

Abstract
Erythroid-specific, high level expression of the β-globin genes is regulated by the locus control region (LCR), composed of multiple DNase I-hypersensitive sites and located far upstream of the genes. Recent studies have shown that LCR core elements recruit RNA polymerase II (pol II). In the present study we demonstrate the following: 1) pol II and other basal transcription factors are recruited to LCR core hypersensitive elements; 2) pol II dissociates from and re-associates with the globin gene locus during replication; 3) pol II interacts with the LCR but not with the β-globin gene prior to erythroid differentiation in embryonic stem cells; and 4) the erythroid transcription factor NF-E2 facilitates the transfer of pol II from immobilized LCR constructs to a β-globin gene in vitro. The data are consistent with the hypothesis that the LCR serves as the primary attachment site for the recruitment of macromolecular complexes involved in chromatin structure alterations and transcription of the globin genes.
The five genes of the human β-globin locus are expressed in erythroid cells in a tissue- and developmental stage-specific manner (1). Appropriate expression of the globin genes is regulated by many DNA elements that are located proximal and distal to the genes. The human β-globin locus control region (LCR)1 is a powerful regulatory DNA element located far upstream of the genes and is required for high level expression of all the globin genes throughout development (1, 2). The LCR, unlike classical enhancer elements, operates in an orientation-dependent manner (3). There is currently no consensus on how the LCR acts to stimulate globin gene transcription, but it is generally believed that it involves some form of communication between the LCR and the globin genes (4–6). The LCR is composed of several regions that exhibit extremely high sensitivity to DNase I in erythroid cells (hypersensitive HS sites 1–5). The core HS sites contain clusters of transcription factor binding sites and are separated from each other by 2–4 kbp (2). The results from analyzing human LCR function at ectopic sites in the context of transgenic mice demonstrate that the HS sites synergistically enhance globin gene transcription (7–12), whereas studies in the endogenous murine locus show that the core HS sites function additively (13–15).
Recent models view the LCR as a holocomplex in which the individual HS sites interact via extensive protein/DNA and protein/protein interactions (7, 16, 17). The LCR holocomplex may provide a highly accessible region for the efficient recruitment of macromolecular complexes involved in chromatin modification and transcription (18). Indeed, it has been shown that RNA polymerase II (pol II) is recruited to LCR HS sites in vitro and in vivo (19–22), suggesting that transcription complexes are first recruited to the LCR and subsequently delivered to the globin genes (18). Sawado et al. (23) recently demonstrated that another important function of the LCR is to regulate transcription elongation at the adult β-globin gene.
An interesting aspect of gene regulation is the question of how transcriptional activity is maintained after replication (24). Activation of at least some gene loci depends on replication, and it is possible that replication could provide a window of opportunity for transcription factors to gain access to regulatory sequences before repressive chromatin is formed. Generally, transcriptionally active regions of the genome replicate early, whereas repressed parts of the genome replicate late. Accordingly, the globin locus replicates late in nonerythroid cells and early in cells expressing the globin genes. It is not known how replication affects the association of transcription factors and pol II with the globin locus.
In this study we analyzed the interaction of transcription complexes with the human and murine β-globin loci in erythroid cells using chromatin immunoprecipitation (ChIP). The data demonstrate that pol II and the basal transcription factors TBP and TFIIB interact with LCR core HS sites but not with a region flanking the cores, indicating that transcription initiation complexes associate with the LCR. The analysis of protein/DNA interactions during S-phase in synchronized cells revealed a dynamic pattern of dissociation and re-association of transcription factors and RNA pol II with the globin locus. In addition, we show that core elements of the LCR adopt a structure characteristic of transcriptionally active chromatin and recruit RNA polymerase II prior to erythroid differentiation in murine embryonic stem cells. Finally, we provide evidence that pol II can be transferred from the LCR to a β-globin gene using a novel in vitro system. This transfer is stimulated by the erythroid transcription factor NF-E2.
EXPERIMENTAL PROCEDURES
Cell Culture and Protein Extracts
K562 cells were grown in RPMI containing 15% fetal bovine serum and 5% antibiotic mixture (penicillin/streptomycin) in 5% CO2 at 37 °C. MEL cells were grown, and protein extracts were prepared as described by Leach et al. (25). Recombinant NF-E2 (p45 tethered to mafg) was expressed in bacteria as described by Leach et al. (25)
Cell Synchronization
K562 cells were synchronized at the border of G1- and S-phase by incubating the cells with complete growth medium in the presence of 2 mm thymidine for 17 h, washing twice with complete medium, incubating in complete medium for 12 h, and blocking the cells in complete medium with 2 mm thymidine for another 17 h (26). Blocked cells were fixed with ethanol, digested with RNase, stained with propidium iodide, and subjected to flow cytometry to verify synchronization. Blocked cells were taken as “time 0” and subjected to ChIP and PCR analysis. The blocked cells were then washed twice with complete medium to release them from the block and were allowed to grow in complete medium. Cells were taken for flow cytometry, ChIP, and PCR analysis at specific time points after release from the arrest.
