

Abstract
The Nlrp3 inflammasome is activated in response to an array of environmental and endogenous molecules leading to caspase-1-dependent IL-1β processing and secretion by myeloid cells. Several identified Nlrp3 inflammasome activators also trigger reactive oxygen species (ROS) production. However, the initial concept that NADPH oxidases are the primary source of ROS production during inflammasome activation is becoming less accepted. Therefore, the importance of mitochondrial-derived ROS has been recently explored. In this study, we explore the impact of mitochondria dysfunction and ROS production on Nlrp3 inflammasome stimulation and IL-1β secretion induced by serum amyloid A (SAA) in primary mouse peritoneal macrophages. To induce mitochondrial dysfunction, we utilized antimycin A, which blocks electron flow at complex III, and carbonyl cyanide-p-trifluoromethoxyphenylhydrazone (FCCP), a mitochondrial oxidative phosphorylation uncoupler. We also utilized a superoxide dismutase (SOD) mimetic, MnTBAP, which targets the mitochondria, as well as the broad spectrum antioxidants DPI (diphenyleneiodonium chloride) and ebselen. Our findings demonstrate that SAA alone induces mitochondrial ROS in a time-dependent manner. We observed that MnTBAP and ebselen blocked IL-1β secretion caused by SAA only when added prior to stimulation, and DPI augmented IL-1β secretion. Surprisingly, these effects were not directly related to intracellular or mitochondrial ROS levels. We also found that mitochondrial-targeted drugs increased IL-1β secretion regardless of their impact on mitochondrial function and ROS levels, suggesting that mitochondrial ROS-dependent and -independent mechanisms play a role in the Nlrp3 inflammasome - IL-1β secretion axis in SAA-stimulated cells. Finally, we found that FCCP significantly sustained the association of the Nlrp3 inflammasome complex, which could explain the most robust effect among the drugs tested in enhancing IL-1β secretion in SAA-treated cells. Overall, our data suggest that the Nlrp3 inflammasome - IL-1β secretion axis is a very highly-regulated inflammatory pathway that is not only susceptible to changes in mitochondrial or intracellular ROS, but also to changes in overall mitochondrial function.
Keywords: Nlrp3 inflammasome, IL-1β, ROS, mitochondria, ATP, MnTBAP, DPI, FCCP, antimycin A, serum amyloid A, mouse peritoneal macrophages
Introduction
The Interleukin 1 (IL-1) family of cytokines is critical to the host response to infection, participating not only in the acute phase response from the liver, but also in alterations of metabolism, induction of fever, and leukocyte activation [1]. Overproduction of IL-1β, in particular, is thought to be responsible for a variety of autoinflammatory syndromes, including familial Mediterranean fever and Muckle-Wells syndrome, and is also a contributing factor in rheumatoid arthritis, gout, multiple sclerosis, Alzheimer’s Disease, and diabetes [2–7]. IL-1β is also a pathogenic mediator in several pulmonary disorders, including infection, asthma, ALI/ARDS, transplant rejection, COPD, PAH, sarcoidosis, asbestosis, and silicosis [8–10]. Setting IL-1β apart from other acute phase cytokines such as IL-6 and TNF-α is the requirement for processing from an inactive pro-form to an active secreted form by caspase-1-dependent cleavage.
The assembly of a cytoplasmic inflammasome complex facilitates the formation of a molecular platform for caspase-1-dependent secretion of IL-1β. Nlrp3 is a component of inflammasomes that are activated in response of to an array of environmental and endogenous molecules [11]. It has been suggested that the mechanism of IL-1β processing and secretion requires sequential steps. The initial event necessary is the synthesis and accumulation of the precursor proteins, including pro–IL-1β and Nlrp3 (“signal 1”), accomplished by several stimuli. These include microbial products such as pathogen-associated molecular pattern molecules (PAMPs), which trigger pathogen recognition receptors (PRRs) such as Toll-like receptors (TLRs). Following PRR triggering, Nlrp3 activation leads to recruitment of the adaptor protein ASC (apoptosis-associated speck-like protein containing a CARD) and the enzyme caspase-1 to form the Nlrp3 inflammasome complex (“signal 2”), which ultimately is responsible for the cleavage and secretion of IL-1β [12]. In addition, the cleavage and secretion of IL-1β can be enhanced by release of endogenous ATP that stimulates the purinergic receptor P2X7 [13]. Interestingly, several identified Nlrp3 inflammasome activators also trigger reactive oxygen species (ROS) production. Furthermore, activation of P2X7 is accompanied by production of ROS, generated at least in part by NADPH oxidases [14, 15]. In the context of redox regulation of target proteins, the cellular location and quantity of ROS generated appears to dictate the response. Overall, several studies using antioxidants support a model in which ROS production induced by Nlrp3 agonists drive inflammasome assembly [16].
The initial concept that NADPH oxidases are the primary source of ROS production during inflammasome activation [14] is becoming less accepted. Two independent studies utilizing mononluclear phagocytes from patients with chronic granulomatous disease demonstrated IL-1β secretion upon stimulation despite an inability of these cells to generate NADPH oxidase-dependent ROS, due to the mutation of p47phox subunit [17, 18]. Since NADPH oxidase is not the only source of ROS in cells, the importance of mitochondrial-derived ROS has been recently explored. The mitochondria is the main source of ROS under physiological condition, however, under conditions of cellular stress, including increases in metabolic rates, hypoxia or cellular disruption, the mitochondria can generate increased amount of ROS [19]. In fact, blockage of key enzymes of the respiratory chain leads to ROS generation and consequent Nlrp3 inflammasome activation [20]. Despite several high-profile publications in the field, the mechanisms of production and the nature of ROS involvement in Nlrp3 inflammasome activation remain the subject of intense scrutiny.
Herein, we investigated the effects of mitochondria-derived ROS on Nlrp3 inflammasome activation in murine macrophages stimulated with serum amyloid A (SAA). Whereas SAA has been known as a biomarker of inflammation in several types of diseases [21], it can also stimulate cells via toll like receptor 2 (TLR2) and evoke a robust signaling cascade in human monocytes [22] and mouse macrophages [23]. More recently, we have demonstrated that SAA instillation into the lungs of mice elicits robust TLR2-, MyD88-, and IL-1–dependent pulmonary neutrophilic inflammation. We also demonstrated that in vitro SAA drives TLR2 and MyD88 dependent production of IL-1α, IL-1β, IL-6, IL-23, and PGE2 by macrophages and dendritic cells, (DC). Furthermore, the production of IL-1β response to SAA requires Nlrp3, ASC and caspase-1 both in vitro and in vivo [24]. In this study, we report that SAA induces mitochondrial ROS and that uncoupling mitochondrial oxidative phosphorylation enhances Nlrp3 activation by preventing disassembly of the Nlrp3 complex.
2. Materials and Methods
2.1. Chemicals and Reagents
Ebselen, antimycin A, diphenyleneiodonium chloride (DPI), carbonyl cyanide p trifluoromethoxyphenylhydrazone (FCCP), digitonin, adenosine 5′-triphosphate, periodate oxidized sodium salt (oxATP), apyrase, 4,4′,4″,4‴-(Porphine-5,10,15,20-tetrayl)tetrakis(benzoic acid) (TBAP), protease inhibitor cocktail, and anti β-actin antibody were purchased from Sigma. Mn(III)tetrakis(4-Benzoic acid)porphyrin Chloride (MnTBAP), a superoxide dismutase (SOD) mimetic, was purchased from Calbiochem. The ROS detection reagent 2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA), the mitochondrial ROS indicator (MitoSOX Red), and the mitochondrion-selective probe (MitoTracker green) were purchased from Molecular Probes/Invitrogen. The mitochondrial membrane potential indicator, 5,5′,6,6′ tetrachloro-1,1′,3,3′-tetraethylbenzimidazol-carbocyanine iodide (JC-1) was purchased from Cayman. The ATP bioluminescence assay kit (StayBrite) was purchased from BioVision. Ultra pure E. coli 0111:B4 lipopolysaccharide (LPS) and human recombinant serum amyloid A (SAA) were purchased from InvivoGen, and Peprotech, respectively. Nlrp3 and ASC antibodies were purchased from Enzo, glutathione antibody was purchased from Virogen, antibodies for caspase-1 and IL-1β detection were purchased from Santa Cruz, and cytochrome c antibody clone 7H8.2C12 was purchased from BD Pharmingen. Secondary antibodies (anti mouse HRP and anti rabbit HRP) were obtained from GE Healthcare. Dynabeads® Protein G for immunoprecipitation was purchased from Invitrogen. IL-1β ELISA kits were purchased from BD Biosciences.
