± 14 | 1,950 ± 414 |
| XID | 748 ± 57 | 157 ± 20 | 288 ± 29 | 237 ± 21 | 509 ± 67 | 110 ± 14 | 1,732 ± 288 | |
| 42 | CBA/CaJ | 297 ± 61 | 1,429 ± 208 | 86 ± 39 | 76 ± 19 | 255 ± 26 | 1,388 ± 672 | 3,297 ± 388 |
| XID | 321 ± 105 | 763 ± 352 | 72 ± 29 | 96 ± 32 | 250 ± 45 | 804 ± 141 | 3,286 ± 290 | |
PI, postinfection.
TGF-β, transforming growth factor β.
Pathogenesis in mice infected with CNH99.
To address the role of C. neoformans strain differences in the XID phenotype, mice were infected with a more virulent strain belonging to serotype A, C. neoformans strain H99 (CNH99). Consistent with CN52D data, CNH99-infected XID mice had a markedly higher (3-log-unit) brain fungal burden than CBA/J controls 3 weeks after infection. XID mice also had higher lung CFU (Fig. 3A and B). Examination of lung cellular infiltrates at this time revealed that, as for CN52D-infected mice, there was no difference in the total number of recruited leukocytes. However, consistent with the known TH2 bias of CNH99-infected mice (37), XID mice had more lung eosinophils and less macrophages than CBA/J controls (Fig. 3C and D), with higher levels of IL-4 and IL-13 and lower levels of gamma interferon (IFN-γ) and IL-17 (Fig. 3E). Thus, XID mice are more susceptible to serotype A (CNH99) and serotype D (CN52D) strains of C. neoformans and exhibit altered lung cellular responses and more C. neoformans dissemination to the brain from the lungs.
FIG 3 .

XID mice exhibit enhanced susceptibility to C. neoformans strain H99. XID and CBA/J mice were infected with the highly virulent C. neoformans strain H99. (A and B) Three weeks after infection, mouse lungs (A) and brains (B) were analyzed for CFU. (C and D) Total numbers of eosinophils (C) and macrophages (D) were analyzed from H&E cytospins of single-cell suspensions. (E) Lung immune polarization was probed by measuring cytokine secretion from lung leukocyte cultures. Values are means ± SEMs (error bars) of two independent experiments (n = 4 to 7). Values that are significantly different are indicated by bars and asterisks as follows: *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Antibody response to CN52D infection.
Having determined that the XID defect increases susceptibility to CN52D and CNH99, we evaluated the role that B-1 B cells play in chronic infection with CN52D, since this strain causes chronic infection. As the absence of B-1a B cells in XID mice results in reduced levels of IgM in naive mice and impaired TI-2 responses (38–40), but B-1b, marginal zone, and B-2 B cells can also produce IgM, we assessed serum IgM and IgG in CN52D-infected mice. One and six weeks after infection, total serum IgM was lower in XID mice than in CBA/CaJ control mice (Fig. 4A) as were levels of specific IgM binding the C. neoformans capsular polysaccharide, glucuronoxylomannan (GXM) (Fig. 4B). Nonetheless, the levels of total and GXM-specific IgM rose in XID mice between weeks 1 and 6 after infection (Fig. 4A and B).
FIG 4 .

Total and antigen-specific immunoglobulin is reduced in C. neoformans strain 52D-infected XID mice compared to CBA/CaJ controls. (A and B) Total IgM and IgG in serum (A) and GXM-specific IgM and IgG reciprocal titer in serum (B). Each symbol represents the value for an individual mouse, with the mean Ig level shown by the horizontal black bars. There were 5 or 10 mice in the groups, and one (day 7 [D7]) or two (day 42 [D42]) independent experiments were performed. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
One and six weeks after infection, total serum IgG was lower in XID mice than in CBA/CaJ control mice (Fig. 4A). However, at week 6, whereas the difference in IgG between XID and CBA/CaJ mice was only 5-fold, IgM differed by 20-fold (Fig. 4A and B). GXM-specific IgG production during infection was minimal. No GXM-specific IgG was detected in XID mice (1:5 titer detection limit), but it was detected in some CBA/CaJ controls (Fig. 4B).
Reconstitution of XID mice with peritoneal B-1 B cells or serum.
To examine the role of B-1 B cells in control of brain fungal burden, we adoptively transferred peritoneal cavity (PerC) B-1 B cells from CBA/CaJ mice intraperitoneally (i.p.) to XID mice prior to infection with CN52D. Six weeks after infection, brain fungal burden in XID mice that received PerC B-1 B cells was similar to XID controls (Fig. 5A). Although peritoneal B-1 B cell numbers and serum IgM were higher in XID mice that received PerC B-1 B cells than in XID controls (Fig. 5B, C), serum IgM levels remained significantly lower than CBA/CaJ mice (Fig. 5C).
FIG 5 .
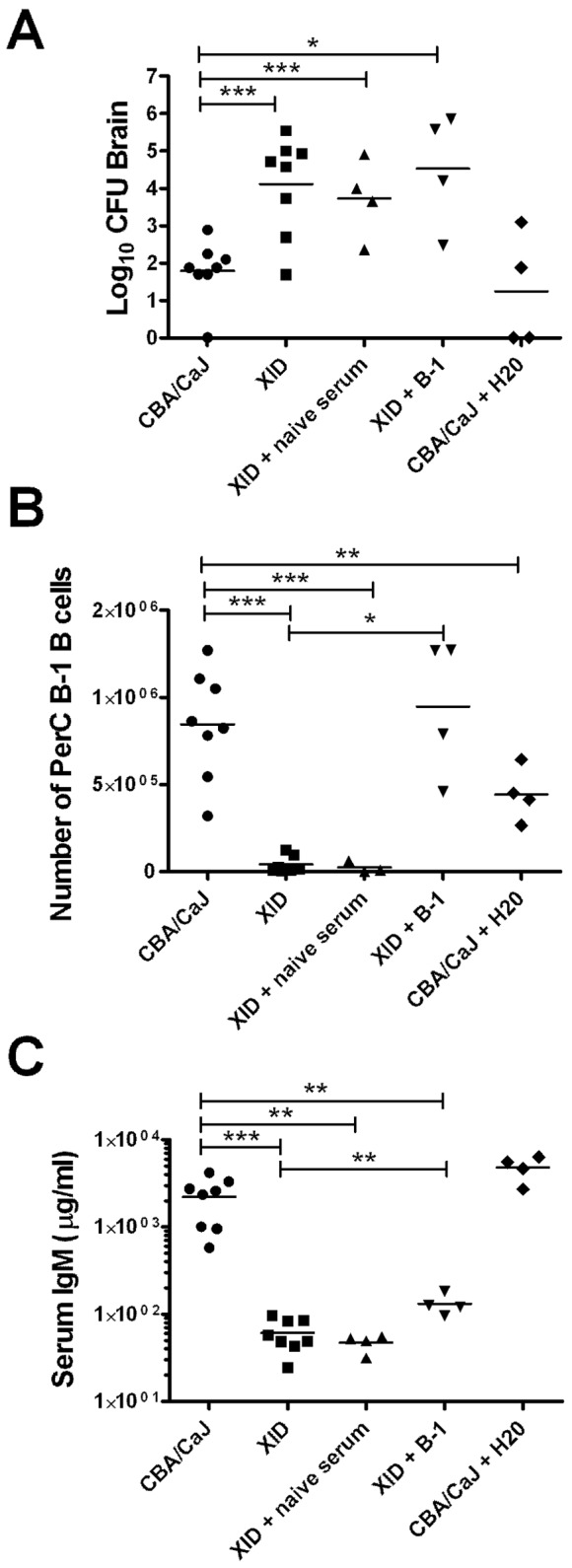
Effects of administration or depletion of peritoneal B-1 B cells and passive transfer of naive sera on fungal burden and B cell and IgM levels 6 weeks after infection. (A) Fungal burden in the brains of C. neoformans strain 52D-infected XID mice that received naive serum or peritoneal B-1 B cells and CBA/CaJ mice that were depleted of peritoneal B-1 cells. (B and C) Peritoneal B-1 B cell numbers (B) and serum IgM levels (C) were also determined. The horizontal black bars show the means for groups (4 or 8 mice per group) from one or two experiments, with each symbol indicating the value for an individual mouse. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