ChIP
ChIP was performed as described by Leach et al. (25). The following DNA primers and antibodies were used in the experiments. The following primers were used: human ∊-globin, US (upstream) 5′-GCTCCTTTATATGAGGCTTTCTTGG-3′ and DS (downstream) 5′-AATGCACCATGATGCCAGG-3′; human β-globin, US 5′-TATCTTAGAGGGAGGGCTGAGGGTTTG-3′ and DS 5′-CCAACTTCATCCACGTTCACCTTGC-3′; human HS2, US 5′-TGTGTGTCTCCATTAGTGACCTCCC-3′ and DS 5′-TTTTGCCATCTGCCCTGTAAGC-3′; human γ-globin, US 5′-CTCAATGCAAATATCTGTCTG-3′ and DS 5′-TCTGGACTAGGAGCTTATTG-3′; human HS2 5′flank, US 5′-TGGGGACTCGAAAATCAAAG-3′ and DS AGTAAGAAGCAAGGGCCACA-3′; mouse βmaj-globin, US 5′-TAATTTGTCAGTAGTTTAAGGTTGC-3′ and DS 5′-CATTGTTCACAGGCAAGAGCAGG; mouse Ey-globin, US 5′-CAAAGAGAGTTTTTGTTGAAGGAGGAG-3′ and DS 5′-AAAGTTCACCATGATGGCAAGTCTGG-3′; mouse HS2, US 5′-TTCCTACACATTAACGAGCCTCTGC-3′ and DS 5′-AACATCTGGCCACACACCCTAAGC-3′; mouse HS2 5′flank, US 5′-CTATTTGCTAACAGTCTGACAATAGAGTAG-3′ and DS 5′-GTTACATATGCAGCTAAAGCCACAAATC-3′; and mouse necdin, US 5′-TTTACATAAGCCTAGTGGTACCCTTCC-3′ and DS 5′-ATCGCTGTCCTGCATCTCACAGTCG-3′. Primers for the human γ-globin genes were published by Schreiber et al. (27). The antibodies used are as follows: TFIIB sc-225, TFIID (TBP) sc-273, pol II (N-20) sc-899, NF-E2 (C-19) sc-291, and USF2 (C-20) sc-862 (all purchased from Santa Cruz Biotechnology) and phospho-pol II 05-623 (Upstate Biotechnology, Inc.). All antibodies were tested in Western blotting experiments using MEL or K562 nuclear extracts as described by Leach et al. (25).
Nuclear Run-on Transcription (NRO) and RT-PCR
The nuclear run-on experiments were performed as described by Greenberg and Bender (28). Globin-specific DNA fragments serving as targets for labeled RNA in slot-blot experiments were generated by PCR. The following primers were used: hHS2 5′-flank, US 5′-GTAACACCTCTTGAGTACTTAGTAT-3′ and DS 5′-GAGGTAATAATCTACTTCTGAGTAT-3′; h∊, US 5′-GTGTCCATCCATCACTGCTGACCCT-3′ and DS 5′-GGAAGTCAGCACCTTCTTGCCATGG-3′; hHS5, 5′flank, US 5′-CGAAGCTTCTGACAAATTATTCTTCC-3′ and DS 5′-CAGACTGTGGGAAAAATCAGAGGGATCCGC-3′; 5′mHS5, US 5′-GGTACCTATATAGGTGACTTACATA-3′ and DS 5′-CACCTAAGACACTGTGGAAGAGCAG-3′; mHS2, US 5′-GGGTCTCTCTAGGAGGAAGTCCACAGG-3′ and DS 5′-CAGATCTAATGACCCTAACTCTAAC-3′; and mβmaj, US 5′-GGTGCACCTGACTGATGCTGAGAAG-3′ and DS 5′-TGGTACTTGTGAGCCAGGGCAGTG. We used pKO916 (Stratagene) as a negative control probe. Slot blot was performed as described in Kang et al. (29). RNA was extracted using the RNeasy kit (Qiagen) according to the protocol provided by the manufacturer.
RNA was isolated for RT-PCR using the Arum Total RNA Mini Kit (Bio-Rad) according to the manufacturer’s protocol. Reverse transcription was performed using 200–250 ng of RNA and the iScript cDNA synthesis kit (Bio-Rad) as described by the manufacturer’s protocol. PCR amplification was performed using the Eppendorf PCR Mastermix (Eppendorf) and primer sequences specific for Flk-1, Epo receptor, GATA-2, β-actin (30), Rex-1 (31), mouse βmaj-globin: US 5′-CACCTTTGCCAGCCTCAGTG-3′ and DS 5′-GGTTTAGTGGTACTTGTGAGCC-3′; mouse Ey, US 5′-AACCCTCATCAATGGCCTGTGG and DS 5′-TCAGTGGTACTTGTGGGACAGC-3′.
Embryonic Stem (ES) Cell Differentiation
Mouse ES cells were differentiated to generate cells of the hematopoietic lineage using the ES/OP9 method established and described by Kitajima et al. (32). Briefly, ESD3 cells (ATCC, CRL-1934) were seeded onto a confluent monolayer of mouse embryonic fibroblasts at a density of 105cells/25 cm2 in ES media (Dulbecco’s modified Eagle’s medium, 4.5 g/liter glucose, 1.5 g/liter sodium bicarbonate, 15% fetal bovine serum, 0.1 mm 2-mercaptoethanol, and 106 units/ml LIF), grown for 2 days, and then passaged (1:6) and grown for another day. An aliquot of the cells (3–4 × 107) was taken at this time (day 0) and subjected to RT-PCR and ChIP analysis. The remaining day 0 cells were then seeded onto confluent OP9 stromal cells in OP9 media (α-minimum Eagle’s medium with ribonucleosides and deoxyribonucleosides; 20% fetal bovine serum) in the absence of LIF at a density of 104 cells/well in 6-well tissue culture dishes. At day 3 Epo was added at a concentration of 2 units/ml for the remainder of the course of induction. On day 5 of induction cells were passaged and reseeded onto fresh OP9 cultures at a density of 105 cells/well. On day 12 cells were collected and subjected to RT-PCR and ChIP analysis.
