

Abstract
Stroke induces a complex web of pathophysiology that may evolve over hours to days and weeks after onset. It is now recognized that inflammation is an important phenomenon that can dramatically influence outcomes after stroke. In this minireview, we explore the hypothesis that inflammatory signals after stroke are biphasic in nature. The high-mobility group box 1 (HMGB1) protein is discussed as an example of this idea. HMGB1 is normally present in the nucleus. Under ischemic conditions, it is released extracellularly from many types of cells. During the acute phase poststroke, HMGB1 promotes necrosis and influx of damaging inflammatory cells. However, during the delayed phase poststroke, HMGB1 can mediate beneficial plasticity and recovery in many cells of the neurovascular unit. These emerging findings support the hypothesis that inflammation after stroke can be both detrimental and beneficial, depending on the cellular situations involved.
Keywords: stroke, inflammation, stroke recovery, HMGB1
Introduction
Mechanisms of cell death and recovery after cerebral ischemia are very complicated. Energy deprivation and excitotoxity are the main causes of the initial tissue damage. Following initial injury, secondary processes of blood–brain barrier (BBB) disruption, neurovascular unit dysfunction, and postischemic inflammation further contribute to ischemic pathophysiology. These are highly heterogeneous. Not all brain cells in the ischemic territory die at the same time after stroke.1 Cellular signals participate in cross-talk between damaged cells and their adjacent tissue and trigger harmful as well as beneficial response to the stress. In the context of these crosstalk signals, poststroke inflammation has emerged as a unifying theme.2 Inflammation can clearly be detrimental as the influx of these inflammatory cells amplify brain cell death. But during recovery, inflammation may also be construed as plastic forms of tissue remodeling. Hence, inflammation can be good or bad, depending on the cellular circumstances involved. In this minireview, we use the protein called high-mobility group box 1 (HMGB1, also known as amphoterin or HMG1) as a “case study” of this concept of biphasic inflammation after stroke.
Traditionally, HMGB1 acts as a nuclear and cellular danger signal.3 HMGB1 can exert different functions depending on its cellular localization. It can be passively released from damaged cells or actively secreted from stimulated cells. Upon release, extra-cellular HMGB1 binds to its putative receptors and induces a series of signaling pathways in response to the original damage. Increasing evidence now suggest that HMGB1 is a key mediator in cerebral ischemic progression.
HMGB1 and its receptors
HMGB1, a highly conserved nonhistone nuclear DNA-binding protein, is widely expressed in most eukaryotic cells including neural cells in several animal species including humans.4 Nuclear HMGB1 is able to bind to DNA to stabilize nucleosome formation and maintains nuclear homeostasis. HMGB1 enables bending of DNA and facilitates transcription to regulate gene expression.5–7 Genetic knockout of HMGB1 causes severe energy deficits, and mice die shortly after birth owing to hypoglycemia caused by impaired glucocorticoid receptor.4 Overall, intracellular HMGB1 plays an essential role in regulating activation of basal transcriptional machinery8,9 and energy homeostasis. Although the function of HMGB1 in the nucleus is not quite understood, the role of extracellular HMGB1 has been extensively studied.
Released HMGB1 has recently been characterized as a key inflammatory mediator in response to infection, injury, and inflammation.10,11 HMGB1 acts as and acts in an autocrine/paracrine fashion. It can be secreted by activated macrophage,12 natural killer cells (NK cells),13 and myeloid dendritic cells14 in response to endotoxin and other inflammatory stimuli. Besides active secretion, HMGB1 can be released into extracellular space from damaged cells or necrotic cells.10,11 In this case, the membranes of necrotic or damaged cells become “leaky” and HMGB1, which is normally bound loosely to chromatin, diffuses from nucleus to cytoplasm, then into the extracellular matrix.
Extracellular HMGB1 acts as a trigger or modulator that affects inflammation, proliferation, migration, and cell survival. High amounts of HMGB1 have been detected in the extracellular space in various inflammatory conditions.15,16 Furthermore, elevated levels of extracellular HMGB1 are detected in cancer, suggesting that HMGB1 acting as a widespread inflammatory mediator.17,18
Release of HMGB1 is observed after traumatic brain injury and ischemic stroke. In rodent middle cerebral artery occlusion models, levels of HMGB1 in the ischemic core are immediately decreased, and in turn, serum HMGB1 is rapidly increased.10,11,19 In clinical stroke patients, HMGB1 is upregulated in serum of up to day 7 after stroke onset.20 HMGB1 is also increased in cerebrospinal fluid of subarachnoid hemorrhage patients on day 3, 7, and 14 after onset.21 In addition, plasma HMGB1 in patients is acutely elevated 30 min after severe trauma in comparison to healthy subjects.22 These findings suggest that HMGB1 is highly relevant to human stroke.
Beside passive release, active secretion of HMGB1 is observed in activated monocytes and macrophages,23 endothelial cells,24 fibroblasts,25 and brain glial cells after stroke.26,27 Whereas, the early acute increase of HMGB1 in serum may be due to the passive release of HMGB1 from necrotic or damaged and leaky cells, the late phase of HMGB1 response may be related to active secretion from functional cells. Secretion of HMGB1 can enhance acute inflammation as well as delayed stroke recovery.