It was previously shown that transplant of B-1 B cells to peritoneal cavities of XID mice did not result in IgM production in XID mice greater than 8 weeks of age (41), but intravenous transfer did restore IgM serum levels in 8- to 12-week-old mice (30, 41). Although XID mice in our studies were between 6 and 8 weeks old at the time of B-1 B cell transfer and the numbers of PerC B-1 B cells were restored (Fig. 5B), we reconstituted XID mice by intravenous transfer of CBA/CaJ PerC cells to confirm that the route of transfer did not impact B-1 B cell IgM production. Six weeks after infection, XID mice that received PerC B-1 cells by intravenous transfer did have higher serum IgM (µg/ml; mean ± SEM) than XID controls (457 ± 115 [XID recipient] and 66 ± 12 [XID]) (P < 0.05; n = 4 or 5), but their levels remained lower than those of CBA/CaJ controls (811 ± 93). The amount of GXM-specific IgM (1/titer) also differed (404 ± 144 [CBA/CaJ], 87 ± 10 [XID plus PerC], and 30 ± 14 [XID]). Fungal burdens (mean log10 CFU ± SEM) in the brains of recipient XID mice (4.5 ± 0.5) were comparable to XID controls (3.7 ± 0.3).
We also determined whether passive transfer of naive serum from CBA/CaJ mice to XID mice would limit fungal dissemination to the brain. The fungal burdens of serum-reconstituted XID mice were similar to those of XID controls (Fig. 5A); however, 6 weeks after infection, serum IgM levels in reconstituted XID mice did not differ from those of XID controls (Fig. 5C).
Depletion of peritoneal B-1 B cells in CBA/CaJ controls.
To examine the role of peritoneal B-1 B cells in controlling C. neoformans dissemination in CBA/CaJ mice, we depleted B-1 B cells by injecting water into the PerC of CBA/CaJ mice prior to and during infection as described previously (26). Six weeks after infection, brain fungal burdens in PerC B-1 B cell-depleted CBA/CaJ mice were similar to those of CBA/CaJ controls (Fig. 5A). B-1 B cell-depleted CBA/CaJ mice had lower numbers of peritoneal B-1 B cells than CBA/CaJ controls (Fig. 5B). Serum IgM was similar in B-1 B depleted CBA/CaJ mice and CBA/CaJ controls (Fig. 5C).
Phagocytosis of CN52D by alveolar macrophages.
Phagocytosis of C. neoformans by alveolar macrophages was previously found to be lower in sIgM−/− mice than in the controls (6). As XID mice are also IgM deficient, we determined whether they were able to phagocytose C. neoformans. One week after infection, the numbers of alveolar macrophages recovered from the airways of XID and CBA/CaJ mice were similar (Fig. 6A), but XID mice had a significantly lower percentage of phagocytosed C. neoformans (Fig. 6B) and fewer C. neoformans per macrophage (phagocytic index) (Fig. 6C) than CBA/CaJ controls.
FIG 6 .
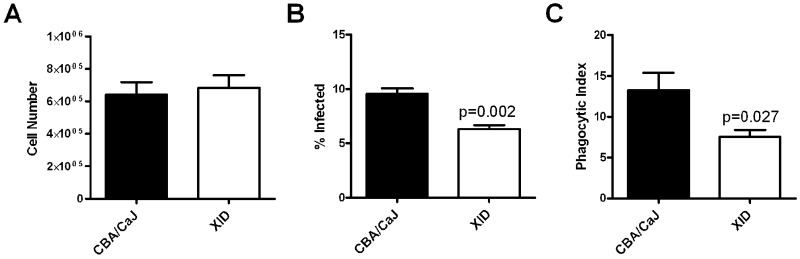
Phagocytosis of C. neoformans by alveolar macrophages is reduced in the lungs of XID mice compared to CBA/CaJ control mice. (A) Lung lavage cell number 7 days after infection with C. neoformans strain 52D. (B) Percent of alveolar macrophages infected with C. neoformans at day 7. (C) Phagocytic index (number of yeasts/total number of uninfected and infected alveolar macrophages × 100) at day 7). Values are means plus SEMs. There were four mice in each group.
Lung histology.
We examined hematoxylin and eosin (H&E)-stained lung sections of CN52D-infected mice 1 and 6 weeks after infection. One week after infection, inflammation severity and pattern were similar in XID and CBA/CaJ control mice (n = 4) (data not shown). Six weeks after infection, lungs of control mice had focal areas of perivascular inflammation (Fig. 7A), whereas XID mice had a diffuse disorganized inflammatory pattern (Fig. 7B). Gomori methenamine silver (GMS) staining revealed that while control mice had numerous small, intracellular yeast cells (Fig. 7C), XID mice had more large, extracellular yeast cells (Fig. 7D).
FIG 7 .

Lungs of XID mice exhibit disorganized inflammation and increased numbers of large, extracellular C. neoformans yeast in lung sections. (A to D) Representative images of H&E-stained lung section (magnification, ×10) from CBA/CaJ (A) and XID mice (B) 6 weeks after infection with C. neoformans strain 52D and Gomori methenamine silver-stained section (magnification, ×40) from CBA/CaJ controls (C) and XID lungs (D) at week 6 (n = 3). The arrows in panel D point to enlarged, extracellular yeast.
Analysis of CN52D size.
To quantify the observed difference in the size of C. neoformans in the lungs of XID and CBA/CaJ mice, we measured the diameter of all silver-stained C. neoformans in lung sections. Greater than 90% of the yeasts were less than 5 µm in both XID and CBA/CaJ mice. However, the percentage (mean ± SEM) of C. neoformans that was 5 to 10 µm in diameter was significantly higher (P < 0.05; n = 3) in XID mice than in control mice (9.1 ± 0.9 [XID] and 3.6 ± 0.4 [CBA/CaJ]), as was the percentage that was 10 to 15 µm in XID lungs (0.5 ± 0.03 [XID] and 0.1 ± 0.05 [CBA/CaJ]).