In Vitro Polymerase Transfer Analysis
A plasmid containing the wild-type human β-globin LCR was linearized and immobilized on streptavidin-coated magnetic beads as described by Leach et al. (19). The immobilized LCR (200 ng) was incubated for 30 min at 30 °C with 50 μg of MEL nuclear protein extract (10 μl) and 15 μl of 2× binding buffer (36 mm HEPES, pH 7.9, 160 mm KCl, 40 mm MgCl2, 4 mm dithiothreitol, 10 mm phenylmethylsulfonyl fluoride, and 20% glycerol) in a total volume of 100 μl. We performed a protein extract titration experiment and determined that 50 μg of the protein extract was optimal in the reactions. The tubes were placed on a magnet and the supernatant was removed, and the beads were washed three times with 1× binding buffer. The beads were then resuspended in 1× binding buffer and incubated for 10 min at 30 °C in the presence or absence of 50 ng of the plasmid pRSβA/X containing the wild-type or mutant β-globin gene (25). The immobilized LCR construct is about four times larger than the β-globin gene construct, so the same molar ratio was used during the transfer. The supernatants were removed, and samples containing the wild-type or mutant β-globin gene were cross-linked by incubation for 10 min in 0.5% formaldehyde at room temperature. The cross-linking reaction was stopped by the addition of 0.125 m glycine and incubation for 5 min at room temperature. The supernatant of reactions not containing the β-globin gene were incubated for 10 min at 30 °C in the presence of pRSβA/X, after which these samples were cross-linked as well. All samples were dialyzed against ChIP dilution buffer and subjected to immunoprecipitation using pol II-specific antibodies and PCR analysis as described by Leach et al. (25). In some experiments recombinant NF-E2 (25) or BSA (0.3 μg/μl) was added to the transfer reactions. The plasmid βmaxi was used as a positive control for the PCR and was generated by ligating an 800-bp Pml1 restriction fragment from the human LCR into the Pml1 site of pRSβA/X. The β-globin PCR primers span the Pml1 site present in the β-globin gene thus generating a PCR product that is 800 bp larger than the PCR product from the wild-type β-globin gene. The TK-HGH plasmid contains the human growth hormone gene under control of the herpes simplex virus thymidine kinase promoter (25). The HS2 plasmid contains the HS2 core plus flanking sequence as described previously (8). The following primers were used in these experiments: β-globin II, US 5′-CCTGAGGAGAAGTCTGCCGTTACTG-3′ and DS 5′-TCCTATGACATGAACTTAACCATAG-3′; TK-HGH, US 5′-GGAGGCTGGAAGATGGCA-3′ and DS 5′-AGTAGTGCGTCATCGTTGTGTG-3′; HS2 as described in the ChIP protocol.
RESULTS
In this study we analyzed the interaction of transcription complexes with the human and murine β-globin gene loci, which are shown as a graph also including the location of primers and fragments used in the ChIP and NRO experiments (Fig. 1).
FIG. 1. Diagrammatic representation of the human and murine β-globin gene locus.

The human β-globin gene locus is located on chromosome 11 and consists of five developmentally regulated genes: the embryonic ∊-globin gene, the two fetal γ-globin genes, and the adult δ- and β-globin genes. The LCR is located upstream of the ∊-globin gene and is composed of at least five HS sites. The murine β-globin locus is located on chromosome 7 and contains four developmentally regulated genes: the Ey-and βh1-globin genes, which are co-expressed in the embryonic stage and the βmaj- and βmin-globin genes, which are expressed during the fetal and adult stages of erythropoiesis. The overall organization of the LCR is highly conserved between the murine and the human β-globin locus (2). PCR fragments analyzed in the ChIP experiments or used as probes in the NRO assays are indicated as lines (horizontal bars) below the respective loci.
Interaction of Transcription Factors and RNA Polymerase II with the β-Globin Locus
We first used ChIP and PCR to analyze the interaction of pol II, TBP, TFIIB, NF-E2 (p45), and acetylated histone H3 with different domains of the human β-globin gene locus in K562 cells, an erythroleukemia cell line expressing the embryonic ∊- but not the adult β-globin gene. As a negative control we also analyzed the interaction of the proteins with the necdin gene, which is not active in erythroid cells. NF-E2 is a hematopoietic-specific transcription factor composed of a large (p45) and a small (maf) subunit (33) and is known to interact with sequences in the human and murine β-globin loci (2).
The data show that pol II is cross-linked to LCR HS2 in K562 cells (Fig. 2a). The interaction of pol II with the globin genes is consistent with the expression pattern in these cells. pol II binding is detectable at ∊- but not at the β-globin gene. pol II does not interact with the HS2 5′-flanking region, demonstrating that pol II specifically interacts with the core of HS2. We observed interactions of phosphorylated pol II with HS2 and the ∊-globin gene promoter, indicating active transcription in these regions. Because extragenic transcripts have been detected over the entire LCR in the human β-globin locus (34), we analyzed the interaction of basal transcription factors that are not associated with the transcription elongation complex. The results show that both TFIIB as well as TBP can be cross-linked to HS2 in K562 cells. This demonstrates that the cross-linking of pol II to HS2 is not solely due to elongating polymerase but is also due to the recruitment of transcription initiation complexes. NF-E2 is clearly detectable at HS2, whereas association with the ∊-globin gene is weak.
FIG. 2. Interaction of transcription factors with and transcription in the human β-globin locus in K562 cells.
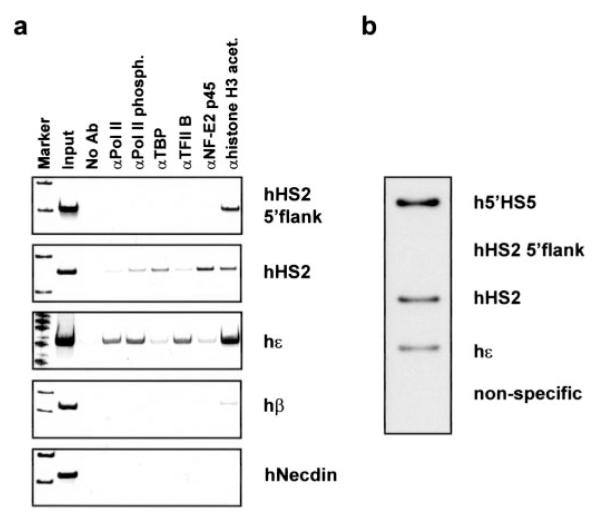
a, interaction of transcription factors and RNA polymerase II with the human β-globin locus and the human necdin (hNecdin) gene. ChIP with antibodies specific for RNA polymerase II (αpol II), RNA polymerase II phosphorylated at the C-terminal domain (αpol II Phosph.), TBP, TFIIB, NF-E2 p45, and acetylated histone H3 (αH3 acet.). DNA purified from the precipitate was analyzed by PCR with primers corresponding to regions in the human β-globin locus as indicated (see Fig. 1). The no antibody (no Ab) lanes represent negative controls. The input lane represents the positive PCR control taken from the no antibody sample. b, nuclear run-on transcription analysis in specific regions of the β-globin locus. The RNA was hybridized to specific DNA fragments, and the position of these fragments is indicated in Fig. 1. The non-specific lane shows hybridization to the negative control plasmid pK0916.