HMGB1 may signal via its putative receptors, such as receptor for advanced glycation end products (RAGE), toll-like receptor-2 (TLR2), and TLR4. These receptors are expressed in brain cells.28–30 RAGE expression usually is low under normal conditions but enhanced expression of RAGE was observed in diabetic vasculature and other inflammatory diseases.29 TLR2 and TLR4 have been shown to play a critical role in infectious diseases.30 Higher constitutive expression levels of TLR4 have been found in brain regions that lack a tight BBB, such as the circumventricular organs and choroid plexus.31,32 Activation of HMGB1 receptors leads to activation of NF-κB and MAP kinase.33 Blockade of either RAGE or TLR4 results in the reduction of cytokine and nitric oxide production and decrease of inflammation,11,30 suggesting that HMGB1 potently contributes to induction of inflammation.
The role of HMGB1 may be more complex. It has been reported that HMGB1 may also possess beneficial actions, such as endothelial activation,34,35 enhancement of neurite outgrowth, and neuronal survival.36,37 In addition, reactive astrocytes release HMGB1 that promotes neurovascular recovery within peri-infarct cortex after focal cerebral ischemia in mice.38 Moreover, others have reported that highly purified recombinant HMGB1 has weak proinflammatory activity, but the formation of specific complexes between HMGB1 and other molecules, such as phospholipids, single-stranded nucleic acids, and cytokines can promote inflammation,39,40 suggesting that HMGB1 may provide multiphasic effects in a time- and local environment-dependent manner in the ischemic brain. This mini-review focuses on the role of HMGB1 in the balance between injury and repair after stroke.
HMGB1 in the acute phase after stroke
Release of HMGB1 as a paracrine signaling has been reported in the stroke patients.2 In animal models of cerebral ischemia, early HMGB1 release is observed from damaged neurons and astrocytes.5,26 Activated astrocytes and immune cells such as microglia may secrete HMGB1 after ischemia.11 Recent data suggest that injection of anti-HMGB1 antibody 6 hours after focal cerebral ischemia is still neuroprotective.41 Blockade of HMGB1 signaling prevents cell death,42,43 suggesting that HMGB1 is a potential therapeutic target for stroke.
Extracellular HMGB1 can bind to its receptors and activate a downstream pathway that leads to the upregulation of cytokines and other pro-inflammatory molecules. HMGB1 induces over-expression of TNFα, IL-1β, ICAM-1, VCAM-1, E-selectin, and iNOS in different cell types.44 Interestingly, most cytokines start to be increased a few hours after stroke,45,46 while HMGB1 release occurs in the ischemic core of striatum as early as 30 min after artery occlusion.11 Furthermore, blockade of HMGB1 reduces inflammation.11,41 These results suggest that HMGB1 is an early mediator that triggers inflammatory cascades.
TLR4, a pivotal receptor for activation of innate immunity, has been shown to play a role in ischemia. TLR4 activation results in extensive axonal and neuronal loss in a model of hypoxic ischemia that normally is below the threshold to induce measurable neurodegeneration.47 A significant reduction of inflammation and infarct area is observed in TLR4 mutant or knockout mice.28–30 TLR2 is also reported to contribute ischemia-induced cell death.30 How HMGB1 may interact with these inflammatory TLR signals should be further explored.
Released HMGB1 also interacts with RAGE. Enhancement of RAGE expression can promote the recruitment of leukocytes across endothelial barriers through its interaction with myeloid cells expressing the β2-integrin macrophage receptor 1.48 In addition, activated macrophages express HMGB1 at the cell surface,35,48 suggesting that cell surface HMGB1 may facilitate recruitment by binding RAGE at the surface of endothelial cells and enabling their translocation into the ischemic brain. Hence, blockade of HMGB1 signaling during the acute phases of stroke may be beneficial via the reduction of inflammatory infiltration.
Finally, others have reported that HMGB1 may also bind to plasminogen and tissue plasminogen activator (tPA) to amplify active plasmin and metalloproteinases (MMPs). Increase of MMPs contributes to BBB disruption and facilitates immune cell migration.49,50 Thus, HMGB1 may also influence neurovascular responses in stroke beyond inflammation per se.
Extracellular HMGB1 contributes to secondary inflammation
For many years, the central nervous system was considered an immune-privileged organ. However, it is now well known that the immune and the nervous system are engaged in bidirectional crosstalk. Accumulating data suggest that inflammation after cerebral ischemia amplifies the initial injury by linking acute responses in glial cells and cytokines to a secondary infiltration of immune cells into ischemic brain tissue.
Within the broad spectrum of immune cells, lymphocytes are known to contribute to the pathogenesis of ischemia-reperfusion (I/R) injury in several vascular beds51,52 by promoting the recruitment of other leukocyte subsets and by enhancing the microvascular dysfunction induced by I/R.51 Supporting the crucial function in I/R injury, T-lymphocytes can also be detected after cerebral ischemia in rats and even humans.53 Recently, the detrimental role of both CD4+ and CD8+ T cells has been described in an experimental stroke model in mice.54 In addition, a delayed infiltration of δγT cells at day 1–3 after cerebral ischemia contributed to promote infarction and brain damage via amplifying the inflammatory cascade in IL-17-dependence mechanism.55 After release from necrotic cells or its secretion by activated macrophages, HMGB1 induces the recruitment of inflammatory cells and mediate signals between natural killer cells (NK cells), dendritic cells (DCs), T cells, and macrophages. Indeed, HMGB1 protein contributes to the maturation of DCs56 and a proliferation of activated T lymphocytes.57 In response to tissue damage, activated NK cells that accumulate in response to HMGB1 then provide an additional source of HMGB1, thus mediating positive feedback. HMGB1 promotes the maturation of DCs. In turn, mature DCs produce HMGB1 and stimulate mature T cells. These types of signaling loops may influence immune response to tissue damage as well as secondary inflammation within the brain after stroke.