2.2. Cell culture and experimental conditions
Mice were housed in an American Association for the Accreditation of Laboratory Animal Care approved facility at University of Vermont, maintained on a 12-h light/dark cycle and were provided food and water ad libitum. All animal studies were approved by the University of Vermont Institutional Animal Care and Use Committee. For the isolation of primary macrophages, C57BL/6 mice were administered 1 ml of 4% thioglycollate by i.p. injection. Ninety-six hours later, mice were euthanized and peritoneal lavage was performed to collect peritoneal exudate cells. For isolation of human monocytes, peripheral blood of healthy donors was freshly drawn and the monocytes were cultured for 24h prior to stimulation. Human monocytes, mouse peritoneal macrophage cell line IC-21 (obtained from ATCC), or primary mouse macrophages were seeded at 106 cells/mL using RPMI 1640 medium supplemented with 10% FBS, penicillin, streptomycin and L-glutamine, and incubated at 37°C in a 5% CO2-supplemented atmosphere for at least 16 h before the appropriate treatments. SAA (1 μg/mL) or LPS (100 ng/mL) were utilized to stimulate primary human and mouse monocytes or IC-21 cells, respectively, at the indicated times. The antioxidants DPI (10 μM), ebselen (10 μM) or MnTBAP (100 μM) or the respective control TBAP (100 μM), and the mitochondria targeting drugs antimycin A (40 μg/mL) and FCCP (10 μM), apyrase (10U/mL) and oxidized ATP (oxATP, 100 μM) were added prior to stimulation or to stimulated cells at the indicated times.
2.3. Immunoprecipitation and Western blot analysis
After respective treatments, tissue culture plates were placed on ice and the attached cells were rinsed once with cold PBS and lysed using RIPA lysis buffer (50 mM Tris, pH 7.4, 150 mM NaCl, 0.1% SDS, 1% NP-40, 0.5% sodium deoxycholate and protease inhibitor cocktail (Sigma)). Lysates were removed from the plate, transferred to microcentrifuge tubes, and immediately frozen in liquid nitrogen to complete the lysis and prevent protein degradation. Alternatively, cell supernatants (medium) were concentrated by precipitation with 10% trichloroacetic acid (TCA) followed by centrifugation at 1400 rpm for 20 min at RT. The precipitates were neutralized and proteins resuspended in loading buffer [25]. Cell lysates or precipitates were equally loaded and proteins were separated on 12% sodium dodecyl sulfate polyacrylamide gels (SDS-PAGE) and transferred from the gel to a nitrocellulose membrane. The membrane was blocked for 1h at RT with 3% milk and then incubated with respective antibodies overnight at 4°C on a rotating platform. The membranes were washed 3 times in PBS containing 0.05% tween-20 (PBST) and incubated with respective horseradish peroxidase-conjugated secondary antibodies. Western blots were developed utilizing chemiluminescence (ECL kit by Pierce), according manufacturer’s instructions.
To analyze the interactions among proteins comprising the Nlrp3-inflammasome complex, cell lysates were prepared as described above and caspase-1 was immunoprecipitated from at least 250 μg of total lysates using 1 μg of antibody. Interactions with Nlrp3 or IL-1β were determined by Western blotting. Alternatively, anti-ASC was utilized for immunoprecipitation and Nlrp3 was detected by Western blotting as above.
2.4. Isolation of mitochondrial fraction and cytochrome c analysis by Western blot
Isolation of the mitochondrial fraction from adherent cells was performed as previously described [26]. Briefly, following treatment, 1–2 × 106 cells were permeabilized in 100 μL of ice-cold plasma-membrane-permeabilization buffer (200μg/mL digitonin, 80 mM KCl in PBS) on ice for 5 min. The cells were scraped from the tissue culture plate and placed in microcentrifuge tubes. The cytosolic fraction (supernatant) was separated by centrifugation at 800 g at 4 °C for 5 min. The pellet (mitochondria-enriched fraction) was resuspended in 100 μL of ice-cold lysis buffer (50 mM Tris HCl, pH 7.4, 150 mM NaCl, 2 mM EGTA, 0.2% Triton X 100, 0.3% Igepal and protease inhibitor cocktail and rocked gently at 4 °C for 10 min followed by centrifugation at 10,000 g at 4 °C for 10 min. The cytosolic and mitochondria-enriched fractions were normalized for protein content by Lowery assay (Bio-Rad) and proteins separated on (SDS-PAGE) and transferred from the gel to a nitrocellulose membrane. The detection of cytochrome c in cytosolic and mitochondrial fractions was performed by Western blotting with cytochrome c antibody.
2.5. Detection of IL-1β in cell supernatant
Mouse IL-1β secreted into cell supernatants was quantitated by Enzyme-linked immunosorbent assay (ELISA) according to the manufacturer’s instruction [24]. Human IL-1β was quantitated from cell supernatnats by Luminex-based Bio-Plex assay (Bio-Rad).
2.6. Detection of intracellular oxidants
The H2DCF-DA fluorescent assay was used to measure the presence of intracellular oxidants [27]. Thirty minutes prior to the end of treatments, 10 μM H2DCF-DA in phenol red free DMEM media was added to the cells. The fluorescence was measured in cell lysates using a microplate fluorimeter (Bio-Tek) with excitation at 480 nm and emission at 530 nm. Protein content from each sample was evaluated by Bradford protein assay (Bio-Rad) and utilized to normalize fluorescence signal intensity. Values are expressed as relative fluorescence units (RFU).
2.7. Detection of mitochondria derived ROS
Mitochondria-derived ROS, mainly superoxide, was detected using the mitochondrial superoxide indicator, MitoSOX Red, a cationic dihydroethidium modified to target the mitochondria. According to the manufacturer, MitoSOX red is a cell-permeative dye that reacts with superoxide to form ethidium, which upon binding to nucleic acids gives a bright red fluorescence. Briefly, cells were seeded onto 8-well CultureSlides (BD Falcon™) and treated as described above. At the end of the respective treatments, the cells were rinsed with PBS and loaded with MitoSOX Red (2.5 μM) for 10 min. For confirmation of ROS localization to mitochondria, cells were subsequently labeled with MitoTracker Green (20 nM) for 20 min. The medium containing fluorescent probes was removed, and the cells were rinsed with PBS, and cells were fixed with 3% paraformaldehyde for 10 min. Images were immediately analyzed by confocal microscopy using a Zeiss LSM 510 META Confocal Laser Scanning Imaging System.
2.8. Analysis of mitochondrial membrane potential (Δψm)
Cells (1–2 × 106 cells/mL) were seeded in 12 well plates and 24 hours later were treated as indicated. After washing with PBS, cells were incubated in fresh medium containing 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazol-carbocyanine iodide (JC-1) for 15 min. The dye was then removed; cells were washed with PBS and fresh medium was added. Live cells were immediately observed in an Olympus IX70 Inverted Fluorescence Microscope coupled to a QImaging Retiga 2000R digital camera. Healthy cells, containing mainly JC-1 aggregates, were observed at 540/570 nm excitation/emission and the apoptotic or unhealthy cells with mainly JC-1 monomers were measured at 485/535 nm excitation/emission.
2.9. Measurement of ATP content
Assessment of relative ATP content was performed using the ATP bioluminescence assay kit following the manufacturer’s instructions. In brief, after the respective treatment 105 cells were lysed in 100 μL of lysis buffer (1% Triton X-100 in PBS) and 10 μL of the lysates were added to 90 μL of Reaction buffer, which contained the enzyme mix and after a delay of 3 s, the luminescence was integrated over 10 s using a TD-20/20 luminometer (Turner Designs). The relative ATP content in the lysates is referred to as intracellular ATP. Alternatively, the extracellular ATP was measured in cell culture supernatants by adding 10 μL of medium to 90 μL of Reaction buffer and the ATP content was measured as described for the lysates. Protein content from lysates was evaluated by Bradford protein assay (Bio-Rad) and utilized to normalize fluorescence signal intensity. Values are expressed as relative fluorescence units (RLU).