The observation that pol II is not detectable in the HS2 5′-flanking region suggests that transcription initiates within the HS2 core enhancer and not upstream of HS2. Therefore, we performed nuclear run-on experiments to determine regions of active transcription in the human β-globin locus (Fig. 2b). Ongoing transcription is detectable in a region upstream of HS5, in the HS2 core, and in the ∊-globin gene but not in the HS2 5′-flanking region. These results are consistent with the ChIP analysis and suggest that transcripts initiating far upstream of HS2 do not elongate beyond the HS2 5′-flanking region, consistent with results from Kim and Dean (35). Transcription upstream of HS5 probably initiates within the long terminal repeat of a retrovirus as described previously (36).
Dissociation and Re-association of RNA Polymerase II with the β-Globin Locus during Early S-phase
We next examined the interaction of transcription complexes with the LCR and globin genes during S-phase in K562 cells (Fig. 3). Cells were synchronized using the double thymidine block method, which arrests the cells at the G1/S-phase boundary (26). By using semiquantitative PCR with primers specific for the human globin locus and, as a control, the necdin gene, we determined that replication of the β-globin locus is completed by 2 h in K562 cells (data not shown).
FIG. 3. Interactions of transcription factors and RNA polymerase II with the human β-globin locus during early S-phase in synchronized K562 cells.

The cells were harvested at time 0 or at specific time points after release from cell cycle arrest (as indicated), and subjected to flow cytometry (shown on top) and ChIP using antibodies specific for RNA polymerase II (αpol II), the p45 subunit of NF-E2 (αNF-E2), and USF2 (αUSF2); time 0 represents the status of the blocked cells at the G1/S-phase boundary. PCR was carried out using primers specific for HS2, the ∊-globin gene, the γ-globin gene, and the β-globin gene as indicated (see Fig. 1). The no antibody (no Ab) and input controls were processed as described in the legend to Fig. 2.
At specific time points after release from cell cycle arrest, cells were subjected to formaldehyde cross-linking and ChIP analysis. In these experiments, we used antibodies against pol II, NF-E2 (p45), and USF2. We have shown previously that USF2 interacts with the human β-globin promoter in K562 cells (25). We analyzed the association of these proteins with four different regions of the human β-globin locus. Cells were cross-linked at time 0, 15, and 45 min and 2 and 6 h after release from cell cycle arrest. At time 0, pol II interacts with HS2, the ∊- and γ-globin genes, but not with the adult β-globin gene. There is no interaction of NF-E2 or USF2 with the globin locus at this point. The only detectable change after 15 min is the association of NF-E2 with HS2 and the γ-globin genes. After 45 min, pol II is no longer detectable at the ∊-globin gene but remains associated with HS2 and the γ-globin genes. After 2 h, pol II no longer interacts with the LCR or the ∊- and γ-globin genes. At this time NF-E2 and USF2 interact with HS2 and the ∊- and γ-globin genes. The interaction of NF-E2 with the β-globin gene is less pronounced. After 6 h, the dissociation of USF2 from the γ-globin genes and HS2 is accompanied by the reappearance of pol II. The different kinetics in the dissociation and re-association of pol II with the ∊- and γ-globin genes was observed in multiple experiments (Fig. 3, and data not shown).
In some experiments we observed interactions between NF-E2 and HS2 at time 0 and re-association of pol II with the ∊-globin gene after 6 h (data not shown). The differences could be due to subtle variations in the status of the cells during synchronization. We documented previously (25) the interaction of USF2 with the β-globin gene locus in unsynchronized K562 cells. At the time points analyzed here, USF2 was not detectable at the β-globin gene suggesting that the interaction takes place at another time during the cell cycle.
RNA Polymerase II Is Recruited to the LCR but Not to the β-Globin Gene in Murine Embryonic Stem Cells Prior to Erythroid Differentiation
We next examined the association of pol II with the murine β-globin gene locus during in vitro differentiation of murine ES cells. In these experiments we utilized the ES/OP9 cell differentiation system described by Kitajima et al. (32). Undifferentiated ES cells (day 0) express the rex1, β-actin, flk1, and gata-2 genes (Fig. 4a). Most importantly, the βmaj-globin gene is not expressed in these cells. Upon erythroid differentiation (day 12), expression of Rex1 is reduced, as anticipated (31), and the globin genes are induced. We analyzed the interaction of pol II and modified histones within the globin locus during the course of differentiation using the ChIP assay (Fig. 4b). We utilized two pol II-specific antibodies, one recognizing both phosphorylated and unphosphorylated pol II (pol II) and one recognizing pol II phosphorylated at serine 5 (pol II Ser-5(P)). We also used antibodies specific for acetylated histone H4 (AcH4) and for dimethylated K4 histone H3 (Me2K4H3). Dimethylation of H3 at lysine 4 is associated with regions permissive for transcription (37).
FIG. 4. Interaction of RNA polymerase II with the murine β-globin gene locus during erythroid differentiation of ES cells.

Mouse ES cells were differentiated to generate cells of the erythroid lineage using the ES/OP9 method described by Kitajima et al. (32). Day 0 represents undifferentiated ES cells grown in the presence of LIF. Day 12 represents differentiated cells grown in the absence of LIF and in the presence of Epo. Cells were collected at day 0 or day 12 and subjected to RT-PCR and ChIP analysis as described under “Experimental Procedures.” a, analysis of gene expression in undifferentiated (day 0) and differentiated (day 12) ES cells by RT-PCR using sets of primers specific for the murine rex1, β-actin, Ey-globin, βmaj-globin, erythropoietin receptor (Epo-R), flk-1, and gata-2 genes as indicated. b, analysis of protein/DNA interactions in undifferentiated (day 0) and differentiated (day 12) ES cells by ChIP using an unspecific antibody directed against chicken IgG (unspec.) or antibodies specific for RNA polymerase II (pol II), RNA polymerase II phosphorylated at serine 5 of the C-terminal domain (pol II Ser5P), histone H3 di-methylated at lysine 4 (Me2K4H3), and acetylated histone H4 (AcH4) as indicated. The precipitated chromatin was analyzed by PCR using primers specific for LCR element HS2, the βmaj promoter region, and the rex1 gene.