Microglial cells are the resident macrophages of the brain and play a critical role as resident immunocomponent and phagocytic cells. Microglia activated under inflammation can transform into phacocytes and release a variety of mediators that are cytotoxic or cytoprotective. Activated microglia after ischemia have the potential of releasing several pro-inflammatory cytokines, such as TNF-α, IL-1β, and IL-6, and nitric oxide, reactive oxygen species.58 On the other hand, microglia may also induce neuroprotection by producing neurotorophic factors such as brain-derived neurotorophic factor (BDNF) or insulin-like growth factor 1 (IGF1). In addition to these mediators, HMGB1 has critical roles for microglial activation after stroke. Within minutes after ischemia, HMGB1 can rapidly activate microglia.11 During the delayed phase, HMGB1 may still be detected in microglia up to days after cerebral ischemia.59 In an earlier study, we reported that early inhibition of microglia expressing HMGB1 by treatment with minocycline significantly reduced infarction,29 suggesting that HMGB1 functions as a cytokine-like mediator in a paracrine and autocrine manner and may lead to secondary brain damage in the postischemic brain.
HMGB1 in recovery and remodeling
Damaged brain can be surprisingly plastic, and crosstalk between various types of remodeling brain cells take place after brain injury.60,61 In the late phase after cerebral ischemia, the generation of new blood vessels underlies a highly coupled neurorestorative process that links neurogenesis, synaptogenesis, and angiogenesis.62,63 These phenomenon are clinically relevant—markers of neuogenesis and angiogenesis have been observed in the adult human brain.64,65
In recent years, emerging data suggest that neurovascular repair may be induced by dynamic interactions between cerebral endothelial cells, glia, neurons, and extracellular matrix from days to weeks after stroke.66 Of these neurovascular cells, astrocytes comprise the most numerous non-neuronal cell type in mammalian brain.67 Traditionally, it was assumed that reactive astrocytes after stroke or brain injury contribute to glial scarring that impedes neuronal remodeling and recovery.68 However, it is now recognized that reactive astrocytes may also release many trophic factors, such as nerve growth factor, basic fibroblast growth factor, platelet-derived growth factor, brain-derived neurotrophic factor, ciliary neurotrophic factor, Neuropilin-1, vascular endothelial growth factor, and others.69–71 Many of these trophic factors may in fact be beneficial by promoting neuronal survival and synaptogenesis, neurogenesis, and angiogenesis after stroke or brain injury.72,73
In contrast to its negative effects as described in the earlier sections, HMGB1 may also possess beneficial actions. HMGB1 signaling can promote endothelial activation35 and sprouting.34 And it has also been reported that HMGB1 may increase neurite outgrowth and cell survival in neurons.26,36,37 Intriguingly, stimulated astrocytes have been shown to induce and release HMGB1 protein into the extracellular medium.26,59 In a recent study, we showed that low levels of IL-1β potently upregulates HMGB1 in astrocytes. The prototypical MAP kinase ERK provided the upstream signal. The nuclear export protein chromosome region maintenance 1 (CRM1) enabled nuclear HMGB1 to translocate from nucleus to cytoplasm before release.27 In an in vivo study, HMGB1 immunoreactivity was also observed in reactive astrocytes, which are concentrated in the ischemic penumbra in an in vivo ischemic model.38,59 Importantly, metabolic inhibition of reactive astrocytes suppressed HMGB1-positive astrocytes in peri-infarct cortex, which disrupted various neurovascular markers, such as CD31, synaptophysin, and PSD95 that are correlated with functional recovery after cerebral ischemia in a mouse focal ischemia model.38 These findings suggest that reactive astrocytes produce HMGB1 that may promote neurovascular repair in brain after stroke.
Other studies under some pathological conditions have also shown a beneficial effect of HMGB1 in terms of its ability to recruit stem cells and promote their proliferation.74 HMGB1 was found to enable endothelial progenitor cells to home to ischemic muscle in animal models of hind limb ischemia.75 If HMGB1 was injected into the infarct area of the heart, it promoted tissue regeneration, and a significant recovery of cardiac performance was indeed mediated by RAGE signaling.76,77 More recently, it was reported that endogenous HMGB1 was crucial for ischemia-induced angiogenesis and that HMGB1 protein administration enhanced collateral blood flow in the ischemic hind limbs of diabetic mice through a VEGF-dependent mechanism.78
How might HMGB1 affect beneficial versus detrimental substrates in the remodeling brain? First, recombinant HMGB1 forms highly inflammatory complexes with ssDNA, lippopolysuccaride, IL-1β, and nucleosomes, which interact with TLR9, TLR4, IL-1R, and TLR2, respectively,79 suggesting that HMGB1 may be a candidate to expand inflammation through forming signaling complexes in the is-chemic early phase. Second, during the ischemic late phase, many kinds of cytokines are decreased in the brain. In contrast, growth factors such as VEGF then become upregulated in astrocytes in peri-infarct cortex days to weeks after stroke.80 VEGF can trigger remodeling responses in both endothelial cells and neurons.81,82 This change of brain circumstance may affect a multitude of extracellular and intracellular molecular signaling events in the neurovascular unit, which may lead to angiogenesis and brain recovery. Third, the HMGB1 receptor, RAGE pathway, may also be biphasic. For example, activation of RAGE promoted trophic effects in the nervous system, whereas hyperactivation of RAGE promoted neuronal apoptosis.36 How RAGE signaling is modulated by HMGB1 during stroke recovery is not fully understood. The biphasic effect of HMGB1 may be induced by both receptor-dependent and/or receptor-independent mechanisms. Taken together, the multifunctional effect of HMGB1 may be modulated by its interaction with brain environment, each activated cells, and its receptors. During stroke recovery, HMGB1 may be a key molecule in enhancing the interaction between neurovascular cells in ischemic brain.