2.10. Statistical Analysis
The data were statistically analyzed by one-way analysis of variance test, followed by Bonferroni’s multiple comparison test, using GraphPad Prizm 5.04 software. The statistical analysis was from a single experiment with three or four replicates. Each experiment was conduct multiple times with similar results; therefore a representative study is presented in the respective figures.
2.11. Image Analysis
The red fluorescence intensity of individual cells (N=10) from images obtained by fluoresce microscopy was quantified by ImageJ software, followed by statistical analysis. Alternatively, red and green fluorescence intensities were quantified separately, and a red over green fluorescence intensity was expressed as R/G ratio.
3. Results
3.1. Stimulated macrophages generate ROS during Nlrp3 inflammasome activation
Bacterial products such as LPS can promote Nlrp3 inflammasome-dependent IL-1β secretion in various human and mouse myeloid cells. We recently demonstrated that SAA, a fibril-like protein considered a biomarker of inflammation, can also stimulate Nlrp3 inflammasome-dependent IL-1β secretion in mouse peritoneal macrophages [24]. We thus sought to further investigate the kinetics of Nlrp3 inflammasome activation in human and mouse myeloid cells. Cells were stimulated with LPS or SAA for indicated times and cellular levels of inflammasome components were analyzed by Western blotting and IL-1β secretion was quantified by ELISA. The mouse peritoneal macrophage cell line (IC-21) demonstrated elevated basal levels of Nlrp3, ASC, pro-caspase-1 and pro-IL-1β. Whereas Nlrp3 and pro-IL-1β intracellular levels were mildly increased upon stimulation with LPS at earlier time points, (Fig. 1A), IL-1β secretion was significantly elevated with prolonged LPS stimulation (Fig. 1B). In unstimulated primary mouse peritoneal macrophages Nlrp3 and pro-IL-1β were found at near to undetectable levels, which increased significantly at 2h and sustained until 24h following SAA stimulation (Fig. 1C). At the 24h time-point, IL-1β secretion was the most prominent from these primary cells (Fig. 1D). Similar to observations in primary mouse macrophages, human monocytes showed increases of NLRP3 intracellular levels at 2h of SAA stimulation; however the active form of caspase-1 was detectable only at 24h of SAA stimulation (Fig. 1E). Different from what was observed in mouse myeloid cells, in human monocytes ASC was induced by SAA (Fig. 1E). Moreover, the kinetics of IL-1β secretion was found to be similar in both human and mouse SAA-stimulated primary cells (Figs. 1D and 1F).
Figure 1.

Time-dependent induction of the synthesis of inflammasome components and IL-1β secretion in inflammatory cells. Transformed mouse peritoneal macrophages, IC 21 cell line (A, B), primary mouse peritoneal macrophages (C, D), and human peripheral blood mononuclear cells (E, F) were stimulated at the indicated times with LPS (100 ng/mL) or SAA (1 μg/mL) and inflammasome components were analyzed by Western blotting from cell lysates (A, C, E). The numbers below the blot images indicate fold induction relative to control. Secreted IL-1β was quantified in the cell supernatants by ELISA (B, D) or Bioplex analysis (F). The data are representative of at least two independent experiments. * = p < 0.05 compared to control.
Elevated levels of ROS have been reported as a common signal in Nlrp3 inflammasome activation [28]. However the kinetics of ROS generation and the respective sources remain to be fully explored. We next investigated the temporal changes in the overall intracellular ROS levels caused by SAA stimulation of murine macrophages using H2DCF-DA, a cell permeable fluorescence probe. Upon stimulation with SAA, mouse primary macrophages demonstrated a trend towards a decrease in intracellular ROS at 2h and a robust increase in intracellular ROS at 24h (Fig. 2A). On the other hand, the LPS-stimulated macrophage cell line showed no early decrease of intracellular ROS, but did display a significant peak in ROS at 4h followed by a subsequent small decrease (Fig. 2B). Since DCF fluorescence detects overall intracellular ROS, but not the source, we next utilized MitoSOX Red a more specific fluorescent probe to detect mitochondrial ROS. Under our experimental conditions, SAA significantly increased mitochondrial ROS at 2h, which gradually returned to the basal level at 8h and diminished further by 24h of stimulation (Fig. 2C and 2D), contrasting with elevated intracellular ROS at this late time point (Fig. 2A). These data demonstrate that upon SAA-stimulation, Nlrp3 and pro-IL-1β are rapidly synthesized in primary cells, but significant IL-1β secretion is not observed until later time points. Different from what was observed for intracellular ROS, the mitochondrial ROS were also increased at early time points. These results suggest that in SAA-stimulated primary macrophages, mitochondria-derived ROS are involved in priming rather than the late step of Nlrp3 activation. However, in the LPS-stimulated macrophage cell line, the elevated basal level of precursors suggests that the actual step of IL-1β secretion is time-dependent, rather than being incumbent upon the synthesis of inflammasome components. Our results also demonstrated that significant increases in intracellular ROS were parallel to IL-1β secretion, supporting the notion that intracellular ROS are involved in Nlrp3 inflammasome activation rather than priming.
Figure 2.

Time-dependent generation of ROS in SAA- or LPS-stimulated cells. Primary mouse peritoneal macrophages (A) or transformed mouse peritoneal macrophages (B) were stimulated with SAA (1μg/mL) or LPS (100 ng/mL) at the indicated times. At the end of incubation, dihydrofluorescein diacetate (DCF-DA) was added to the live cells for 30 min and intracellular ROS levels were analyzed in a fluorometric microplate reader (A, B). Data are expressed as relative fluorescence units (RFU), mean ± S.D. (n=3), normalized by protein content. The data are representative of at least two independent experiments. * = p < 0.05 compared to control. Mitochondrial ROS (C) was assessed at the end of the incubation by adding MitoSox green to the live cells for 10 min. followed by prompt analysis using a fluorescence confocal microscope at 630x magnification. The fluorescence intensity (D) of individual cells (N=10) from images displayed in Fig. 2C was analyzed by ImageJ software. The values represent the mean ±S.D* = p < 0.05 compared to control.
3.2. ROS generated during Nlrp3 inflammasome activation are involved in IL-1β secretion
Several groups have reported that ROS scavengers or inhibitors added prior to or concomitant with Nlrp3 stimulation diminish or abolish IL-1β secretion, implying a direct involvement of ROS in the Nlrp3 inflammasome/IL-1β secretion axis [14, 29]. Therefore, we next utilized inhibitors or scavengers to target different sources or species of ROS at different time points and investigated the impact on Nlrp3 inflammasome priming and IL-1β secretion in mouse peritoneal macrophages. To investigate the impact of diverse ROS on Nlrp3 inflammasome priming, we utilized DPI a broad spectrum NADPH oxidase inhibitor, ebselen a peroxide scavenger, and MnTBAP a SOD mimetic that is known to substitute in vivo in mice lacking mitochondrial MnSOD [30] prior to SAA or LPS stimulation. We also investigated the possibility for nitric oxide (NO) to be involved in the Nlrp3-inflammasome/IL-1β secretion axis, by utilizing a broad spectrum nitric oxide synthase inhibitor, L-NG-Nitroarginine methyl ester (L-NAME). To investigate the ROS participation during “signal 1” (priming), we added the respective inhibitors prior to the stimulants, SAA and LPS. When DPI was administered before SAA stimulation, this inhibitor surprisingly augmented intracellular ROS (Fig. 3A) and IL-1-β secretion (Fig. 3B) in comparison to SAA alone. On the other hand, ebselen, and to a lesser extent MnTBAP, mildly diminished intracellular ROS (Fig. 3A) and strongly inhibited IL-1β secretion (Fig. 3B) when added prior to SAA. L-NAME did not have any effect on IL-1β secretion when compared with SAA alone (Fig. 3B, insert). In LPS-stimulated macrophages, DPI again showed a pro- rather than antioxidant effect, but had no impact on IL-1β secretion. Ebselen exerted no significant impact on intracellular ROS or on LPS-induced IL-1β secretion, whereas MnTBAP appeared to have a slight pro-oxidant effect (non-significant) and modestly but significantly decreased IL-1β secretion in comparison to LPS alone (Figs. 3C and 3D). Importantly, treatment with inhibitors alone did not induce IL-1β secretion from any cell type tested (data not shown). These data indicated that when added prior to SAA (before priming), MnTBAP and Ebselen had a negative impact on IL-1β secretion, but this action appeared to be independent of the levels of intracellular ROS. However, when levels of intracellular ROS were increased by treatment with DPI prior to SAA, the secretion of IL-1β was also increased. When LPS was utilized as a stimulant, the presence of inhibitors prior to stimulation had minimal impact on IL-1β secretion, even when intracellular ROS were greatly increased, as was seen with DPI treatment.