The results (Fig. 4b) show that pol II is present at the LCR but not at the β-globin gene in undifferentiated ES cells. The presence of H3 dimethylated at Lys-4 is indicative of active transcription in this region. The β-globin gene is associated with acetylated histone H4 but not H3 dimethylated at Lys-4, suggesting that the chromatin structure is open but not transcriptionally active. The rex1 gene is associated with a chromatin structure characteristic of an open transcriptionally active domain. The primers used for rex1 amplify a region in the body of the gene and thus do not assay for interaction of pol II with the promoter.
In differentiated erythroid cells (day 12), pol II is associated with the LCR and the β-globin gene. The enrichment of H3 dimethylated at Lys-4 in these regions is compatible with an open, transcriptionally active chromatin structure. In contrast, the rex1 gene is repressed at this stage and this is accompanied by a reduction in Lys-4 dimethylated H3 and acetylated H4. These results demonstrate that prior to erythroid differentiation, the LCR lies within a transcriptionally competent chromatin structure and is associated with pol II.
In Vitro Transfer of pol II from the LCR to the β-Globin Gene
To examine the possibility that pol II can be recruited to the LCR first and then transferred to the β-globin gene, we established a novel in vitro system utilizing immobilized LCR templates and nuclear extracts from MEL cells (Fig. 5a). A plasmid containing the entire human β-globin LCR was linearized, biotinylated, and immobilized on streptavidin-coated magnetic beads as described by Leach et al. (19). After incubation with MEL nuclear extract, the protein-DNA complex was washed several times in binding buffer. Next, a plasmid bearing the wild-type β-globin gene was added. After incubation, the supernatant containing the β-globin gene was removed from the immobilized LCR and subjected to cross-linking with formaldehyde and ChIP analysis using pol II-specific antibodies as described under “Experimental Procedures” (Fig. 5b, lane 2). Another sample was processed in the same manner except that no pol II-specific antibodies were added; this sample served as a negative control (Fig. 5b, lane 4). To control for diffusion, we incubated the assembled protein-LCR complex in the absence of the β-globin template in binding buffer. The reaction was placed on a magnet, and the supernatant was removed and incubated afterward with the β-globin template. The sample was then cross-linked and subjected to ChIP analysis with pol II antibodies (Fig. 5b, lane 1). As a positive control, we incubated the β-globin gene with MEL protein extract and subjected the sample to cross-linking and ChIP analysis (Fig. 5b, lane 5). After reversal of the cross-link and purification of the DNA, the samples were analyzed by PCR using primers specific for the coding region of the β-globin gene. The βmaxi gene was added after the immunoprecipitation and served as an internal PCR control. Fig. 5b (lane 2) shows that pol II is efficiently transferred from the LCR to a β-globin gene template. Diffusion does not contribute significantly to the transfer of pol II from the LCR to the β-globin gene (Fig. 5b, lane 1). The inclusion of recombinant NF-E2 consistently enhanced the transfer of pol II from the LCR to the β-globin gene (Fig. 5, b, lane 3, c, lane 4, and e), supporting previous conclusions that NF-E2 is required for the recruitment of pol II to the globin genes (38). The addition of BSA at the same concentration as NF-E2 did not enhance pol II transfer (Fig. 5c, lane 5), indicating that stimulation of pol II transfer by NF-E2 is not an unspecific effect due to increased protein content. To examine whether pol II transfer is specific for the β-globin gene, we have performed transfer experiments in the presence of a β-globin gene lacking the TATA box. We have shown previously that this mutant is inefficiently transcribed in vitro (25). We found that the β-globin TATA box is necessary for pol II recruitment (Fig. 5c, lane 6) demonstrating that pol II is specifically transferred to the β-globin gene and not to sequences elsewhere in the plasmid. To test further the specificity of the transfer, we performed an experiment in which we simultaneously incubated LCR-protein complexes with two different plasmids, one containing LCR element HS2 and another containing the β-globin gene. The results in Fig. 5d show that pol II is preferentially transferred to HS2 (lane 2), which could be due to interactions between HS2 and the LCR-protein complex. Most importantly, we observed that in the presence of NF-E2, pol II is preferentially transferred to the β-globin gene and not to HS2 (Fig. 5d, lane 3), demonstrating that NF-E2 switches the specificity of pol II transfer from HS2 to the β-globin gene promoter. We also analyzed the transfer to the thymidine kinase promoter and found that pol II is not transferred to this template (compare Fig. 5d, lanes 1 and 4). This again demonstrates that the transfer is specific for the β-globin gene. The graph in Fig. 5e summarizes the results of multiple independent experiments.
FIG. 5. Analysis of RNA polymerase II transfer from the LCR to the β-globin gene in vitro.