Conclusions
Extensive data from molecular, cellular, and animal models support a potent proinflammatory effect of HMGB1 in acute ischemic stroke. In fact, the therapeutic effect of the monoclonal HMGB1 antibody has been described in several animal models of cerebral ischemia. However, it is now important to realize that like other inflammatory mediators, HMGB1 may play complex and biphasic roles after stroke (Fig. 1). During delayed phases of stroke recovery, extracellular HMGB1 may enhance the crosstalk among neurovascular cells, such as neurons, astrocytes, and endothelial cells. Moreover, HMGB1 may contribute to stem cell migration into the ischemic region and promote stroke recovery through neurogenesis, vasculogenesis, and angiogenesis. Ultimately, a more nuanced approach to modifying the HMGB1 response after stroke may be needed in order to optimize inhibition during acute stages of injury without interfering with beneficial endogenous mechanisms of neurovascular remodeling.
Figure 1.
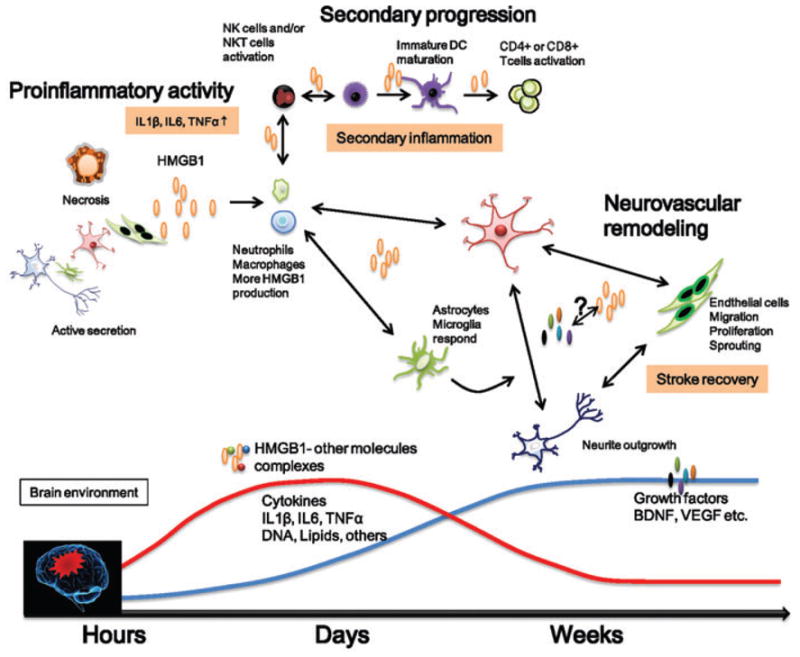
Schematic to summarize the multiphasic roles of HMGB1 after stroke. Acute effects involve cytokine up-regulation and infiltration of neutrophils and macrophages into the brain. HMGB1 can form complexes with other molecules, such as DNA, lipids, and IL-1β. Secondary inflammation may be caused by stimulated T lymphocytes. Delayed actions involve neurovascular remodeling and neurovasculogenesis that may contribute to stroke recovery, as beneficial growth factors such as BDNF and VEGF become elevated. Dissecting the milleu of cytokines, trophic factors, attendant cells, and neurovascular homeostasis may eventually allow us to understand the regulation and transition of detrimental versus beneficial signaling actions of HMGB1 and other inflammatory signals in the brain after stroke.
Acknowledgments
Supported in part by NIH grants, the American Heart Association, the Deane Institute, the Mochida Memorial Foundation for Medical and Pharmaceutical Research, and a Postdoctoral Fellowship for Research Abroad from the Japan Society for the Promotion of Science.
Footnotes
Conflicts of interest
Authors declare no conflicts of interest.