Figure 3.
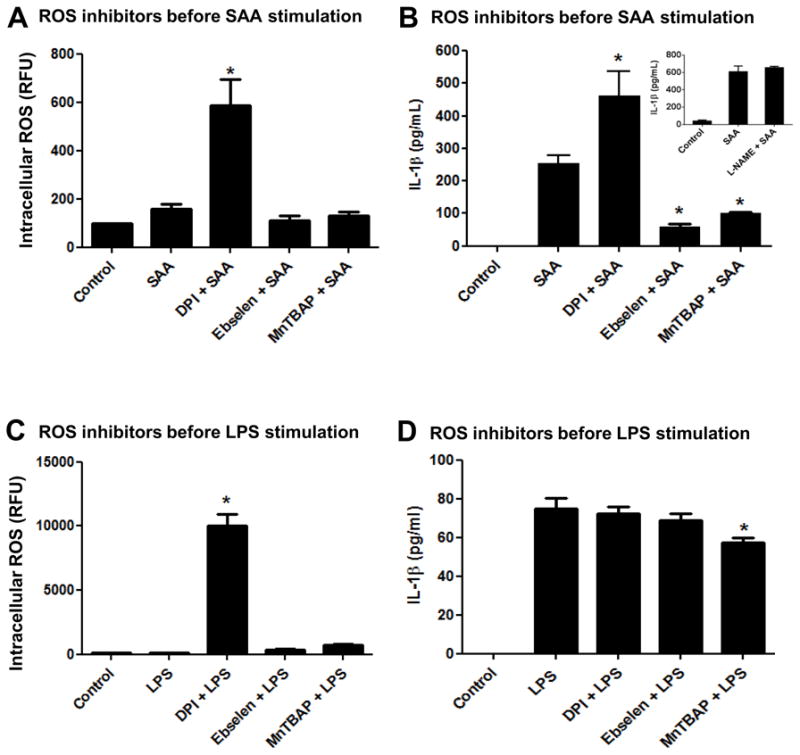
Effect of ROS inhibitors added prior to SAA or LPS stimulation on IL-1β secretion and intracellular ROS levels. Primary mouse peritoneal macrophages (A, B) or transformed mouse peritoneal macrophages (C, D) were left to adhere for at least 24h. Flavoprotein inhibitor DPI (10 μM), ebselen (10 μM) or the MnSOD mimetic MnTBAP (100 μM) were added for 30 min prior to SAA (1 μg/mL) or LPS (100 ng/mL) stimulation, respectively. After 24h of simultaneous treatment, intracellular ROS levels were analyzed as described in Fig. 2 (A, C). IL-1β secretion was quantified by ELISA (B, D). The data are representative of at least two independent experiments. * = p < 0.05 compared to SAA (A,B) or LPS (C, D).
We next determined whether intracellular ROS were involved in Nlrp3 inflammasome activation and IL-1β secretion after priming (“signal 1”). Therefore, we chose to add the inhibitors after 8h of stimulation with LPS or SAA, when Nlrp3 and other inflammasome components were peaked, but before IL-1β had been secreted (Fig. 1). When these drugs were added after 8h of SAA-stimulation, DPI again demonstrated augmentation of intracellular ROS (Fig. 4A), consistent with increases in IL-1β secretion (Fig. 4B). However, MnTBAP, but not the respective control TBAP (Fig. 4A, insert) was able to strongly increase IL-1β secretion (Fig. 4B) without having a significant positive impact on intracellular ROS (Fig. 4A). Ebselen did not have any impact on intracellular ROS (Fig. 1A) or on IL-1β secretion when utilized after priming with SAA (Fig. 4B). When LPS was utilized as an Nlrp3 inflammasome stimulator, the presence of the inhibitors DPI and MnTBAP significantly increased intracellular ROS (Fig. 4C). However all inhibitors tested exerted a mild but significant impact on increasing IL-1β secretion (Fig. 4D) when LPS was used as stimulant. Collectively, our data demonstrate that ROS-modifying drugs, whether added prior to or after Nlrp3 stimulation, can have a pro-oxidant rather than an antioxidant effect. However, depending on the stimuli, this dual effect may have no impact on IL-1β secretion. Our results revealed that DPI has a significant pro oxidant effect and, at least in SAA treated cells, also a stimulatory effect on IL-1β secretion independent of when it is administered. Both Ebselen and MnTBAP demonstrated a strong inhibitory effect on IL-1β secretion when added prior to SAA-stimulation, suggesting that their action is more important for preventing priming (“signal 1”). However, when MnTBAP was added after SAA-stimulation, this drug demonstrated a strong stimulatory effect on IL-1β secretion.
Figure 4.

Effect of ROS inhibitors added after SAA or LPS stimulation on IL-1β secretion and intracellular ROS levels. Primary mouse peritoneal macrophages (A, B) or transformed mouse peritoneal macrophages (C, D) were left to adhere for at least 24h and stimulated with SAA (1 μg/mL) or LPS (100 ng/mL) respectively for 8h. DPI (10 μM), ebselen (10 μM), MnTBAP (100 μM) or respective control (TBAP 100 μM, insert) were added to stimulated cells for another 16h. After a total 24h of simultaneous treatment, intracellular ROS levels were analyzed as described in Fig. 2 (A, C) and IL-1β secretion was quantified by ELISA (B, D). The data are representative of at least two independent experiments. * = p < 0.05 compared to SAA (A, B) or LPS (C, D).
3.3 Mitochondrial ROS modulate Nlrp3 inflammasome activation and IL-1β secretion
Recent publications have implicated that dysfunctional mitochondria generate ROS that sustain inflammasome activation and IL-1β secretion [20, 31, 32]. Since SAA alone (Fig. 2C) and in the presence of MnTBAP (Fig. 4B) reveal the potential involvement of mitochondrial ROS in the activation of Nlrp3, we next utilized mitochondria-targeted drugs in SAA stimulated cells to investigate in more detail the participation of the mitochondria on Nlrp3 inflammasome activation and IL-1β secretion. Therefore, we utilized a mitochondrial respiratory chain inhibitor, antimycin A, which blocks electron flow at complex III and is well known for increasing mitochondrial ROS [33]. We also utilized an oxidative phosphorylation uncoupler, FCCP, which disrupts the proton gradient and has been postulated to decrease ROS production by the mitochondria [34]. In mouse peritoneal macrophages, antimycin A increased overall intracellular ROS in SAA-stimulated cells, whereas FCCP did not have any effect (Fig. 5A). Surprisingly, under the same experimental conditions, opposite effects on IL-1β secretion were observed. Whereas, antimycin A caused only a marginal increase in IL-1β secretion, FCCP significantly increased IL-1β secretion in comparison with SAA alone (Fig. 5B). Again, none of the inhibitors induced IL-1β secretion from non-stimulated cells (data not shown). Analyzing the ROS levels in the mitochondria using MitoSOX Red, we found that the treatment of SAA-stimulated cells with antimycin A for a long period (16h, Fig. 5C) did not significantly increase mitochondrial ROS (Fig. 5E, left panel), but appeared to cause the fluorescence probe to relocate to the nuclear compartment. This nuclear staining by the mitochondrial probe can be indicative of apoptosis [35]. Similarly, FCCP (16h) also caused probe relocation (Fig. 5C); however, the fluorescence intensity was significantly greater than was observed in SAA-exposed cells treated with antimycin A (Fig. 5E, left panel), suggesting a greater amount of mitochondrial ROS. Because the long period of treatment with mitochondria-targeted drugs (16h) might have caused some apoptosis, we next decided to assess mitochondria derived ROS 1h after these drugs were added to SAA-stimulated cells. At this early time point, antimycin A disrupted the mitochondria network, as seen as increased fluorescence and “dotted pattern” (Fig. 5D), but again this drug did not significantly impact fluorescence intensity (Fig. 5E, right panel). Regardless of some nuclear staining, the red fluorescence also co-localized to mitochondria that were counter stained with the mitochondrial marker, MitoTracker green (inserts in Fig. 5C and 5D). Interestingly, the treatment with MnTBAP significantly diminished the fluorescence intensity at 16h (Fig. 5C and Fig. 5E, left panel) or 1h (Fig. 1D and Fig. 5E, right panel) incubation times, reinforcing its MnSOD mimetic activity in the mitochondria.