a and b, a depicts a schematic representation of the experimental system as described under the “Experimental Procedures.” The plasmid pLCR was linearized, biotin-labeled, and immobilized on streptavidin-coated magnetic beads as described by Leach et al. (19). After incubation with MEL protein extracts, the LCR was placed on a magnet, and all material not interacting with the LCR was removed. The immobilized LCR-protein complex was washed twice in binding buffer (see “Experimental Procedures”), resuspended in binding buffer, and incubated in the presence or absence of the β-globin gene. After placing the samples on the magnet, the supernatant was removed, and the sample containing the β-globin gene was cross-linked and subjected to ChIP analysis using RNA polymerase II-specific antibodies (b, lane 2). The supernatant of the sample incubated without the β-globin gene was incubated with pRSβA/X and subjected to cross-linking and ChIP analysis using a pol II-specific antibody (b, lane 1). Lane 3 represents a reaction processed in the same manner as described for lane 2 except that recombinant NF-E2 was added to the reaction. Lane 4 represents a negative control in which no antibody was added. Lane 5 represents the positive control in which the β-globin gene was incubated with MEL protein extract and subjected to ChIP and PCR. The βmaxi gene was added after immunoprecipitation and amplified with the same β-globin primers as the wild-type globin gene. PCR was performed as described under “Experimental Procedures” with 22 cycles. c, transfer of RNA polymerase II to the β-globin gene in the presence of NF-E2 or BSA. Samples were prepared and analyzed as described in a and b except that NF-E2, lane 4, or BSA, lane 5, were added during the transfer step. Lane 6 represents transfer to a mutant (mut) β-globin gene template lacking the TATA box. d, analysis of NF-E2 mediated transfer to the β-globin gene. Samples were processed as described in a and b except that in lanes 2 and 3 the transfer was carried out in the presence of both β-globin and HS2 containing plasmids. Lane 3, NF-E2 was added to the transfer reaction. Lane 4, transfer to the thymidine kinase/human growth hormone gene (TK-HGH) construct was analyzed. Lane 1 represents the reaction of the no antibody control containing all three plasmids (β-globin, HS2, and TK-HGH). Lanes 5–7 represent positive PCR controls for β-globin (lane 5), HS2 (lane 6), and TK-HGH (lane 7). e, quantitative summary of the transfer experiments. The graphs represent fold increases over the no antibody control of two (BSA and TATA mutant) to three (transfer, transfer plus NF-E2) experiments. Statistical analysis of all experimental data demonstrates that the NF-E2-mediated increase in pol II transfer is significant with a p value of 0.04.
DISCUSSION
Expression of the β-globin genes is regulated by the LCR (1, 39). The LCR and the genes appear to be in close proximity in cells expressing the globin genes, suggesting physical contacts between activities associated with the LCR and proteins interacting with the promoter (40, 41). How this contact stimulates transcription is not known but could involve the transfer of activities from the LCR to the globin genes (18). We tested this hypothesis using three different experimental systems. First, we analyzed the recruitment of pol II to the globin locus during S-phase, and we show that pol II dissociates and re-associates with the globin locus during replication. Second, we analyzed the association of pol II with the globin locus during ES cell differentiation and show that it is already present at the LCR but not at the genes in undifferentiated ES cells. Finally, we developed a novel in vitro system and demonstrate that pol II can be transferred from immobilized LCR templates to a β-globin gene in a process facilitated by the erythroid transcription factor NF-E2. These data are consistent with the hypothesis that transcription complexes are first recruited to a highly accessible LCR holocomplex and subsequently transferred to the globin genes.
We began our studies by addressing the question of whether RNA pol II is recruited to the LCR independent from its known interactions with the globin genes in human K562 cells. We show here that pol II interacts with the HS2 core region together with other components of the transcription initiation complex. The results from the NRO analysis are consistent with the ChIP data and demonstrate that transcription is ongoing at places where transcription complexes are bound, supporting the notion that pol II is recruited to the LCR independent from interactions with the promoter regions. It is also important to note that whereas HS2 is transcribed in K562 cells, the HS2 5′-flanking region in K562 cells is not, consistent with the lack of pol II binding to this region. This suggests that extragenic transcripts over the LCR do not represent long continuous transcripts but are due to multiple transcription initiation sites in HS2, HS3, and possibly other regions consistent with results recently published by Johnson et al. (22).
The experiments using synchronized cells did not reveal temporal differences in the recruitment of pol II to the LCR or the globin genes during S-phase. This could suggest that pol II is recruited simultaneously to the genes and to the LCR in these experiments; perhaps the LCR and the genes cooperate in recruiting pol II after replication of the globin locus. On the other hand, it is also possible that the process of recruitment is dynamic making it unlikely to select a time point at which pol II is detectable first at the LCR. We thus decided to analyze pol II recruitment during differentiation of ES cells into the erythroid lineage. The results clearly demonstrate that pol II and dimethyl-Lys-4 H3 are present at the LCR in undifferentiated ES cells. The presence of dimethyl-Lys-4 H3 suggests ongoing transcription in the LCR at this stage. After differentiation, pol II and dimethyl-Lys-4 H3 are associated with the LCR and the transcribed β-globin gene.
The summary data are consistent with a model proposing that an important function of the LCR is to act as a reservoir of transcription and maybe other protein complexes, which will be provided to the globin genes during cell cycle and erythroid differentiation. NF-E2 may play a major role in directing these activities to the globin locus. In K562 cells, NF-E2 interacts directly or indirectly with HS2 and both ∊- and γ-globin genes during S-phase. The interaction of NF-E2 with the globin locus precedes the re-association of pol II suggesting a role for NF-E2 in recruiting pol II to the globin locus. Other data demonstrate that NF-E2 interacts with the human β-globin LCR before chromatin is re-modeled (42), consistent with NF-E2 playing an important role in maintaining transcriptional activity during replication of the globin locus.
Previous studies (25, 43) have shown that USF interacts with LCR core elements and with the β-globin promoter. Our data demonstrate that USF2 only interacts with HS2 and the globin genes at a stage when pol II is not bound. It is conceivable that USF2, in conjunction with NF-E2, prepares specific regions in the globin locus for the re-association of transcription complexes after replication.
It was proposed that one function of the LCR could be to recruit macromolecular protein complexes including pol II, which are subsequently transferred to individual globin genes in a developmental stage-specific manner (18, 19). Similar conclusions were derived from studies on the TCRβ locus, which show that coactivators and components of the transcription initiation complex are first recruited to an enhancer element and subsequently delivered to the promoter (44). We have developed an in vitro system to test the polymerase transfer hypothesis, and we show that pol II can be efficiently transferred from an immobilized LCR template to the β-globin gene (Fig. 5). The transfer of pol II is sensitive to mutations in the β-globin promoter and enhanced by recombinant NF-E2. The results suggest that LCRs, like classical enhancer elements (45), can act in trans to assist in the recruitment of transcription complexes to the globin genes. Although this in vitro system ignores nucleosome and higher order chromatin structure, we believe that it does reveal aspects of LCR function. We have shown previously that specific characteristics of HS site formation and function, including the recruitment of transcription complexes, can be recapitulated in vitro (19). This demonstrates that chromatin may not be directly involved in recruiting transcription complexes to the globin locus but rather regulates a preceding step, which is to render regulatory and functional sites available or unavailable for the interaction with transcription factors and polymerase.