As GMS does not stain the C. neoformans capsule, we performed India ink staining of C. neoformans isolated directly from the lungs of CBA/CaJ (Fig. 8A) and XID (Fig. 8B) mice 6 weeks after infection and determined total yeast (cell and capsule) diameters (Fig. 8C). There were more C. neoformans with diameters greater than 30 µm in the lungs of XID mice than in CBA/CaJ mice (12.4% versus 2.5% [P < 0.05]) (Fig. 8D). On day 3 after infection, the percentages of yeast (mean ± SEM) greater than 30 µm were similar in XID and CBA/CaJ mice (7.1 ± 4.08 [XID] and 9.8 ± 1.2 [CBA/CaJ]; n = 3).
FIG 8 .

Diameters of C. neoformans yeast recovered from lungs 6 weeks after infection with C. neoformans strain 52D. (A and B) Representative images (magnification, ×10) of India ink preparations of yeast recovered from the lungs of CBA/CaJ (A) and XID (B) mice after lysis of host cells. Bars, 50 µm. (C) Histogram of C. neoformans yeast diameter, including capsule, from CBA (gray) and XID (white) mice. The number of yeast cells with a diameter within a 5-µm range were used to prepare the plot. The number of C. neoformans (CN) is shown on the y axis. (D) Graph of the percentage of C. neoformans yeast greater than the indicated diameter. Values are means plus SEMs from two independent experiments.
DISCUSSION
Our data show that XID mice were unable to control yeast dissemination to the brain during chronic infection with CN52D or acute infection with CNH99. Further analysis in the chronic pulmonary infection model with CN52D revealed that compared to controls, XID mice had less total and specific serum IgM, reduced macrophage phagocytosis of C. neoformans, and a disordered lung inflammatory pattern with more extracellular yeasts that were larger than those in the lungs of control mice. These findings link reduced IgM to an increase in C. neoformans size, impaired phagocytosis, and a failure to contain C. neoformans in the lungs.
The phenotype of C. neoformans-infected XID mice in this study resembled that of previously reported C. neoformans-infected sIgM−/− (IgM-deficient) mice (6). Reduced levels of IgM and C. neoformans phagocytosis in XID mice provide additional evidence that IgM plays a major role in promoting the containment of C. neoformans in the lungs and limiting dissemination to the brain. As XID mice had higher brain fungal burdens after infection with both CN52D and CNH99, the XID defect impaired control of fungal dissemination despite C. neoformans strain differences in serotype and immunopathogenesis. Consistent with studies in which XID mice exhibited exacerbated allergic responses (42), CNH99-infected XID mice had a TH2-biased response. C. neoformans-induced TH2 responses enhance disease severity in mice (35, 37, 43–46), perhaps explaining higher fungal burdens in CNH99-infected XID mice.
In addition to causing B cell defects, the absence of Btk signaling can affect myeloid cell functions in XID mice. Btk deficiency has been linked to impaired production of reactive oxygen species (ROS) and nitric oxide (NO) in macrophages and neutrophils (47, 48). However, we found a trend toward increased transcription of inducible nitric oxide synthase in the lungs of XID mice compared to those of controls 6 weeks after infection (data not shown). A recent study demonstrated similarly contradictory data showing that neutrophils from XID mice produced greater amounts of ROS in response to Toll-like receptor (TLR) activation (49). The same study also showed a higher sensitivity of neutrophils to activation-induced cell death, but we found more neutrophil infiltration into the lungs of C. neoformans-infected XID than in control mice. Thus, at present, the role of Btk in myeloid cell activation seems to be controversial, and our data do not support a key role for Btk-associated myeloid cells in the phenotype of C. neoformans-infected XID mice.
Passive transfer of naive control serum did not promote control of C. neoformans infection in XID mice, but C. neoformans-specific IgM and/or a larger amount of natural IgM might be needed for protection. For example, passive transfer of naive IgM restored resistance to bacterial sepsis and reduced fungal burden in mice infected with Pneumocystis murina (16, 50), but specific IgM was needed to protect against West Nile virus (51). Both natural and antigen-induced IgM were required for protection from influenza in mice (52). However, our data do not exclude a protective role for natural IgM because naive serum did not reconstitute serum IgM in XID mice. While the lower level of IgG in XID mice could also have contributed to the inability of these mice to control pulmonary C. neoformans, the disparity between XID and control IgM was much greater than that for IgG, and antigen-specific IgG production was very minimal in control mice.
The B cell subset that produces IgM which protects against C. neoformans has not been established. Given that XID mice did produce some GXM-specific IgM despite their lack of B-1a B cells, albeit in a much smaller amount than controls, non-B-1a B cells must also produce antigen-specific IgM. We found that transferred PerC B-1 B cells did not reconstitute XID IgM to the level of CBA/CaJ controls and PerC B-1 B cell depletion had no effect on IgM in CBA/CaJ controls. Hence, PerC B-1 B cells are not likely to be the major source of protective IgM in our model. Given that there were fewer B-2 B cells in the lungs of XID mice than in the lungs of control mice 1 and 6 weeks after CN52D infection, B-2 cell-derived IgM could mediate protection against C. neoformans. In support of this, both B-1 and B-2 cell-derived IgM were necessary for protection against influenza in mice (53). C. neoformans-specific B-2 B cell levels increased as early as 3 days after infection in C57BL/6 mice (26), and splenic follicular B cells that harbor B-2 B cells are reduced in XID mice (54). Bone marrow or splenic B-1 B cells and/or plasmablasts that reside in the bone marrow could also contribute to IgM production during infection (13, 55, 56). Irrespective of the exact B cell source, our data suggest that IgM deficiency impairs containment of pulmonary C. neoformans. The inability of adoptively transferred sera and peritoneal B-1 B cells to promote fungal containment could have been due to an inability of serum IgM to enter the lungs or insufficient B cell homing to the lungs. Indeed, in a previous study, IgM was administered intranasally (6), and in our study, the lung B-1 B cell levels in reconstituted XID recipients were not increased 6 weeks after infection (data not shown). Antibody-independent functions of B-1 B cells, such as their capacity to differentiate into macrophage-like phagocytes (14, 27), could also contribute to fungal containment in the lungs of control mice.
A recent study demonstrated that depletion of PerC B-1 B cells in C57BL/6 mice led to an increase in lung and brain fungal burdens 3 days after C. neoformans infection (26). In our study, depletion of B-1 B cells did not affect fungal burdens in the lungs or brains in CBA/CaJ mice (data not shown). Thus, in CBA/CaJ mice, B-1 B cells do not contribute significantly to the early response to C. neoformans infection. Given that mice on the CBA background are more resistant to CN52D (57), the discrepancy between this and the aforementioned study could reflect differences in disease pathogenesis in CBA and C57BL/6 mice.