Figure 5.

Effect of mitochondria targeted drugs (antimycin A and FCC) and a ROS inhibitor (MnTBAP) on intracellular and mitochondrial ROS, and IL-1β secretion in SAA-stimulated cells. Primary mouse peritoneal macrophages were stimulated with SAA (1 μg/mL) for 8h. Antimycin A (40 μg/mL), FCCP (10 μM), or MnTBAP (100 μM) were added to SAA-stimulated cells for another 16h. After a total 24h of treatment, intracellular ROS levels (A) and mitochondrial ROS (C, D) were analyzed as described in Fig. 2 and IL-1β secretion was quantified by ELISA (B). Alternatively, antimycin A, FCCP or MnTBAP were added for just 1h (D) after 8h of SAA stimulation and mitochondrial ROS was also detected as described in Fig. 2C. The indicated merged images in C and D represent the same field in which mitochondria were stained with MitoSOX red and Mitotracker green as a counter stain. The images were captured with a confocal microscope at 630x magnification. The data are representative of at least two independent experiments. * =p < 0.05 compared to SAA. The red fluorescence intensity (E) of individual cells (N=10) from images displayed in Fig. 5C (E, left panel) or in Fig. 5D (E, right panel) was analyzed by ImageJ software. The values represent the mean ± S.D (D). * = p < 0.05 compared to control.
Since the mitochondria targeted drugs can cause mitochondrial dysfunction, leading to mitochondrial depolarization and energy depletion, we next assessed the impact of these drugs on mitochondrial membrane potential (Δψ), cytochrome c release and ATP content. Not surprisingly, all mitochondria targeted-drugs alone caused changes in mitochondrial membrane potential, indicated by increased green and diminished red florescence (Fig. 6A, top panel). Whereas DPI is not generally considered to be a mitochondria targeting-drug, it has been reported to can cause mitochondrial membrane depolarization and apoptosis in a human promyelocytic leukemia cell line (HL-60 cells) [36]. In our experimental conditions, DPI alone also caused loss in mitochondrial membrane potential, although to a lesser extent than the other drugs tested (Fig. 6A, top panel). Surprisingly, SAA alone also affected the mitochondrial membrane potential and the concomitant treatment with mitochondria-targeted drugs or DPI appeared to have no additional impact on the membrane potential loss caused by these drugs alone (Fig. 6A bottom panel). Another indication of loss of mitochondrial permeability is release of cytochrome c from the mitochondrial membrane to the cytoplasm [37], characterized by the appearance of an approximately 15kDa band. Regardless of the effect on mitochondrial membrane potential exerted by each of the drugs tested (Fig. 6A), only in antimycin A plus SAA-treated cells was the cytochrome c 15 kDa band undetected in the mitochondrial fraction (Fig. 6B top panel). However, no accumulation of the 15 kDa band was detected in the cytoplasmic fraction under the same experimental condition (Fig. 6B bottom panel). Since depolarization of the mitochondrial membrane can have a direct impact on ATP production by the mitochondria, we also measured intracellular and extracellular levels of ATP. Accumulation of extracellular ATP is considered one of the most potent signals capable of inducing Nlrp3 inflammasome activation [28]. SAA treatment alone did not significantly affect intracellular ATP levels (Fig. 6C, insert), regardless of its effect on mitochondrial depolarization (Fig. 6A, bottom panel). Not surprisingly, in SAA treated-cells the presence of antimycin A and FCCP, as well as DPI, decreased the intracellular levels of ATP significantly. Under the same experimental condition, MnTBAP did not affect intracellular ATP levels when compared to SAA alone (Fig. 6B). On the other hand, the extracellular levels of ATP were dramatically decreased by SAA treatment alone (Fig. 6C, insert). In contrast to antimycin A and DPI, FCCP did not cause any changes in extracellular ATP in SAA-treated cells. Moreover, MnTBAP was the only drug that significantly increased the extracellular levels of ATP under our experimental conditions (Fig. 6C). Taken together, our results suggest that regardless of the impact on mitochondrial ROS production, mitochondrial function and ATP levels, the drugs tested have different mechanisms of action for increasing SAA-induced IL-1β secretion.
Figure 6.

Effect of mitochondria-targeted drugs (FCCP and, antimycin A), and ROS inhibitors (DPI and MnTBAP) on mitochondria dysfunction in SAA-stimulated cells. Primary mouse peritoneal macrophages were stimulated with SAA (1 μg/mL) for 8h and antimycin A (40 μg/mL), FCCP (10 μM), DPI (10 μM) or MnTBAP (100 μM) were added to SAA-stimulated cells for another 16h. Change in the mitochondrial membrane potential (Δψm) (A) was analyzed by (JC-1) fluorescence imaging. After 24h of total treatment, JC-1 dye was added to live cells and after 15 min of incubation the images were immediately captured using an Olympus IX70 Inverted Fluorescence Microscope coupled to a QImaging Retiga 2000R digital camera at 400x magnification. Green fluorescence indicates depolarized (monomeric form of JC 1, unhealthy mitochondria) and red indicates hyperpolarized (J aggregate, healthy mitochondria) Red and green fluorescence intensity was analyzed separately using ImageJ and red over green ratio is displayed on the top left of each merged picture. Cytochrome c release from the mitochondria to cytoplasm was analyzed by Western blotting (B). Intracellular (C) and extracellular (D) ATP relative content in cell lysates and cell supernatants was analyzed with an ATP bioluminescence assay kit. Data are expressed as relative luminescence units (RLU), mean ± S.D. (n=3), normalized by protein content. * = p < 0.05 compared to SAA.