References
- 1.Thomas JO. HMG1 and 2: architectural DNA-binding proteins. Biochem Soc Trans. 2001;29:395–401. doi: 10.1042/bst0290395. [DOI] [PubMed] [Google Scholar]
- 2.Goldstein RS, Gallowitsch-Puerta M, Yang L, et al. Elevated high-mobility group box 1 levels in patients with cerebral and myocardial ischemia. Shock. 2006;25:571–574. doi: 10.1097/01.shk.0000209540.99176.72. [DOI] [PubMed] [Google Scholar]
- 3.Lotze MT, Tracy KJ. High-mobility group box 1 protein (HMGB1): nuclear weapon in the immune arsenal. Nat Rev Neurosci. 2005;5:331–342. doi: 10.1038/nri1594. [DOI] [PubMed] [Google Scholar]
- 4.Yang H, Wang H, Czura CJ, Tracey KJ. The cytokine activity of HMGB1. J Leukoc Biol. 2005;78:1–8. doi: 10.1189/jlb.1104648. [DOI] [PubMed] [Google Scholar]
- 5.Pasqualini JR, Sterner R, Mercat P, Allfrey VG. Estradiol enhanced acetylation of nuclear high mobilitygroup proteins of the uterus of newborn guinea pigs. Biochem Biophys Res Commun. 1989;161:1260–1266. doi: 10.1016/0006-291x(89)91378-8. [DOI] [PubMed] [Google Scholar]
- 6.Prendergast P, Onate SA, Christensen K, Edwards DP. Nuclear accessory factors enhance the binding of progesterone receptor to specific target DNA. J Steroid Biochem Mol Biol. 1994;48:1–13. doi: 10.1016/0960-0760(94)90245-3. [DOI] [PubMed] [Google Scholar]
- 7.Zhang CC, Krieg S, Shapiro DJ. HMG-1 stimulates estrogen response element binding by estrogen receptor from stably transfacted HeLa cells. Mol Endocrinol. 1999;13:632–643. doi: 10.1210/mend.13.4.0264. [DOI] [PubMed] [Google Scholar]
- 8.Ellwood KB, Yen YM, Johnson RC, Carey M. Mechanism for specificity by HMG-1 in enhanceosome assembly. Mol Cell Biol. 2000;20:4359–4370. doi: 10.1128/mcb.20.12.4359-4370.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Verrijdt G, Haelens A, Schoenmakers E, et al. Comparative analysis of the influence of the high-mobility group box 1 protein on DNA binding and transcriptional activation by the androgen, glucocorticoid, progesterone and mineralcorticoid receptors. Biochem J. 2002;361:97–103. doi: 10.1042/0264-6021:3610097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Qiu J, Nishimura M, Wang Y, et al. Early release of HMGB-1 from neurons after the onset of brain ischemia. J Cereb Blood Flow Metab. 2008;28:927–928. doi: 10.1038/sj.jcbfm.9600582. [DOI] [PubMed] [Google Scholar]
- 11.Kim JB, Sig J, Choi, Yu YM, et al. HMGB1, a novel cytokine-like mediator linking acute neuronal death and delayed neuroinflammation in the postischemic brain. J Neurosci. 2006;26:6413–6421. doi: 10.1523/JNEUROSCI.3815-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bonaldi T, Talamo F, Scaffidi P, et al. Monocytic cells hyperacetylate chromatin protein HMGB1 to redirect it towards secretion. EMBO J. 2003;22:5551–5560. doi: 10.1093/emboj/cdg516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Semino C, Angelini G, Poggi A, Rubartelli A. NK/iDC interaction results in IL-18 secretion by DCs at the synaptic cleft followed by NK cell activation and release of the DC maturation factor HMGB1. Blood. 2005;106:609–616. doi: 10.1182/blood-2004-10-3906. [DOI] [PubMed] [Google Scholar]
- 14.Dumitriu IE, Bianchi ME, Bacchi M, et al. The secretion of HMGB1 is required for the migration of maturing dendritic cells. J Leukoc Biol. 2007;81:84–91. doi: 10.1189/jlb.0306171. [DOI] [PubMed] [Google Scholar]
- 15.Wang H, Bloom O, Zhang M, et al. HMG-1 as a late mediator of endotoxin lethality in mice. Science. 1999;285:248–251. doi: 10.1126/science.285.5425.248. [DOI] [PubMed] [Google Scholar]
- 16.Tsung A, Sahai R, Tanaka H, et al. The nuclear factor HMGB1 mediates hepatic injury after murine liver ischemia-reperfusion. J Exp Med. 2005;201:1135–1143. doi: 10.1084/jem.20042614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hatada T, Wada H, Nobori T, et al. Plasma concentrations and importance of high mobility group box protein in the prognosis of organ failure in patients with disseminated intravascular coagulation. Thromb Haemost. 2005;94:975–979. doi: 10.1160/TH05-05-0316. [DOI] [PubMed] [Google Scholar]
- 18.Pachot A, Monneret G, Voirin N, et al. Longitudinal study of cytokine and immune transcription factor mRNA expression in septic shock. Clin Immunol. 2005;114:61–69. doi: 10.1016/j.clim.2004.08.015. [DOI] [PubMed] [Google Scholar]
- 19.Hayakawa K, Mishima K, Nozako M, et al. Delayed treatment with minocycline improves neurological impairment via activated microglia expressing high-mobility group box1 inhibiting mechanism. Stroke. 2008;39:951–958. doi: 10.1161/STROKEAHA.107.495820. [DOI] [PubMed] [Google Scholar]
- 20.Vogelgesang A, V, May E, Grunwald U, et al. Functional status of peripheral blood T cells in ischemic stroke patients. PLoS One. 2010;5:e8718. doi: 10.1371/journal.pone.0008718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nakahara T, Tsuruta R, Kaneko T, et al. High-mobility group box1 protein in CSF of patients with sub-arachnoid hemorrhage. Neurocrit Care. 2009;11:362–368. doi: 10.1007/s12028-009-9276-y. [DOI] [PubMed] [Google Scholar]