CN52D infection resulted in quantitative differences in lung cellular subsets and qualitative differences in pathology. The lungs of XID mice contained more neutrophils and eosinophils during the chronic, but not the early, phase of infection. Given that B cells can regulate neutrophil influx into lungs during infection (58, 59), the higher number of neutrophils in XID lungs could stem from the reduced number of B-2 B cells and/or local concentration of IgM. The lungs of XID mice also displayed loose, disorganized, diffuse pulmonary infiltrates, a pattern that has previously been linked to C. neoformans progression (37, 60). Similarly, this pattern of lung inflammation was also observed in XID mice infected with Mycobacterium bovis BCG (33).
Compared to control mice, C. neoformans dissemination to the brain in XID mice was associated with the presence of a significantly greater number of large, extracellular C. neoformans in their lungs 6 weeks after infection. When grown in vitro, C. neoformans yeast cells average 4 to 10 µm in size but transition to an enlarged state in vivo (61–63). However, to our knowledge, this is the first report of a host immune defect leading to alteration in the size of C. neoformans. We found yeast as large as 60 µm in the lungs of XID mice 6 weeks after infection. This population consisted of yeast with larger cell bodies and larger capsules. Capsular enlargement in C. neoformans is thought to confer an advantage to the pathogen, since the capsule promotes resistance to phagocytosis and oxidative stress (64, 65). Large C. neoformans cells with larger cell body volume, capsule size, and resistance to oxidative and nitrosative stress that exhibit multiple ploidy have been termed “giant” or “titan cells” (66, 67). Serotype A and serotype D strains can each form titan cells in mice (66). These cells inhibit C. neoformans phagocytosis of enlarged titan as well as normal-sized progeny (68) and promote fungal dissemination (69). Although studies of lung IgM were not undertaken, in aggregate, our data suggest that local IgM production plays a role in controlling C. neoformans phagocytosis and size in the lungs. The most direct proof of this hypothesis would be with IgM chimeras, as for influenza (52), but these studies are beyond the scope of the current study.
Factors that promote cryptococcal enlargement in lungs are not known, and the phenotype can be partially induced only in vitro (66, 67). Specific monoclonal anti-GXM IgM isotype binds to enlarged cryptococcal yeast in tissue sections (61). One mechanism by which cryptococcal size could be restrained is by IgM-mediated phagocytosis of C. neoformans, which may limit the number of extracellular yeast capable of expanding. Additionally, enlarged C. neoformans may act as a sieve for IgM, effectively reducing the local concentration of IgM in the lung microenvironment. This would limit binding of IgM to the smaller progeny, which could then escape from the lung. Btk deficiency could have had a direct effect on phagocytic uptake of C. neoformans by myeloid cells, as Btk promotes uptake of apoptotic cells by macrophages (70) and is associated with impaired phagocytosis in individuals with X-linked agammaglobulinemia with no or reduced Btk (71). However, phagocytosis of bacteria by XID macrophages was not found to be impaired (47). Another hypothesis for the role that IgM plays in C. neoformans enlargement is that its binding induces a transcriptional program that inhibits C. neoformans enlargement. Binding of monoclonal IgM to C. neoformans was shown to directly induce transcriptional changes in the organism (1). Future studies will examine the mechanism(s) by which IgM promotes fungal containment.
In summary, our data suggest that IgM is crucial to prevent C. neoformans dissemination from the lungs to the brain in a chronic pulmonary infection model. Our data also suggest that protective IgM could be derived from B cell subsets other than PerC B-1 B cells, while providing a novel clue that a mechanism by which IgM mediates protection is to restrict C. neoformans size in the lungs, thereby promoting fungal containment. Although the association between IgM, fungal containment, and fungal size reported herein does not establish direct causality, our data strongly reinforce previous work linking IgM to resistance to C. neoformans in mice and humans (6, 11). Furthermore, as Btk is critical for survival of human B cells and has been identified as a target for therapy of B cell lymphomas (72–74), our data raise the possibility that such therapy could pose a risk for the development of cryptococcosis.
MATERIALS AND METHODS
Mice.
For infections with CN52D, male 6- to 8-week-old CBA/CaJ controls or CBA/CaHN-Btk-XID (XID) mice were purchased from Jackson Laboratories (Bar Harbor, ME). Mice were housed under specific-pathogen-free conditions in the Institute for Animal Studies at the Albert Einstein College of Medicine (AECOM). All mouse experiments were conducted with prior approval from the Animal Care and Use Committee of AECOM following established guidelines. For infections with C. neoformans H99, XID and CBA/J controls were bred at the University of Michigan Medical School. Both CBA/Ca and CBA mice are used as approximate controls for XID mice, which arose from a spontaneous mutation in Btk in the CBA/Ca strain (19, 25, 32, 42, 75).
Cryptococcal infection model.
Clinical isolate C. neoformans strains, 52D (ATCC 24067), a serotype D strain, and H99 (ATCC 208821), a serotype A strain, were used to infect XID and control mice. C. neoformans strains were stored at −80°C in 15% glycerol until needed. Thawed aliquots were grown in Difco Sabouraud dextrose broth (Becton Dickinson, Franklin Lakes, NJ) for 48 h at 37°C prior to infection. For the intranasal CN52D infection, mice were anesthetized with isoflurane (Halocarbon, River Edge, NJ), and a volume of 20 µl containing 5 × 105 CFU of C. neoformans was administered via the nares. Intravenous injections of 1 × 105 CN52D in 150 µl of phosphate-buffered saline (PBS) were performed into the retro-orbital sinus where indicated. CNH99 was delivered intratracheally with 1 × 104 yeast cells as described previously (45). For all infections, inocula were verified by plating.
Measurement of tissue fungal burden.
The lungs and brains were removed from the mice and homogenized in 1 ml PBS. The numbers of CFU were determined by plating onto Sabouraud dextrose agar plates (BBL, Sparks, MD) in duplicate.
Determination of serum antibody concentrations.
Concentrations of serum antibodies were determined by an enzyme-linked immunosorbent assay (ELISA). EIA/RIA 96-well plates (Costar, Corning, NY) were coated with 10 µg/ml of goat anti-mouse IgM or IgG (Southern Biotech, Birmingham, AL). Mouse serum and IgM or IgG standards (Southern Biotech) were added and serially diluted 1:3 with PBS containing 1% bovine serum albumin (1% BSA−PBS). Alkaline phosphatase-labeled anti-IgM or anti-IgG (Southern Biotech) was added at a concentration of 1:2,500. Plates were developed with 1 mg/ml p-nitrophenyl phosphate (Sigma-Aldrich) dissolved in bicarbonate buffer (pH 9).
Determination of GXM-specific IgM.
Plates were coated with 10 µg/ml CN52D GXM. After the addition of serum samples, GXM-specific IgM was detected by the addition of goat anti-mouse IgM as described previously (6). The titer for GXM-specific IgM was defined as the point at which the titration curve crossed an optical density (OD) of 0.1 after subtraction of the background.
Analysis of yeast phagocytosis by alveolar macrophages.