3.4. Mitochondria-targeted drugs affect Nlrp3 inflammasome complex assembly
As expected, the short concomitant incubation of these drugs with SAA did not affect the levels of inflammasome components when compared with SAA alone (data not shown). However, when FCCP was added to SAA-stimulated cells a robust decrease in Nlrp3, ASC, pro-caspase-1 and to a lesser extent pro- IL-1β intracellular levels were observed. Moreover, antimycin A added to SAA-stimulated cells also showed a trend towards decreasing the levels of inflammasome components. On the other hand, MnTBAP did not affect the intracellular levels of inflammasome components in SAA-stimulated cells (Fig. 7A). Since we observed substantial diminution of the intracellular levels of Nlrp3 inflammasome components upon treatment with FCCP, we investigated whether these components were being released from the cells. In the supernatants of SAA-treated cells we observed a significant increase in Nlrp3 and pro-IL-1β, and to a much lesser extent caspase-1, especially the active form (p10), in comparison with supernatants of non-treated cells (Fig. 7B). Supernatants of SAA/antimycin A treated cells showed a significant increase in Nlrp3 (Fig. 6B, upper panel) and caspase 1 (Fig. 7B, middle panel), whereas SAA/MnTBAP treatment seemed to increase only pro-IL-1β (Fig. 7C, bottom panel) in the cell supernatants when compared with supernatants from cells treated with SAA alone. It is important to mention that regardless of the increases in pro-caspase-1 under some treatment conditions, the levels of active caspase-1 (p10) in cell supernatants did not seem to be different than that observed in supernatants from cells treated with SAA alone (Fig. 7B, middle panel). Assembly of the NLRP3 inflammasome complex, which occurs after priming, is a crucial step for complete IL-1β processing and secretion [38]. Therefore after analyzing the intracellular and extracellular levels of Nlrp3 inflammasome components, we investigated whether SAA alone or in combination with mitochondria targeted drugs could affect assembly of the Nlrp3 inflammasome complex. Using immunoprecipitation techniques, we observed that the association between Nlrp3 and ASC was present at modest levels in SAA-stimulated cells (Fig. 7C, upper panel). However, in the presence of FCCP this association was substantially augmented (Fig. 7C, upper panel, the band of interest is indicated by the arrow). Furthermore, we also observed that SAA alone induced Nlrp3/caspase-1 interaction, which was again robustly enhanced by the presence of FCCP (Fig. 7A, middle pane, the band of interest is indicated by the arrow). Finally, we also investigated the association between caspase-1 and pro-IL-1β. Interestingly, the only condition under which pro-IL-1β and caspase-1 appeared to interact was when FCCP was added to SAA-stimulated cells. This interaction is indicated by a substantial band corresponding to the unprocessed IL-1β, near to the lower chain (Fig. 7C, lower panel, the band of interest is indicated by the arrow). It is important to mention that none of the mitochondria-targeted drugs alone showed any effect on inducing Nlrp3 inflammasome components (priming) or IL-1β secretion (data not shown). Collectively, these data indicate that in SAA-treated cells FCCP, but not antimycin A or MnTBAP, significantly enhances the interaction among the Nlrp3 complex members and as a consequence might contribute to augmented IL-1β secretion in these experimental conditions.
Figure 7.

Effect of mitochondria-targeting drugs (antimycin A and FCCP) and a ROS inhibitor (MnTBAP) on the synthesis of Nlrp3 inflammasome components and Nlrp3 inflammasome complex assembly in SAA-stimulated cells. Primary mouse peritoneal macrophages were stimulated with SAA (1 μg/mL) for 8h and antimycin A (40 μg/mL), FCCP (10 μM), or MnTBAP (100 μM) were added to SAA-stimulated cells for another 16h. After 24h of total treatment, inflammasome components were analyzed in total cell lysates (A) or supernatants (B) by Western blotting. The inflammasome complex assembly was analyzed in the total cell lysates (C) by immunoprecipitation using ASC or caspase-1 antibody followed by Western blotting, using Nlrp3 or pro IL-1β antibodies, as indicated. The arrows indicate the bands of interest. HC = heavy chain; LC= light chain. The data are representative of two independent experiments.
4. Discussion
In this study we report that stimulated macrophages generate ROS during Nlrp3 inflammasome activation, in the absence of exogenous ATP. We also demonstrated the participation of mitochondria derived ROS in the activation of the Nlrp3 inflammasome in SAA-stimulated murine primary macrophages. Furthermore, our study provides the first evidence that mitochondrial dysfunction can cause disturbance of Nlrp3 inflammasome disassembly, evidenced by enhanced interaction among the Nlrp3 complex members, which ultimately promotes sustained Nlrp3 inflammasome activation and enhanced IL-1β secretion. Our time course studies revealed that intracellular ROS are peaked at 24h of SAA and at 4h of LPS stimulation in murine macrophages. In addition, ROS manipulations utilizing classical ROS inhibitors appeared to affect to a greater extent the Nlrp3/IL-1β secretion axis in SAA- rather than in LPS-stimulated cells. These apparent discrepancies might be explained, at least in part, by the differences in the receptors triggered by these agonists as well as in differences in antioxidant status of primary cells as opposed to cell lines. Cell stimulation leading to IL-1β secretion in response to SAA requires engaging the cell surface pattern recognition receptor TLR2 [22, 24], whereas LPS is a classical activator of TLR4 [39] and TLR2 (due to the presence of contaminants [40]), both of which can be associated with ROS generation by NADPH oxidases [41]. The different redox status between primary myeloid cells and cell lines can also reflect on the various responses to ROS inhibitors or stimulators. It has been reported that resting human monocytes display a balanced redox state, with low production of ROS and consequently low antioxidant defenses [25]. Conversely, the equivalent human cell line, THP-1, have upregulated antioxidant systems that counterbalance the ROS production induced by TLR triggering and reduce the subsequent response [25]. Because of the more robust stimulation of the Nlrp3/IL-1β axis in primary cells than the respective cell line, we conducted the majority of our experiments in primary mouse peritoneal macrophages.
It has been shown that the major sources of ROS participating in Nlrp3 inflammasome stimulation and activation are NADPH oxidases. These studies were mainly based upon data obtained utilizing the broad spectrum flavoprotein inhibitor, DPI, prior to LPS treatment and then followed by short-term incubation with high concentrations of ATP [13], a potent extracellular inducer of Nlrp3 activity [28]. We have reported that IL-1β secretion induced by SAA does not require the addition of ATP [24], therefore all experiments in this current study were performed in absence of supplemental exogenous ATP. Surprisingly, in the absence of exogenous ATP, DPI enhanced IL-1β secretion in conjunction with increases in intracellular ROS in primary mouse peritoneal macrophages, regardless whether it was added prior to or after SAA-stimulation. This robust effect on IL-1β secretion in SAA-treated cells suggested that DPI is exerting its effect beyond its classical action as flavoprotein inhibitor and consequently a ROS suppressor, but instead a ROS generator, and therefore contributing to de novo translation of Nlrp3 and pro-IL-1β (priming), which ultimately results in increased IL-1β secretion. This pro-oxidant effect is consistent with previous findings in which DPI can increase the generation of superoxide by the mitochondria, induce membrane depolarization and cause apoptosis [36]. In our experimental conditions, ebselen and MnTBAP did not affect intracellular ROS when added before or after SAA stimulation. However both were able to substantially block IL-1β secretion only when added prior to SAA stimulation. Ebselen is a cell-permeable superoxide dismutase and glutathione peroxidase mimetic [42]. Because ebselen exerts broad-spectrum antioxidant properties by effectively scavenging organic hydroperoxides, especially lipid hydroperoxides [43], our results suggest that in macrophages these lipophilic ROS play an important role in Nlrp3 inflammasome priming (“signal 1”) but not in Nlrp3 inflammasome activation (“signal 2”). We also explored the nature of these ROS by using MnTBAP, a cell-permeable MnSOD mimetic [42]. Although MnTBAP is considered a catalytic antioxidant targeting, in general, superoxide from overall intracellular sources, this compound also exerts its activity in the mitochondria, both in vitro [44] and in vivo [30]. Similar to what was observed for ebselen, we demonstrated that MnTBAP is a potent inhibitor of Nlrp3 inflammasome stimulation but not Nlrp3 inflammasome activation, and this inhibitory effect can be associated with the classical antioxidant activity described for this MnSOD mimetic, in particular targeting the mitochondria. In fact, the presence of MnTBAP significantly decreased mitochondrial ROS in SAA-treated cells in comparison to SAA alone (Figs. 5C, D and E). Because MnTBAP targets the mitochondria and its inhibitory effect is noticeable only prior Nlrp3 inflammasome stimulation, these observations led us to implicate that mitochondrial ROS are important at least in part in the priming step of Nlrp3 inflammasome.