- 22.Cohen MJ, Brohi K, Calfee CS, et al. Early release of high mobility group box nuclear protein 1 after severe trauma in humans: role of injury severity and tissue hypoperfusion. Crit Care. 2009;13:R174. doi: 10.1186/cc8152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bonaldi T, Talamo F, Scaffidi P, et al. Monocytic cells hyperacetylate chromatin protein HMGB1 to redirect it towards secretion. EMBO J. 2003;22:5551–5560. doi: 10.1093/emboj/cdg516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kawahara K, Hashiguch T, Kikuchi K, et al. Induction of high mobility group box 1 release from serotonin-stimulated human umbilical vein endothelial cells. Int J Mol Med. 2008;22:639–644. [PubMed] [Google Scholar]
- 25.Feghali K, Iwasaki K, Tanaka K, et al. Human gingival fibroblasts release high-mobility group box-1 protein through active and passive pathways. Oral Microbial Immunol. 2009;24:292–298. doi: 10.1111/j.1399-302X.2009.00508.x. [DOI] [PubMed] [Google Scholar]
- 26.Passalacqua M, Patrone M, Picotti GB, et al. Stimulated astrocytes release high-mobility group 1 protein, an inducer of LAN-5 neuroblastoma cell differentiation. Neuroscience. 1998;82:1021–1028. doi: 10.1016/s0306-4522(97)00352-7. [DOI] [PubMed] [Google Scholar]
- 27.Hayakawa K, Arai K, Lo EH. Role of ERK map kinase and CRM1 in IL-1beta-stimulated release of HMGB1 from cortical astrocytes. Glia. 2010;58:1007–1015. doi: 10.1002/glia.20982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Caso JR, Pradillo JM, Hurtado O, et al. Toll-like receptor 4 is involved in brain damage and inflammation after experimental stroke. Circulation. 2007;115:1599–1608. doi: 10.1161/CIRCULATIONAHA.106.603431. [DOI] [PubMed] [Google Scholar]
- 29.Hua F, Ma J, Ha T, et al. Activation of Toll-like receptor 4 signaling contributes to hippocampal neuronal death following global cerebral ischemia/reperfusion. J Neuroimmunol. 2007;190:101–111. doi: 10.1016/j.jneuroim.2007.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tang SC, Arumugam TV, Xu X, et al. Pivotal role for neuronal Toll-like receptors in ischemic brain injury and functional deficits. Proc Natl Acad Sci USA. 2007;104:13798–13803. doi: 10.1073/pnas.0702553104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mishra BB, Mishra PK, Teale JM. Expression and distribution of Toll-like receptors in the brain during murine neurocysticercosis. J Neuroimmunol. 2006;181:46–56. doi: 10.1016/j.jneuroim.2006.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Laflamme N, Echchannaoui H, Landmann R, Rivest S. Cooperation between toll-like receptor 2 and 4 in the brain of mice challenged with cell wall components derived from gram-negative and gram-positive bacteria. Eur J Immunol. 2003;33:1127–1138. doi: 10.1002/eji.200323821. [DOI] [PubMed] [Google Scholar]
- 33.Huttunen HJ, Fages C, Rauvala H. Receptor for advanced glycation end products (RAGE)-mediated neurite outgrowth and activation of NF-kappaB require the cytoplasmic domain of the receptor but different downstream signaling pathways. J Biol Chem. 1999;274:19919–19924. doi: 10.1074/jbc.274.28.19919. [DOI] [PubMed] [Google Scholar]
- 34.Schlueter C, Weber H, Meyer B, et al. Angiogenetic signaling through hypoxia: HMGB1: an angiogenetic switch molecule. Am J Pathol. 2005;166:1259–1263. doi: 10.1016/S0002-9440(10)62344-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Treutiger CJ, Mullins GE, Johansson AS, et al. High mobility group 1 B-box mediates activation of human endothelium. J Intern Med. 2003;254:375–385. doi: 10.1046/j.1365-2796.2003.01204.x. [DOI] [PubMed] [Google Scholar]
- 36.Huttunen HJ, Kuja-Panula J, Sorci G, et al. Coregulation of neurite outgrowth and cell survival by amphoterin and s100 proteins through receptor for advanced glycation end products (RAGE) activation. J Biol Chem. 2000;275:40096–40105. doi: 10.1074/jbc.M006993200. [DOI] [PubMed] [Google Scholar]
- 37.Huttunen HJ, Kuja-Panula J, Rauvala H. Receptor for advanced glycation end products (RAGE) signaling induces CREB-dependent chromogranin expression during neuronal differentiation. J Biol Chem. 2002;41:38635–38646. doi: 10.1074/jbc.M202515200. [DOI] [PubMed] [Google Scholar]
- 38.Hayakawa K, Nakano T, Irie K, et al. Inhibition of reactive astrocytes with fluorocitrate retards neurovascular remodeling and recovery after focal cerebral ischemia in mice. J Cereb Blood Flow Metab. 2010;30:871–882. doi: 10.1038/jcbfm.2009.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rouhiainen A, Tumova S, Valmu L, et al. Analysis of proinflammatory activity of highly purified eukaryotic recombinant HMGB1 (Amphoterin) J Leukoc Biol. 2006;81:49–58. doi: 10.1189/jlb.0306200. [DOI] [PubMed] [Google Scholar]