Mouse alveolar macrophages were recovered by bronchoalveolar lavage. Lavage fluid samples were washed, resuspended in 500 µl of RPMI 1640 (Mediatech) with 10% fetal bovine serum (FBS) (HyClone, Logan, UT), plated into each well of 4-well glass chamber slides (Nunc, Rochester, NY), and incubated at 37°C and 5% CO2 for 2 h. The nonadherent cells from bronchoalveolar lavage fluid samples were washed away with PBS. The remaining adherent cells were fixed with methanol and then stained with Giemsa (Sigma-Aldrich). A minimum of 100 adherent alveolar macrophages per sample were counted, and the number of yeast within each macrophage recorded. The phagocytic index was calculated as the total number of yeast divided by the total number of adherent macrophages counted multiplied by 100.
Analysis of lung leukocyte populations.
Lungs were treated with collagenase (Roche, Indianapolis, IN) and then dissociated using the gentleMACS dissociator (Miltenyi Biotec, Auburn, CA). Recovered lung cells were incubated with CD16/32 and stained with combinations of the following antibodies [the antibody is shown first and then the conjugate(s)]: CD45-Pacific Blue or CD45-Alexa Fluor 700, Ly6G-APC-Cy7 (APC stands for allophycocyanin), CD11b-PerCP-Cy5.5 (PerCP stands for peridinin chlorophyll protein) or CD11b-APC-Cy7, CD11c-PE-Cy7 (PE stands for phycoerythrin), Ly6C-FITC (FITC stands for fluorescein isothiocyanate), F4/80-Alexa Fluor 647, CD19-PE-Cy7, B220-PerCP-Cy5.5, IgD-Alexa Fluor 647, IgM-FITC, CD5-PE, CD49b-APC, CD4-APC-Cy7, CD8-Pacific Blue, and CD3-Alexa Fluor 647. Antibodies were purchased from BD Biosciences (Franklin Lakes, NJ), eBioscience (San Diego, CA), and BioLegend (San Diego, CA). Data were collected on an LSRII (BD Biosciences) and analyzed with FlowJo software (Tree Star, Ashland, OR). The absolute number of each lung leukocyte population was calculated by multiplying the hemocytometer lung cell count by the relative percentage, subsequent to gating on CD45+ leukocytes. For infections with CNH99, eosinophils and macrophages were enumerated from lung cytospin preparations.
Determination of cytokine levels.
Lung homogenates were centrifuged at 3,000 × g for 30 min at 4°C, followed by centrifugation of the supernatant at 13,000 × g at 4°C for an additional 10 min to remove any remaining debris. Samples were immediately stored at −80°C prior to use. Cytokine concentrations were determined using ELISA Duosets (R&D Systems, Minneapolis, MN).
Histology.
Lungs and brains were fixed in 10% neutral buffered formalin. Following 48 to 72 h of fixation, samples were sent to the Histopathology Core of AECOM for routine processing into paraffin blocks. Five-micron lung and brain tissue sections were routinely stained with H&E or GMS and examined with a Zeiss AxioScope II microscope (Carl Zeiss, Thornwood, NY).
Measurement of C. neoformans size.
Gomori methenamine silver-stained lung sections were used to compare the size of C. neoformans yeast in lung sections. Images of 10 sections per lung at a magnification of ×40 were acquired using a Zeiss AxioScope II microscope and camera. ImageJ software (http://rsbweb.nih.gov/ij/) was then used to quantitate the number and measure the diameter of each silver-stained yeast cell in the tissue sections. For measurement of yeast diameter by India ink staining, single-cell lung suspensions were prepared from collagenase-digested lung tissues. Host cells were then lysed by incubation with water for 30 min at 4°C. India ink preparations of the remaining C. neoformans cells were made and examined with a Zeiss AxioScope II microscope at a magnification of ×10 and analyzed with ImageJ software.
Serum reconstitution of XID mice.
Blood samples were collected from 20 naive, anesthetized CBA/CaJ control mice by retro-orbital puncture. Serum samples were obtained after allowing the blood samples to clot for 1 h at 37°C. Pooled serum samples were heat inactivated at 56°C for 30 min and then stored at −80°C until use. One day prior to infection and at weekly intervals thereafter, 200 µl of pooled serum was administered to XID mice intraperitoneally (i.p.), as described previously (51).
B-1 B cell adoptive transfer.
Peritoneal cavity (PerC) cells from donor CBA/CaJ mice were collected by lavage. B-1 B cells were enriched for by removing adherent peritoneal macrophages after 2 h of culture in RPMI 1640 supplemented with 10% FBS. The nonadherent B-1 B cells were washed and resuspended in PBS. A total of 1 × 106 B-1 B cells in 500 µl PBS were injected intraperitoneally (i.p.) as performed previously (33). Alternatively, 3 × 106 PerC cells from naive CBA/CaJ mice were transferred intravenously to XID mice as performed previously (30). Mice were infected 1 week after PerC cells were transferred.
B-1 B cell depletion.
Peritoneal B-1 B cells were depleted as described previously (26). Briefly, 1 ml of sterile H2O was administered i.p. to CBA/CaJ mice for 3 weeks prior to infection at 4- or 5-day intervals to induce osmotic lysis of PerC cells. Mice were infected intranasally (i.n.) with C. neoformans 1 day after the last injection. During infection, PerC cell depletion was maintained by injection of H2O at 5- or 6-day intervals.
Statistical analysis.
Mouse survival data were evaluated by comparing Kaplan-Meier survival curves with a log rank (Mantel-Cox) test. Student’s t test or Mann-Whitney U test was used to compare differences in XID mice and control mice, after determination of normality. For comparisons between multiple groups of mice, analysis of variance (ANOVA) or Kruskal-Wallis test was performed followed by Holm-Sidak or Dunn’s posttest, respectively. P values of <0.05 were considered significant. All statistical tests were performed using the advisory statistical program SigmaStat, version 3 (Systat, San Jose, CA).
ACKNOWLEDGMENTS
We thank Rani Sellers and the Albert Einstein Histopathology Core Facility for performing the histological analysis.
This work was supported by the Einstein/Montefiore Center for AIDS Research (P30AI051519) and the AECOM Flow Cytometry Core Facility under the support of the AECOM National Cancer Institute (P30CA013330). Contributing investigators were supported by National Institutes of Health grants R01 AI 0970906 (L.P.); VA Merit Review Grant (M.O.); Arthritis Foundation, the Dryer Foundation, and NIH-NIAMS grant K01-AR053846 (S.K.L.); institutional AIDS training grant 5T32A1007501 and by the Geographic Medicine and Emerging Infections training grant 5T32AI701175 (W.A.S.); and NIH T32-HL07749-19 Training Grant in Pulmonary and Critical Care Medicine (M.J.D.).
Footnotes
Citation Szymczak WA, Davis MJ, Lundy SK, Dufaud C, Olszewski M, Pirofski L. 2013. X-linked immunodeficient mice exhibit enhanced susceptibility to Cryptococcus neoformans infection. mBio 4(4):e00265-13. doi:10.1128/mBio.00265-13.