In summary, we propose that transcription complexes are first recruited to a highly accessible LCR holocomplex (Fig. 6). The genes are subsequently brought in contact with the LCR holocomplex, consistent with data from conformational studies (40, 41), and transcription complexes are transferred from the LCR to the promoter regions. This process is mediated by NF-E2 and facilitated by bringing strong globin gene promoters (with high affinities for the transcription complex) in close proximity to LCR bound transcription complexes.
FIG. 6. Model of transcription complex recruitment to the β-globin gene.

According to a recent model, the LCR and other HS sites interact to form a chromatin hub (41). In the active chromatin hub, the expressed genes interact with the HS sites. We propose that transcription and other protein complexes are first recruited to a highly accessible LCR holocomplex in the context of the proposed chromatin hub. The genes come in close proximity to the LCR holocomplex by as yet unknown mechanisms that may involve local remodeling of chromatin structure at the active promoters. Transcription complexes are then transferred from the LCR to high affinity binding sites at the globin gene promoters. This transfer is facilitated by NF-E2 and/or related proteins.
Acknowledgments
We thank Takeesha Roland for technical assistance and our colleagues Drs. Christof Dame, Kelly M. Leach, Mike Kilberg, and Linda Bloom for helpful discussions. We thank Drs. Nakano (Osaka, Japan) and Ohneda (Tsukuba, Japan) for providing us with detailed protocols for ES cell differentiation. We appreciate the effort of Keiji Tanimoto (Tsukuba, Japan), Kazuhiko Igarashi (Hiroshima, Japan), and Thomas Yang (UF) for critically reading the manuscript.
Footnotes
This work was supported by National Institutes of Health Grants DK058209 and DK0522356 (to J. B.).
The abbreviations used are: LCR, locus control region; HS, hypersensitive; RT, reverse transcription; Epo, erythropoietin; ChIP, chromatin immunoprecipitation; BSA, bovine serum albumin; ES, embryonic stem; NRO, nuclear run-on transcription; TBP, TATA binding protein; MEL, mouse erythroleukemia; USF, upstream stimulatory factor; LIF, leukemia inhibitory factor.
REFERENCES
- 1.Grosveld F. Curr. Opin. Genet. Dev. 1999;9:152–157. doi: 10.1016/S0959-437X(99)80023-9. [DOI] [PubMed] [Google Scholar]
- 2.Hardison RC, Slightom JL, Gumucio DL, Goodman M, Stojanovich N, Miller W. Gene (Amst.) 1997;51:73–94. doi: 10.1016/s0378-1119(97)00474-5. [DOI] [PubMed] [Google Scholar]
- 3.Tanimoto K, Liu Q, Bungert J, Engel JD. Nature. 1999;398:344–348. doi: 10.1038/18698. [DOI] [PubMed] [Google Scholar]
- 4.Bulger M, Groudine M. Genes Dev. 1999;13:2465–2477. doi: 10.1101/gad.13.19.2465. [DOI] [PubMed] [Google Scholar]
- 5.Engel JD, Tanimoto K. Cell. 2000;100:499–502. doi: 10.1016/s0092-8674(00)80686-8. [DOI] [PubMed] [Google Scholar]
- 6.McDowell JC, Dean A. Mol. Cell. Biol. 1999;19:7600–7609. doi: 10.1128/mcb.19.11.7600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bungert J, Dave U, Lim K-C, Lieuw KH, Shavit JA, Liu Q, Engel JD. Genes Dev. 1995;9:3083–3096. doi: 10.1101/gad.9.24.3083. [DOI] [PubMed] [Google Scholar]
- 8.Bungert J, Tanimoto K, Patel S, Liu Q, Fear M, Engel JD. Mol. Cell. Biol. 1999;19:3062–3072. doi: 10.1128/mcb.19.4.3062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.May C, Rivella S, Callegari J, Heller G, Gaensler KM, Luzzatto L, Sadelain M. Nature. 2000;406:82–86. doi: 10.1038/35017565. [DOI] [PubMed] [Google Scholar]
- 10.Milot E, Strouboulis J, Trimborn T, Wijgerde M, de Boer E, Langeveld A, Tan-un K, Vergeer W, Yannoutsos N, Grosveld F, Fraser P. Cell. 1996;87:105–114. doi: 10.1016/s0092-8674(00)81327-6. [DOI] [PubMed] [Google Scholar]
- 11.Molete JM, Petrykowska H, Bouhassira EE, Feng Y-Q, Miller W, Hardison RC. Mol. Cell. Biol. 2001;21:2969–2980. doi: 10.1128/MCB.21.9.2969-2980.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Navas PA, Peterson KR, Li Q, Skarpidi E, Rohde A, Shaw SE, Clegg CH, Asano H, Stamatoyannopoulos G. Mol. Cell. Biol. 1998;17:4188–4196. doi: 10.1128/mcb.18.7.4188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bender MA, Bulger M, Close J, Groudine M. Mol. Cell. 2000;5:387–393. doi: 10.1016/s1097-2765(00)80433-5. [DOI] [PubMed] [Google Scholar]
- 14.Fiering S, Epner E, Robinson K, Zhuang Y, Telling A, Hu M, Martin DIK, Enver T, Ley TJ, Groudine M. Genes Dev. 1995;9:2203–2213. doi: 10.1101/gad.9.18.2203. [DOI] [PubMed] [Google Scholar]
- 15.Hug B, Wesselschmidt RL, Fiering S, Bender MA, Groudine M, Ley TJ. Mol. Cell. Biol. 1996;16:2906–2912. doi: 10.1128/mcb.16.6.2906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wijgerde M, Grosveld F, Fraser P. Nature. 1995;377:209–213. doi: 10.1038/377209a0. [DOI] [PubMed] [Google Scholar]