- 40.Bianchi ME. DAMPs, PAMPs and alarmins: all we need to know about danger. J Leukoc Biol. 2007;81:1–5. doi: 10.1189/jlb.0306164. [DOI] [PubMed] [Google Scholar]
- 41.Liu K, Mori S, Takahashi HK, et al. Anti-high mobility group box1 monoclonal antibody ameliorates brain infarction induced by transient ischemia in rats. FASEB J. 2007;21:3904–3916. doi: 10.1096/fj.07-8770com. [DOI] [PubMed] [Google Scholar]
- 42.Scaffidi P, Misteli T, Bianchi ME. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature. 2002;418:191–195. doi: 10.1038/nature00858. [DOI] [PubMed] [Google Scholar]
- 43.Kim JS. Cytokines and adhesion molecules in stroke and related diseases. Review. J Neurol Sci. 1996;137:69–78. doi: 10.1016/0022-510x(95)00338-3. [DOI] [PubMed] [Google Scholar]
- 44.Horie Y, Wolf R, Chervenak RP, et al. T-lymphocytes contribute to hepatic leukostasis and hypoxic stress induced by gut ischemia-reperfusion. Microcirculation. 1999;6:267–280. [PubMed] [Google Scholar]
- 45.Maddahi A, Edvinsson L. Cerebral ischemia induces microvascular pro-inflammatory cytokine expression via the MEK/ERK pathway. J Neuroinflammation. 2010;7:14. doi: 10.1186/1742-2094-7-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cai Hongxin, Luan Y, Wang X, Xia Z. Changes in tumor necrosis factor alpha and myeloper-oxidase in mouse models of local cerebral infarction induced by photochemical mothod. Neural Regen Res. 2007;2:38–41. [Google Scholar]
- 47.Lehnardt S, Massillon L, Follett P, et al. Activation of innate immunity in the CNS triggers neurodegeneration through a Toll-like receptor 4-dependent pathway. Proc Natl Acad Sci USA. 2003;100:8514–8519. doi: 10.1073/pnas.1432609100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rouhiainen A, Kuja-Panula J, Wilkman E, et al. Regulation of monocyte migration by amphoterin (HMGB1) Blood. 2004;104:1174–1182. doi: 10.1182/blood-2003-10-3536. [DOI] [PubMed] [Google Scholar]
- 49.Parkkinen J, Rauvala H. Interactions of plasmino-gen and tissue plasminogen activator (t-PA) with amphoterin. Enhancement of t-PA-catalyzed plasminogen activation by amphoterin. J Biol Chem. 1991;266:16730–16735. [PubMed] [Google Scholar]
- 50.Taguchi A, Blood DC, del Toro G, et al. Blockade of RAGE-amphoterin signalling suppresses tumour growth and metastases. Nature. 2000;405:354–360. doi: 10.1038/35012626. [DOI] [PubMed] [Google Scholar]
- 51.Shigematsu T, Wolf RE, Granger DN. T-lymphocytes modulate the microvascular and inflammatory responses to intestinal ischemia-reperfusion. Microcirculation. 2002;9:99–109. doi: 10.1038/sj/mn/7800126. [DOI] [PubMed] [Google Scholar]
- 52.Yokota N, Daniels F, Crosson J, et al. Protective effect of T cell depletion in murine renal ischemia-reperfusion injury. Transplantation. 2002;74:759–763. doi: 10.1097/00007890-200209270-00005. [DOI] [PubMed] [Google Scholar]
- 53.Jander S, Kraemer M, Schroeter M, et al. Lymphocytic infiltration and expression of intercellular adhesion molecule-1 in photochemically induced ischemia of the rat cortex. J Cereb Blood Flow Metab. 1995;15:42–51. doi: 10.1038/jcbfm.1995.5. [DOI] [PubMed] [Google Scholar]
- 54.Yilmaz G, Arumugam TV, Stokes KY, Granger DN. Role of T lumphocytes and interferon-gamma in is-chemic stroke. Circulation. 2006;113:2105–2112. doi: 10.1161/CIRCULATIONAHA.105.593046. [DOI] [PubMed] [Google Scholar]
- 55.Sichita T, Sugiyama Y, Ooboshi H, et al. Pivotal role of cerebral interleukin-17-producing gammadelta T cells in the delayed phase of ischemic brain injury. Nat Med. 2009;15:946–950. doi: 10.1038/nm.1999. [DOI] [PubMed] [Google Scholar]
- 56.Blanco P, Palucka K, Pascual V, Banchereau J. Dendritic cells and cytokines in human inflammatory and autoimmune disease. Review. Cytokine Growth Factor Rev. 2008;19:41–52. doi: 10.1016/j.cytogfr.2007.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sundberg E, Fasth AER, Palmblad K, et al. High mobility group box chromosomal protein 1 acts as a proliferation signal for activated T lymphocytes. Immunobiology. 2009;214:303–309. doi: 10.1016/j.imbio.2008.09.006. [DOI] [PubMed] [Google Scholar]
- 58.Lucas SM, Rothwell NJ, Gibson RM. Brain inflammation and adult neurogenesis: the dual role of microglia. Br J Pharmacol. 2006;147:S232–S240. doi: 10.1038/sj.bjp.0706400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kim JB, Lim CM, Yu YM, Lee JK. Induction ad subcellular localization of high-mobility group box-1 (HMGB1) in the postischemic rat brain. J Neurosci Res. 2008;86:1125–1131. doi: 10.1002/jnr.21555. [DOI] [PubMed] [Google Scholar]
- 60.Chen Y, Swanson RA. Astrocytes and brain injury. J Cereb Blood Flow Metab. 2003;23:137–149. doi: 10.1097/01.WCB.0000044631.80210.3C. [DOI] [PubMed] [Google Scholar]
- 61.Chopp M, Zhang ZG, Jiang Q. Neurogenesis, angiogenesis, and MRI induces of functional recovery from stroke. Stroke. 2007;38:827–831. doi: 10.1161/01.STR.0000250235.80253.e9. [DOI] [PubMed] [Google Scholar]