REFERENCES
- 1. McClelland EE, Nicola AM, Prados-Rosales R, Casadevall A. 2010. Ab binding alters gene expression in Cryptococcus neoformans and directly modulates fungal metabolism. J. Clin. Invest. 120:1355–1361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Martinez LR, Moussai D, Casadevall A. 2004. Antibody to Cryptococcus neoformans glucuronoxylomannan inhibits the release of capsular antigen. Infect. Immun. 72:3674–3679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Rivera J, Mukherjee J, Weiss LM, Casadevall A. 2002. Antibody efficacy in murine pulmonary Cryptococcus neoformans infection: a role for nitric oxide. J. Immunol. 168:3419–3427 [DOI] [PubMed] [Google Scholar]
- 4. Miller GP, Kohl S. 1983. Antibody-dependent leukocyte killing of Cryptococcus neoformans. J. Immunol. 131:1455–1459 [PubMed] [Google Scholar]
- 5. Feldmesser M, Mednick A, Casadevall A. 2002. Antibody-mediated protection in murine Cryptococcus neoformans infection is associated with pleotrophic effects on cytokine and leukocyte responses. Infect. Immun. 70:1571–1580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Subramaniam KS, Datta K, Quintero E, Manix C, Marks MS, Pirofski LA. 2010. The absence of serum IgM enhances the susceptibility of mice to pulmonary challenge with Cryptococcus neoformans. J. Immunol. 184:5755–5767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rachini A, Pietrella D, Lupo P, Torosantucci A, Chiani P, Bromuro C, Proietti C, Bistoni F, Cassone A, Vecchiarelli A. 2007. An anti-beta-glucan monoclonal antibody inhibits growth and capsule formation of Cryptococcus neoformans in vitro and exerts therapeutic, anticryptococcal activity in vivo. Infect. Immun. 75:5085–5094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sundstrom JB, Cherniak R. 1992. The glucuronoxylomannan of Cryptococcus neoformans serotype A is a type 2 T-cell-independent antigen. Infect. Immun. 60:4080–4087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Maitta RW, Datta K, Chang Q, Luo RX, Witover B, Subramaniam K, Pirofski LA. 2004. Protective and nonprotective human immunoglobulin M monoclonal antibodies to Cryptococcus neoformans glucuronoxylomannan manifest different specificities and gene use profiles. Infect. Immun. 72:4810–4818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Taborda CP, Casadevall A. 2001. Immunoglobulin M efficacy against Cryptococcus neoformans: mechanism, dose dependence, and prozone-like effects in passive protection experiments. J. Immunol. 166:2100–2107 [DOI] [PubMed] [Google Scholar]
- 11. Subramaniam K, Metzger B, Hanau LH, Guh A, Rucker L, Badri S, Pirofski LA. 2009. IgM(+) memory B cell expression predicts HIV-associated cryptococcosis status. J. Infect. Dis. 200:244–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ha SA, Tsuji M, Suzuki K, Meek B, Yasuda N, Kaisho T, Fagarasan S. 2006. Regulation of B1 cell migration by signals through Toll-like receptors. J. Exp. Med. 203:2541–2550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Choi YS, Dieter JA, Rothaeusler K, Luo Z, Baumgarth N. 2012. B-1 cells in the bone marrow are a significant source of natural IgM. Eur. J. Immunol. 42:120–129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ghosn EE, Russo M, Almeida SR. 2006. Nitric oxide-dependent killing of Cryptococcus neoformans by B-1-derived mononuclear phagocyte. J. Leukoc. Biol. 80:36–44 [DOI] [PubMed] [Google Scholar]
- 15. Kantor AB, Stall AM, Adams S, Herzenberg LA, Herzenberg LA. 1992. Differential development of progenitor activity for three B-cell lineages. Proc. Natl. Acad. Sci. U. S. A. 89:3320–3324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rapaka RR, Ricks DM, Alcorn JF, Chen K, Khader SA, Zheng M, Plevy S, Bengtén E, Kolls JK. 2010. Conserved natural IgM antibodies mediate innate and adaptive immunity against the opportunistic fungus Pneumocystis murina. J. Exp. Med. 207:2907–2919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Thurnheer MC, Zuercher AW, Cebra JJ, Bos NA. 2003. B1 cells contribute to serum IgM, but not to intestinal IgA, production in gnotobiotic Ig allotype chimeric mice. J. Immunol. 170:4564–4571 [DOI] [PubMed] [Google Scholar]
- 18. Haas KM, Poe JC, Steeber DA, Tedder TF. 2005. B-1a and B-1b cells exhibit distinct developmental requirements and have unique functional roles in innate and adaptive immunity to S. pneumoniae. Immunity 23:7–18 [DOI] [PubMed] [Google Scholar]
- 19. Cole LE, Yang Y, Elkins KL, Fernandez ET, Qureshi N, Shlomchik MJ, Herzenberg LA, Vogel SN. 2009. Antigen-specific B-1a antibodies induced by Francisella tularensis LPS provide long-term protection against F. tularensis LVS challenge. Proc. Natl. Acad. Sci. U. S. A. 106:4343–4348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yang Y, Ghosn EE, Cole LE, Obukhanych TV, Sadate-Ngatchou P, Vogel SN, Herzenberg LA, Herzenberg LA. 2012. Antigen-specific antibody responses in B-1a and their relationship to natural immunity. Proc. Natl. Acad. Sci. U. S. A. 109:5382–5387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Parra D, Rieger AM, Li J, Zhang YA, Randall LM, Hunter CA, Barreda DR, Sunyer JO. 2012. Pivotal advance: peritoneal cavity B-1 B cells have phagocytic and microbicidal capacities and present phagocytosed antigen to CD4+ T cells. J. Leukoc. Biol. 91:525–536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Baumgarth N. 2011. The double life of a B-1 cell: self-reactivity selects for protective effector functions. Nat. Rev. Immunol. 11:34–46 [DOI] [PubMed] [Google Scholar]
- 23. Ghosn EE, Yamamoto R, Hamanaka S, Yang Y, Herzenberg LA, Nakauchi H, Herzenberg LA. 2012. Distinct B-cell lineage commitment distinguishes adult bone marrow hematopoietic stem cells. Proc. Natl. Acad. Sci. U. S. A. 109:5394–5398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yang Y, Ghosn EE, Cole LE, Obukhanych TV, Sadate-Ngatchou P, Vogel SN, Herzenberg LA, Herzenberg LA. 2012. Antigen-specific memory in B-1a and its relationship to natural immunity. Proc. Natl. Acad. Sci. U. S. A. 109:5388–5393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Alugupalli KR, Gerstein RM, Chen J, Szomolanyi-Tsuda E, Woodland RT, Leong JM. 2003. The resolution of relapsing fever borreliosis requires IgM and is concurrent with expansion of B1b lymphocytes. J. Immunol. 170:3819–3827 [DOI] [PubMed] [Google Scholar]