- 17.Yoshida C, Tokumasu F, Hohmura KI, Bungert J, Hayashi N, Nagasawa T, Engel JD, Yamamoto M, Takeyasu K, Igarashi K. Genes Cells. 1999;4:643–655. doi: 10.1046/j.1365-2443.1999.00291.x. [DOI] [PubMed] [Google Scholar]
- 18.Levings PP, Bungert J. Eur. J. Biochem. 2002;269:1589–1599. doi: 10.1046/j.1432-1327.2002.02797.x. [DOI] [PubMed] [Google Scholar]
- 19.Leach KM, Nightingale K, Igarashi K, Levings PP, Engel JD, Becker PB, Bungert J. Mol. Cell. Biol. 2001;21:2629–2640. doi: 10.1128/MCB.21.8.2629-2640.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tuan D, Kong S, Hu K. Proc. Natl. Acad. Sci. U. S. A. 1992;89:11219–11223. doi: 10.1073/pnas.89.23.11219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Routledge SJ, Proudfoot NJ. J. Mol. Biol. 2002;323:601–611. doi: 10.1016/s0022-2836(02)01011-2. [DOI] [PubMed] [Google Scholar]
- 22.Johnson KD, Grass JA, Parker C, Im H, Choi K, Bresnick EH. Mol. Cell. Biol. 2003;23:6484–6493. doi: 10.1128/MCB.23.18.6484-6493.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sawado T, Halow J, Bender MA, Groudine M. Genes Dev. 2003;17:1009–1018. doi: 10.1101/gad.1072303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gilbert DM. Cur. Opin. Cell Biol. 2002;14:377–383. doi: 10.1016/s0955-0674(02)00326-5. [DOI] [PubMed] [Google Scholar]
- 25.Leach KM, Vieira KF, Kang SH, Aslanian A, Teichmann M, Roeder RG, Bungert J. Nucleic Acids Res. 2003;31:1292–1301. doi: 10.1093/nar/gkg209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dubrez L, Goldwasser F, Genne P, Pommier Y, Solary E. Leukemia (Baltimore) 1995;9:1013–1024. [PubMed] [Google Scholar]
- 27.Schreiber R, Goncalves MS, Junqueria ML, Saad ST, Krieger JE, Costa FF. Braz. J. Med. Biol. Res. 2001;34:489–492. doi: 10.1590/s0100-879x2001000400008. [DOI] [PubMed] [Google Scholar]
- 28.Greenberg ME, Bender TP. In: Current Protocols in Molecular Biology. Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K, editors. John Wiley and Sons, Inc.; New York: 1997. pp. 4.10.1–4.10.11. [Google Scholar]
- 29.Kang SH, Kiefer CM, Yang TP. Mol. Cell. Biol. 2003;23:4150–4161. doi: 10.1128/MCB.23.12.4150-4161.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Elefanty AG, Robb L, Birner R, Begley CG. Blood. 1997;90:1435–1447. [PubMed] [Google Scholar]
- 31.Scherer CA, Chen J, Nachabeh A, Hopkins N, Ruley HE. Cell Growth & Differ. 1996;7:1393–1401. [PubMed] [Google Scholar]
- 32.Kitajima K, Tanaka M, Zheng J, Sakai-Ogawa E, Nakano T. Methods Enzymol. 2003;365:72–83. doi: 10.1016/s0076-6879(03)65005-6. [DOI] [PubMed] [Google Scholar]
- 33.Motohashi H, Shavit JA, Igarashi K, Yamamoto M, Engel JD. Nucleic Acids Res. 1997;25:2953–2959. doi: 10.1093/nar/25.15.2953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ashe HL, Monks J, Wijgerde M, Fraser P, Proudfoot NJ. Genes Dev. 1997;11:2494–2509. doi: 10.1101/gad.11.19.2494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim A, Dean A. Proc. Natl. Acad. Sci. U. S. A. 2004;101:7028–7033. doi: 10.1073/pnas.0307985101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Long Q, Bengra C, Li C, Kutlar F, Tuan D. Genomics. 1998;54:542–555. doi: 10.1006/geno.1998.5608. [DOI] [PubMed] [Google Scholar]
- 37.Schübeler D, MacAlpine DM, Scalzo D, Wirbelauer C, Kooperberg C, van Leeuwen F, Gottschling DE, O’Neill LP, Turner BM, Delrow J, Bell SP, Groudine M. Gene Dev. 2004;18:1263–1271. doi: 10.1101/gad.1198204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Johnson K, Christensen H, Zhao B, Bresnick EH. Mol. Cell. 2001;8:465–471. doi: 10.1016/s1097-2765(01)00309-4. [DOI] [PubMed] [Google Scholar]
- 39.Grosveld F, van Assendelft GB, Greaves DR, Kollias G. Cell. 1987;51:975–985. doi: 10.1016/0092-8674(87)90584-8. [DOI] [PubMed] [Google Scholar]
- 40.Carter D, Chakalova L, Osborne CS, Dai YF, Fraser P. Nat. Genet. 2002;32:623–626. doi: 10.1038/ng1051. [DOI] [PubMed] [Google Scholar]
- 41.Tolhuis B, Palstra RJ, Splinter E, Grosveld F, de Laat W. Mol. Cell. 2002;10:1453–1465. doi: 10.1016/s1097-2765(02)00781-5. [DOI] [PubMed] [Google Scholar]
- 42.Onishi Y, Kiyama R. J. Biol. Chem. 2003;278:8163–8171. doi: 10.1074/jbc.M209612200. [DOI] [PubMed] [Google Scholar]
- 43.Bresnick EH, Felsenfeld G. J. Biol. Chem. 1993;268:18824–18834. [PubMed] [Google Scholar]
- 44.Spicuglia S, Kumar S, Yeh J-H, Vachesz E, Chasson L, Gorbatch S, Cautres J, Ferrier P. Mol. Cell. 2002;10:1479–1487. doi: 10.1016/s1097-2765(02)00791-8. [DOI] [PubMed] [Google Scholar]
- 45.Muller HP, Schaffner W. Trends Genet. 1990;6:300–3004. doi: 10.1016/0168-9525(90)90236-y. [DOI] [PubMed] [Google Scholar]