- 62.Zhang ZG, Zhang L, Jiang Q, et al. VEGF enhances angiogenesis and promotes blood-brain barrier leakage in the ischemic brain. J Clin Invest. 2000;106:829–838. doi: 10.1172/JCI9369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sun Y, Jin K, Xie L, et al. VEGF-induced neuropro-tection, neurogenesis, and angiogenesis after focal cerebral ischemia. J Clin Invest. 2003;111:1843–1851. doi: 10.1172/JCI17977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Krupinski J, Kaluza J, Kumar P, et al. Some remarks on the growth-rate and angiogenesis of microvessels in ischemic stroke. Morphometric and immunocytochemical studies. Patol Pol. 1993;44:203–209. [PubMed] [Google Scholar]
- 65.Minger SL, Ekonomou A, Carta EM, et al. Endogeneous neurogenesis in the human brain following cerebral ischemia. Regen Med. 2007;2:69–74. doi: 10.2217/17460751.2.1.69. [DOI] [PubMed] [Google Scholar]
- 66.Arai K, Jin G, Navaratna D, Lo EH. Brain angio-genesis in developmental and pathological processes: neurovascular injury and angiogenic recovery after stroke. FEBS J. 2009;276:4644–4652. doi: 10.1111/j.1742-4658.2009.07176.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tower DB, Young OM. The activities of butyryl-cholinesterase and carbonic anhydrase, the rate of anaerobic glycolysis, and the question of a constant density of glial cells in cerebral cortices of various mammalian species from mouse to whale. J Neurosci. 1973;20:269–278. doi: 10.1111/j.1471-4159.1973.tb12126.x. [DOI] [PubMed] [Google Scholar]
- 68.Silver J, Miller JH. Regeneration beyond the glial scar. Nat Rev Neurosci. 2004;5:146–156. doi: 10.1038/nrn1326. [DOI] [PubMed] [Google Scholar]
- 69.Strauss S, Otten U, Joggerst B, et al. Increased levels of nerve growth factor (NGF) protein and mRNA and reactive gliosis following kainic acid injection into the rat striatum. Neurosci Lett. 1994;168:193–196. doi: 10.1016/0304-3940(94)90448-0. [DOI] [PubMed] [Google Scholar]
- 70.Mocchetti I, Wrathall JR. Neurotrophic factors in central nervous system trauma. J Neurotrauma. 1995;12:853–870. doi: 10.1089/neu.1995.12.853. [DOI] [PubMed] [Google Scholar]
- 71.Tokita Y, Keino H, Matsui F, et al. Regulation of neuregulin expression in the injured rat brain and cultured astrocytes. J Neurosci. 2001;21:1257–1264. doi: 10.1523/JNEUROSCI.21-04-01257.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Horner PJ, Gage FH. Regenerating the damaged central nervous system. Nature. 2000;407:963–970. doi: 10.1038/35039559. [DOI] [PubMed] [Google Scholar]
- 73.Panickar KS, Norenberg MD. Astrocytes in cerebral ischemic injury: morphological and general considerations. Glia. 2005;50:287–298. doi: 10.1002/glia.20181. [DOI] [PubMed] [Google Scholar]
- 74.Palumbo R, Bianchi ME. High mobility group box 1 protein, a cue for stem cell recruitment. Review. Biochem Pharmacol. 2004;68:1165–1170. doi: 10.1016/j.bcp.2004.03.048. [DOI] [PubMed] [Google Scholar]
- 75.Chavakis E, Hain A, Vinci M, et al. High-mobility group box 1 activates integrin-dependent homing of endothelial progenitor cells. Circ Res. 2007;100:204–212. doi: 10.1161/01.RES.0000257774.55970.f4. [DOI] [PubMed] [Google Scholar]
- 76.Limana F, Germani A, Zacheo A, et al. Exogenous high-mobility group box 1 protein induces myocardial regeneration after infarction via enhanced cardiac C-kit+ cell proliferation and differentiation. Circ Res. 2005;97:e73–e83. doi: 10.1161/01.RES.0000186276.06104.04. [DOI] [PubMed] [Google Scholar]
- 77.Germani A, Limana F, Capogrossi MC. Pivotal advances: high-mobility group box 1 protein—a cytokine with a role in cardiac repair. J Leukoc Biol. 2007;81:41–45. doi: 10.1189/jlb.0306165. [DOI] [PubMed] [Google Scholar]
- 78.Biscetti F, Straface G, De Cristofaro R, et al. High-mobility group box-1 protein promotes angiogenesis after peripheral ischemia in diabetic mice through a VEGF-dependent mechanism. Diabetes. 2010;59:1496–1505. doi: 10.2337/db09-1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bianchi ME. HMGB1 loves company. Review. J Leukoc Biol. 2009;86:573–576. doi: 10.1189/jlb.1008585. [DOI] [PubMed] [Google Scholar]
- 80.Zhao BQ, Wang S, Kim HY, et al. Role of matrix metalloproteinases in delayed cortical responses after stroke. Nat Med. 2006;12:441–445. doi: 10.1038/nm1387. [DOI] [PubMed] [Google Scholar]
- 81.Hansen TM, Moss AJ, Brindle NP. Vascular endothelial growth factor and angiopoietins in neurovascular regeneration and protection following stroke. Curr Neurovasc Res. 2008;5:235–244. doi: 10.2174/156720208786413433. [DOI] [PubMed] [Google Scholar]
- 82.Fagan SC, Hess DC, Hohnadel EJ, et al. Targets for vascular protection after acute ischemic stroke. Stroke. 2004;35:2220–2225. doi: 10.1161/01.STR.0000138023.60272.9e. [DOI] [PubMed] [Google Scholar]