- 26. Rohatgi S, Pirofski LA. 2012. Molecular characterization of the early B cell response to pulmonary Cryptococcus neoformans infection. J. Immunol. 189:5820–5830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Almeida SR, Aroeira LS, Frymuller E, Dias MA, Bogsan CS, Lopes JD, Mariano M. 2001. Mouse B-1 cell-derived mononuclear phagocyte, a novel cellular component of acute non-specific inflammatory exudate. Int. Immunol. 13:1193–1201 [DOI] [PubMed] [Google Scholar]
- 28. Rawlings DJ, Saffran DC, Tsukada S, Largaespada DA, Grimaldi JC, Cohen L, Mohr RN, Bazan JF, Howard M, Copeland NG, et al. 1993. Mutation of unique region of Bruton’s tyrosine kinase in immunodeficient XID mice. Science 261:358–361 [DOI] [PubMed] [Google Scholar]
- 29. Khan WN, Alt FW, Gerstein RM, Malynn BA, Larsson I, Rathbun G, Davidson L, Müller S, Kantor AB, Herzenberg LA, Rosen FS, Siderase P. 1995. Defective B cell development and function in Btk-deficient mice. Immunity 3:283–299 [DOI] [PubMed] [Google Scholar]
- 30. Prior L, Pierson S, Woodland RT, Riggs J. 1994. Rapid restoration of B-cell function in XID mice by intravenous transfer of peritoneal cavity B cells. Immunology 83:180–183 [PMC free article] [PubMed] [Google Scholar]
- 31. Amsbaugh DF, Hansen CT, Prescott B, Stashak PW, Barthold DR, Baker PJ. 1972. Genetic control of the antibody response to type 3 pneumococcal polysaccharide in mice. I. Evidence that an X-linked gene plays a decisive role in determining responsiveness. J. Exp. Med. 136:931–949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Marquis G, Montplaisir S, Pelletier M, Mousseau S, Auger P. 1985. Genetic resistance to murine cryptococcosis: increased susceptibility in the CBA/N XID mutant strain of mice. Infect. Immun. 47:282–287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Russo RT, Mariano M. 2010. B-1 cell protective role in murine primary Mycobacterium bovis bacillus Calmette-Guerin infection. Immunobiology 215:1005–1014 [DOI] [PubMed] [Google Scholar]
- 34. Roussel L, Houle F, Chan C, Yao Y, Berube J, Olivenstein R, Martin JG, Huot J, Hamid Q, Ferri L, Rousseau S. 2010. IL-17 promotes p38 MAPK-dependent endothelial activation enhancing neutrophil recruitment to sites of inflammation. J. Immunol. 184:4531–4537 [DOI] [PubMed] [Google Scholar]
- 35. Szymczak WA, Sellers RS, Pirofski LA. 2012. IL-23 dampens the allergic response to Cryptococcus neoformans through IL-17-independent and -dependent mechanisms. Am. J. Pathol. 180:1547–1559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wozniak KL, Hardison SE, Kolls JK, Wormley FL. 2011. Role of IL-17A on resolution of pulmonary C. neoformans infection. PLoS One 6:e17204. 10.1371/journal.pone.0017204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jain AV, Zhang Y, Fields WB, McNamara DA, Choe MY, Chen GH, Erb-Downward J, Osterholzer JJ, Toews GB, Huffnagle GB, Olszewski MA. 2009. Th2 but not Th1 immune bias results in altered lung functions in a murine model of pulmonary Cryptococcus neoformans infection. Infect. Immun. 77:5389–5399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Briles DE, Nahm M, Schroer K, Davie J, Baker P, Kearney J, Barletta R. 1981. Antiphosphocholine antibodies found in normal mouse serum are protective against intravenous infection with type 3 Streptococcus pneumoniae. J. Exp. Med. 153:694–705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mond JJ, Lieberman R, Inman JK, Mosier DE, Paul WE. 1977. Inability of mice with a defect in B-lymphocyte maturation to respond to phosphorycholine on immunogenic carriers. J. Exp. Med. 146:1138–1142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Briles DE, Nahm M, Marion TN, Perlmutter RM, Davie JM. 1982. Streptococcal group A carbohydrate has properties of both a thymus-independent (TI-2) and a thymus-dependent antigen. J. Immunol. 128:2032–2035 [PubMed] [Google Scholar]
- 41. Julius P, Jr, Kaga M, Palmer Y, Vyas V, Prior L, Delice D, Riggs J. 1997. Recipient age determines the success of intraperitoneal transplantation of peritoneal cavity B cells. Immunology 91:383–390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lundy SK, Berlin AA, Martens TF, Lukacs NW. 2005. Deficiency of regulatory B cells increases allergic airway inflammation. Inflamm. Res. 54:514–521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hernandez Y, Arora S, Erb-Downward JR, McDonald RA, Toews GB, Huffnagle GB. 2005. Distinct roles for IL-4 and IL-10 in regulating T2 immunity during allergic bronchopulmonary mycosis. J. Immunol. 174:1027–1036 [DOI] [PubMed] [Google Scholar]
- 44. Piehler D, Stenzel W, Grahnert A, Held J, Richter L, Köhler G, Richter T, Eschke M, Alber G, Müller U. 2011. Eosinophils contribute to IL-4 production and shape the T-helper cytokine profile and inflammatory response in pulmonary cryptococcosis. Am. J. Pathol. 179:733–744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Arora S, Hernandez Y, Erb-Downward JR, McDonald RA, Toews GB, Huffnagle GB. 2005. Role of IFN-gamma in regulating T2 immunity and the development of alternatively activated macrophages during allergic bronchopulmonary mycosis. J. Immunol. 174:6346–6356 [DOI] [PubMed] [Google Scholar]
- 46. Stenzel W, Muller U, Kohler G, Heppner FL, Blessing M, McKenzie AN, Brombacher F, Alber G. 2009. IL-4/IL-13-dependent alternative activation of macrophages but not microglial cells is associated with uncontrolled cerebral cryptococcosis. Am. J. Pathol. 174:486–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Mangla A, Khare A, Vineeth V, Panday NN, Mukhopadhyay A, Ravindran B, Bal V, George A, Rath S. 2004. Pleiotropic consequences of Bruton tyrosine kinase deficiency in myeloid lineages lead to poor inflammatory responses. Blood 104:1191–1197 [DOI] [PubMed] [Google Scholar]
- 48. Mukhopadhyay S, Mohanty M, Mangla A, George A, Bal V, Rath S, Ravindran B. 2002. Macrophage effector functions controlled by Bruton’s tyrosine kinase are more crucial than the cytokine balance of T cell responses for microfilarial clearance. J. Immunol. 168:2914–2921 [DOI] [PubMed] [Google Scholar]
- 49. Honda F, Kano H, Kanegane H, Nonoyama S, Kim ES, Lee SK, Takagi M, Mizutani S, Morio T. 2012. The kinase Btk negatively regulates the production of reactive oxygen species and stimulation-induced apoptosis in human neutrophils. Nat. Immunol. 13:369–378 [DOI] [PubMed] [Google Scholar]