The idea that mitochondrial-derived ROS are involved in Nlrp3 priming is also consistent with our data demonstrating that mitochondrial ROS peak at 2h of SAA-stimulation, which is the equivalent time point at which the levels of intracellular pro-IL-1β and Nlrp3 begin to be induced. More recent publications reinforce the idea that mitochondrial ROS, especially generated as a consequence of mitochondrial dysfunction, are the main source of ROS participating in Nlrp3 inflammasome activation [32], In our study, we demonstrated that mitochondrial disturbance caused after Nlrp3 inflammasome stimulation by antimycin A, FCCP and MnTBAP enhanced the responsiveness to SAA in primary macrophage. However this apparently similar effect cannot be attributed only to changes in mitochondrial ROS, since we observed that antimycin A and FCCP showed different potencies in affecting mitochondrial ROS and MnTBAP demonstrated an opposite effect in decreasing mitochondrial ROS in SAA-stimulated cells. Therefore, the levels of mitochondrial ROS are poorly correlated with effects in enhancing IL-1β secretion, at least after Nlrp3 inflammasome stimulation. Analyzing other aspects of mitochondrial function, we found that all tested drugs caused some degree of mitochondrial dysfunction, but in different manners. All tested drugs alone caused changes in mitochondrial membrane potential, including SAA. In addition, the intracellular ATP levels were also affected by almost all the drugs tested in SAA-treated cells, with exception of MnTBAP. Interestingly, only MnTBAP dramatically increased extracellular ATP in SAA-treated cells. Although, ATP is considered a potent Nlrp3 activator after priming [28], this dramatic effect on enhancing extracellular ATP did not explain the increases in IL-1β secretion caused by MnTBAP, since the concomitant utilization of an ATP receptor (P2X7) blocker (oxATP) or enzymatic decomposition of ATP by apyrase [45] with MnTBAP did not prevent this effect in SAA-stimulated cells (data not shown). Therefore, MnTBAP appeared to exert its effects of increasing IL-1β secretion through a ROS- and extracellular ATP-independent mechanism. Another important observation regarding mitochondria dysfunction was the fact that only antimycin A had an effect on cytochrome c release in SAA-treated cells, although the typical 15 kDa band was not observed in the cytosolic fraction. This apparent discrepancy related to the relocation of cytochrome c could be due to the late time point assessed, after 24h of total treatment. Because of the overall effects on mitochondrial function, we do not exclude the possibility that antimycin A or FCCP may induce apoptosis or necrosis in SAA-treated cells, as suggested by the dramatic depletion of intracellular ATP, alteration in the mitochondrial network, or increases in mitochondrial ROS. However the potential toxicity of antimycin A or FCCP does not appear to play a role in increasing IL-1β secretion from SAA-stimulated macrophages, since we did not observe any leakage of other cellular protein contents.
Under our experimental conditions, we demonstrated that FCCP has a much more robust effect than antimycin A on IL-1β secretion despite their similar effects on mitochondrial membrane potential and intracellular ATP levels in SAA-stimulated cells. These observations suggest that the mechanism by which FCCP increases IL-1β secretion might be different than that of antimycin A. One striking observation is that FCCP was the only mitochondria-targeted drug that strongly sustained the association of the Nlrp3 complex, in particular, the association between caspase-1 and pro-IL-1β. The prolonged proximity of the protease, caspase-1, and its substrate, pro-IL-1β, may in part explain the high levels of secreted IL-1β induced by FCCP in SAA-stimulated cells. A recent publication demonstrating that activation of selective autophagy by inflammatory signals limits IL-1β production by targeting inflammasome components for destruction via ubiquitination [46] led us to speculate that another potential mechanism by which FCCP contributes to augmentation of IL-1β secretion is the opposite of that seen in selective autophagy, i.e. by preventing the destruction of the Nlrp3 inflammasome complex or its subunits. Therefore the potential non-destruction of the Nlrp3 inflammasome complex by FCCP could prolong the association among the Nlrp3 inflammasome components and contribute to sustained IL-1β secretion in SAA-treated cells
In conclusion, our data demonstrated that the Nlrp3 inflammasome/IL-1β secretion axis is a very intricate and highly-regulated inflammatory pathway that is susceptible to modulation by changes in mitochondrial redox status, as observed in our time course studies. Furthermore, depending on whether the changes to mitochondrial redox status occur prior to (Fig. 8 top diagram) or after Nlrp3 inflammasome stimulation (Fig. 8 bottom diagram) the impact on IL-1β secretion is different, suggesting that there are additional mechanisms involving mitochondrial dysfunction participating in the sustained Nlrp3 inflammasome activation. Further studies are necessary to determine the potential target proteins in this pathway that are susceptible to redox regulation as well as the kinetics of these potential post translational modifications.
Figure 8.

Proposed models demonstrating the different mechanisms by which mitochondria-targeted drugs, FCCP and antimycin A, and the classical antioxidants ebselen, DPI and MnTBAP, affect IL-1β secretion in mouse macrophages. (1) SAA stimulated mitochondria ROS to induce priming. When (2) ebselen and (2b) MnTBAP were added before SAA (top diagram), these drugs attenuated IL-1β secretion, perhaps due to their antioxidant activities against peroxide and mitochondrial ROS, respectively. On the other hand, (3) DPI enhanced IL-1β secretion associated with increases in intracellular ROS. (4) When mitochondrial-targeted drugs were added after SAA (bottom diagram), overproduction of mitochondrial ROS by FCCP or intracellular ROS by antimycin A or DPI was correlated with increases in IL-1β secretion. (4b) The robust effect of FCCP on IL-1β secretion was associated with sustained interaction among Nlrp3 inflammasome components; perhaps by decreasing their degradation. (5) After priming, MnTBAP appeared to exert its effects in increasing IL-1β secretion by a ROS independent mechanism.
Highlights.
Nlrp3 inflammasome activation caused by SAA involves mitochondrial reactive oxygen species.
A mechanism by which mitochondrial dysfunction cause sustained IL-1β is proposed.
The mechanism is based on impaired Nlrp3 complex disassembly.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Dinarello CA. Immunological and inflammatory functions of the interleukin-1 family. Annu Rev Immunol. 2009;27:519–550. doi: 10.1146/annurev.immunol.021908.132612. [DOI] [PubMed] [Google Scholar]
- 2.Clutterbuck AL, Mobasheri A, Shakibaei M, Allaway D, Harris P. Interleukin-1beta-induced extracellular matrix degradation and glycosaminoglycan release is inhibited by curcumin in an explant model of cartilage inflammation. Ann N Y Acad Sci. 2009;1171:428–435. doi: 10.1111/j.1749-6632.2009.04687.x. [DOI] [PubMed] [Google Scholar]
- 3.Daheshia M, Yao JQ. The interleukin 1beta pathway in the pathogenesis of osteoarthritis. J Rheumatol. 2008;35:2306–2312. doi: 10.3899/jrheum.080346. [DOI] [PubMed] [Google Scholar]
- 4.Griffin WS, Liu L, Li Y, Mrak RE, Barger SW. Interleukin-1 mediates Alzheimer and Lewy body pathologies. J Neuroinflammation. 2006;3:5. doi: 10.1186/1742-2094-3-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gris D, Ye Z, Iocca HA, Wen H, Craven RR, Gris P, Huang M, Schneider M, Miller SD, Ting JP. NLRP3 plays a critical role in the development of experimental autoimmune encephalomyelitis by mediating Th1 and Th17 responses. J Immunol. 2010;185:974–98. doi: 10.4049/jimmunol.0904145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhou R, Tardivel A, Thorens B, Choi I, Tschopp J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat Immunol. 2010;11:136–140. doi: 10.1038/ni.1831. [DOI] [PubMed] [Google Scholar]