- 50. Boes M, Prodeus AP, Schmidt T, Carroll MC, Chen J. 1998. A critical role of natural immunoglobulin M in immediate defense against systemic bacterial infection. J. Exp. Med. 188:2381–2386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Diamond MS, Sitati EM, Friend LD, Higgs S, Shrestha B, Engle M. 2003. A critical role for induced IgM in the protection against West Nile virus infection. J. Exp. Med. 198:1853–1862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Baumgarth N, Herman OC, Jager GC, Brown LE, Herzenberg LA, Chen J. 2000. B-1 and B-2 cell-derived immunoglobulin M antibodies are nonredundant components of the protective response to influenza virus infection. J. Exp. Med. 192:271–280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Donahue AC, Hess KL, Ng KL, Fruman DA. 2004. Altered splenic B cell subset development in mice lacking phosphoinositide 3-kinase p85alpha. Int. Immunol. 16:1789–1798 [DOI] [PubMed] [Google Scholar]
- 54. Choi YS, Baumgarth N. 2008. Dual role for B-1a cells in immunity to influenza virus infection. J. Exp. Med. 205:3053–3064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Foote JB, Mahmoud TI, Vale AM, Kearney JF. 2012. Long-term maintenance of polysaccharide-specific antibodies by IgM-secreting cells. J. Immunol. 188:57–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Racine R, McLaughlin M, Jones DD, Wittmer ST, MacNamara KC, Woodland DL, Winslow GM. 2011. IgM production by bone marrow plasmablasts contributes to long-term protection against intracellular bacterial infection. J. Immunol. 186:1011–1021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Carroll SF, Loredo Osti JC, Guillot L, Morgan K, Qureshi ST. 2008. Sex differences in the genetic architecture of susceptibility to Cryptococcus neoformans pulmonary infection. Genes Immun. 9:536–545 [DOI] [PubMed] [Google Scholar]
- 58. Maglione PJ, Xu J, Chan J. 2007. B cells moderate inflammatory progression and enhance bacterial containment upon pulmonary challenge with Mycobacterium tuberculosis. J. Immunol. 178:7222–7234 [DOI] [PubMed] [Google Scholar]
- 59. Kondratieva TK, Rubakova EI, Linge IA, Evstifeev VV, Majorov KB, Apt AS. 2010. B cells delay neutrophil migration toward the site of stimulus: tardiness critical for effective bacillus Calmette-Guérin vaccination against tuberculosis infection in mice. J. Immunol. 184:1227–1234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Wozniak KL, Ravi S, Macias S, Young ML, Olszewski MA, Steele C, Wormley FL. 2009. Insights into the mechanisms of protective immunity against Cryptococcus neoformans infection using a mouse model of pulmonary cryptococcosis. PLoS One 4:e6854. 10.1371/journal.pone.0006854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Feldmesser M, Kress Y, Casadevall A. 2001. Dynamic changes in the morphology of Cryptococcus neoformans during murine pulmonary infection. Microbiology 147:2355–2365 [DOI] [PubMed] [Google Scholar]
- 62. Cruickshank JG, Cavill R, Jelbert M. 1973. Cryptococcus neoformans of unusual morphology. Appl. Microbiol. 25:309–312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Love GL, Boyd GD, Greer DL. 1985. Large Cryptococcus neoformans isolated from brain abscess. J. Clin. Microbiol. 22:1068–1070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Zaragoza O, Taborda CP, Casadevall A. 2003. The efficacy of complement-mediated phagocytosis of Cryptococcus neoformans is dependent on the location of C3 in the polysaccharide capsule and involves both direct and indirect C3-mediated interactions. Eur. J. Immunol. 33:1957–1967 [DOI] [PubMed] [Google Scholar]
- 65. Zaragoza O, Chrisman CJ, Castelli MV, Frases S, Cuenca-Estrella M, Rodríguez-Tudela JL, Casadevall A. 2008. Capsule enlargement in Cryptococcus neoformans confers resistance to oxidative stress suggesting a mechanism for intracellular survival. Cell. Microbiol. 10:2043–2057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Zaragoza O, García-Rodas R, Nosanchuk JD, Cuenca-Estrella M, Rodríguez-Tudela JL, Casadevall A. 2010. Fungal cell gigantism during mammalian infection. PLoS Pathog. 6:e1000945. 10.1371/journal.ppat.1000945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Okagaki LH, Strain AK, Nielsen JN, Charlier C, Baltes NJ, Chrétien F, Heitman J, Dromer F, Nielsen K. 2010. Cryptococcal cell morphology affects host cell interactions and pathogenicity. PLoS Pathog. 6:e1000953. 10.1371/journal.ppat.1000953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Okagaki LH, Nielsen K. 2012. Titan cells confer protection from phagocytosis in Cryptococcus neoformans infections. Eukaryot. Cell 11:820–826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Crabtree JN, Okagaki LH, Wiesner DL, Strain AK, Nielsen JN, Nielsen K. 2012. Titan cell production enhances the virulence of Cryptococcus neoformans. Infect. Immun. 80:3776–3785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Byrne JC, Ní Gabhann J, Stacey KB, Coffey BM, McCarthy E, Thomas W, Jefferies CA. 2013. Bruton’s tyrosine kinase is required for apoptotic cell uptake via regulating the phosphorylation and localization of calreticulin. J. Immunol. 190:5207–5215 [DOI] [PubMed] [Google Scholar]
- 71. Amoras AL, Kanegane H, Miyawaki T, Vilela MM. 2003. Defective Fc-, CR1- and CR3-mediated monocyte phagocytosis and chemotaxis in common variable immunodeficiency and X-linked agammaglobulinemia patients. J. Investig. Allergol. Clin. Immunol. 13:181–188 [PubMed] [Google Scholar]
- 72. Advani RH, Buggy JJ, Sharman JP, Smith SM, Boyd TE, Grant B, Kolibaba KS, Furman RR, Rodriguez S, Chang BY, Sukbuntherng J, Izumi R, Hamdy A, Hedrick E, Fowler NH. 2013. Bruton tyrosine kinase inhibitor ibrutinib (PCI-32765) has significant activity in patients with relapsed/refractory B-cell malignancies. J. Clin. Oncol. 31:88–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Chang BY, Huang MM, Francesco M, Chen J, Sokolove J, Magadala P, Robinson WH, Buggy JJ. 2011. The Bruton tyrosine kinase inhibitor PCI-32765 ameliorates autoimmune arthritis by inhibition of multiple effector cells. Arthritis Res. Ther. 13:R115. 10.1186/ar3400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Kil LP, de Bruijn MJ, van Nimwegen M, Corneth OB, van Hamburg JP, Dingjan GM, Thaiss F, Rimmelzwaan GF, Elewaut D, Delsing D, van Loo PF, Hendriks RW. 2012. Btk levels set the threshold for B-cell activation and negative selection of autoreactive B cells in mice. Blood 119:3744–3756 [DOI] [PubMed] [Google Scholar]
- 75. Rohrer J, Conley ME. 1999. Correction of X-linked immunodeficient mice by competitive reconstitution with limiting numbers of normal bone marrow cells. Blood 94:3358–3365 [PubMed] [Google Scholar]