- 7.Masters SL, Dunne A, Subramanian SL, Hull RL, Tannahill GM, Sharp FA, Becker C, Franchi L, Yoshihara E, Chen Z, Mullooly N, Mielke LA, Harris J, Coll RC, Mills KH, Mok KH, Newsholme P, Nunez G, Yodoi J, Kahn SE, Lavelle EC, O’Neill LA. Activation of the NLRP3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced IL-1beta in type 2 diabetes. Nat Immunol. 2012;11:897–904. doi: 10.1038/ni.1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wanderer AA. Interleukin-1beta targeted therapy in severe persistent asthma (SPA) and chronic obstructive pulmonary disease (COPD): proposed similarities between biphasic pathobiology of SPA/COPD and ischemia-reperfusion injury. Isr Med Assoc J. 2008;10:837–842. [PubMed] [Google Scholar]
- 9.Dorfmuller P, Perros F, Balabanian K, Humbert M. Inflammation in pulmonary arterial hypertension. Eur Respir J. 2003;22:358–363. doi: 10.1183/09031936.03.00038903. [DOI] [PubMed] [Google Scholar]
- 10.Soon E, Holmes AM, Treacy CM, Doughty NJ, Southgate L, Machado RD, Trembath RC, Jennings S, Barker L, Nicklin P, Walker C, Budd DC, Pepke-Zaba J, Morrell NW. Elevated levels of inflammatory cytokines predict survival in idiopathic and familial pulmonary arterial hypertension. Circulation. 2010;122:920–927. doi: 10.1161/CIRCULATIONAHA.109.933762. [DOI] [PubMed] [Google Scholar]
- 11.Stutz A, Golenbock DT, Latz E. Inflammasomes: too big to miss. J Clin Invest. 2009;119:3502–3511. doi: 10.1172/JCI40599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Martinon F, Mayor A, Tschopp J. The inflammasomes: guardians of the body. Annu Rev Immunol. 2009;27:229–265. doi: 10.1146/annurev.immunol.021908.132715. [DOI] [PubMed] [Google Scholar]
- 13.Piccini A, Carta S, Tassi S, Lasiglie D, Fossati G, Rubartelli A. ATP is released by monocytes stimulated with pathogen-sensing receptor ligands and induces IL-1beta and IL-18 secretion in an autocrine way. Proc Natl Acad Sci U S A. 2008;105:8067–8072. doi: 10.1073/pnas.0709684105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cruz CM, Rinna A, Forman HJ, Ventura AL, Persechini PM, Ojcius DM. ATP activates a reactive oxygen species-dependent oxidative stress response and secretion of proinflammatory cytokines in macrophages. J Biol Chem. 2007;282:2871–2879. doi: 10.1074/jbc.M608083200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dostert C, Petrilli V, Van Bruggen R, Steele C, Mossman BT, Tschopp J. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science. 2008;320:674–677. doi: 10.1126/science.1156995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tschopp J, Schroder K. NLRP3 inflammasome activation: The convergence of multiple signalling pathways on ROS production? Nat Rev Immunol. 2010;10:210–215. doi: 10.1038/nri2725. [DOI] [PubMed] [Google Scholar]
- 17.van de Veerdonk FL, Smeekens SP, Joosten LA, Kullberg BJ, Dinarello CA, van der Meer JW, Netea MG. Reactive oxygen species-independent activation of the IL-1beta inflammasome in cells from patients with chronic granulomatous disease. Proc Natl Acad Sci U S A. 2010;107:3030–3033. doi: 10.1073/pnas.0914795107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Meissner F, Seger RA, Moshous D, Fischer A, Reichenbach J, Zychlinsky A. Inflammasome activation in NADPH oxidase defective mononuclear phagocytes from patients with chronic granulomatous disease. Blood. 2010;116:1570–1573. doi: 10.1182/blood-2010-01-264218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brookes PS, Yoon Y, Robotham JL, Anders MW, Sheu SS. Calcium, ATP, and ROS: a mitochondrial love hate triangle. Am J Physiol Cell Physiol. 2004;287:C817–833. doi: 10.1152/ajpcell.00139.2004. [DOI] [PubMed] [Google Scholar]
- 20.Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469:221–225. doi: 10.1038/nature09663. [DOI] [PubMed] [Google Scholar]
- 21.Noh KT, Park YM, Cho SG, Choi EJ. GSK3-beta-induced ASK1 stabilization is crucial in LPS-induced endotoxin shock. Exp Cell Res. 2011;317:1663–1668. doi: 10.1016/j.yexcr.2011.03.022. [DOI] [PubMed] [Google Scholar]
- 22.Prinarakis E, Chantzoura E, Thanos D, Spyrou G. S-glutathionylation of IRF3 regulates IRF3-CBP interaction and activation of the IFN beta pathway. EMBO J. 2008;27:865–875. doi: 10.1038/emboj.2008.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wong SW, Kwon MJ, Choi AM, Kim HP, Nakahira K, Hwang DH. Fatty acids modulate Toll-like receptor 4 activation through regulation of receptor dimerization and recruitment into lipid rafts in a reactive oxygen species-dependent manner. J Biol Chem. 2009;284:27384–27392. doi: 10.1074/jbc.M109.044065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ather JL, Ckless K, Martin R, Foley KL, Suratt BT, Boyson JE, Fitzgerald KA, Flavell RA, Eisenbarth SC, Poynter ME. Serum Amyloid A Activates the NLRP3 Inflammasome and Promotes Th17 Allergic Asthma in Mice. J Immunol. 2011;187:64–73. doi: 10.4049/jimmunol.1100500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Carta S, Tassi S, Pettinati I, Delfino L, Dinarello CA, Rubartelli A. The rate of IL-1{beta} secretion in different myeloid cells varies with the extent of redox response to Toll-like receptor triggering. J Biol Chem. 2011;286:27069–80. doi: 10.1074/jbc.M110.203398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Waterhouse NJ, Steel R, Kluck R, Trapani JA. Assaying cytochrome C translocation during apoptosis. Methods Mol Biol. 2004;284:307–313. doi: 10.1385/1-59259-816-1:307. [DOI] [PubMed] [Google Scholar]
- 27.Myhre O, Andersen JM, Aarnes H, Fonnum F. Evaluation of the probes 2′,7′-dichlorofluorescin diacetate, luminol, and lucigenin as indicators of reactive species formation. Biochem Pharmacol. 2003;65:1575–1582. doi: 10.1016/s0006-2952(03)00083-2. [DOI] [PubMed] [Google Scholar]
- 28.Gross O, Thomas CJ, Guarda G, Tschopp J. The inflammasome: an integrated view. Immunol Rev. 2011;243:136–151. doi: 10.1111/j.1600-065X.2011.01046.x. [DOI] [PubMed] [Google Scholar]
- 29.Tassi S, Carta S, Delfino L, Caorsi R, Martini A, Gattorno M, Rubartelli A. Altered redox state of monocytes from cryopyrin associated periodic syndromes causes accelerated IL-1beta secretion. Proc Natl Acad Sci U S A. 2010;107:9789–9794. doi: 10.1073/pnas.1000779107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Melov S, Schneider JA, Day BJ, Hinerfeld D, Coskun P, Mirra SS, Crapo JD, Wallace DC. A novel neurological phenotype in mice lacking mitochondrial manganese superoxide dismutase. Nat Genet. 1998;18:159–163. doi: 10.1038/ng0298-159. [DOI] [PubMed] [Google Scholar]
- 31.Bulua AC, Simon A, Maddipati R, Pelletier M, Park H, Kim KY, Sack MN, Kastner DL, Siegel RM. Mitochondrial reactive oxygen species promote production of proinflammatory cytokines and are elevated in TNFR1-associated periodic syndrome (TRAPS) J Exp Med. 2011;208:519–533. doi: 10.1084/jem.20102049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tschopp J. Mitochondria: Sovereign of inflammation? Eur J Immunol. 2011;41:1196–1202. doi: 10.1002/eji.201141436. [DOI] [PubMed] [Google Scholar]
- 33.Chen Q, Vazquez EJ, Moghaddas S, Hoppel CL, Lesnefsky EJ. Production of reactive oxygen species by mitochondria: central role of complex III. J Biol Chem. 2003;278:36027–36031. doi: 10.1074/jbc.M304854200. [DOI] [PubMed] [Google Scholar]
- 34.Aronis A, Melendez JA, Golan O, Shilo S, Dicter N, Tirosh O. Potentiation of Fas mediated apoptosis by attenuated production of mitochondria derived reactive oxygen species. Cell Death Differ. 2003;10:335–344. doi: 10.1038/sj.cdd.4401150. [DOI] [PubMed] [Google Scholar]
- 35.Mukhopadhyay P, Rajesh M, Hasko G, Hawkins BJ, Madesh M, Pacher P. Simultaneous detection of apoptosis and mitochondrial superoxide production in live cells by flow cytometry and confocal microscopy. Nat Protoc. 2007;2:2295–2301. doi: 10.1038/nprot.2007.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li N, Ragheb K, Lawler G, Sturgis J, Rajwa B, Melendez JA, Robinson JP. DPI induces mitochondrial superoxide-mediated apoptosis. Free Radic Biol Med. 2003;34:465–477. doi: 10.1016/s0891-5849(02)01325-4. [DOI] [PubMed] [Google Scholar]
- 37.Waterhouse NJ, Goldstein JC, von Ahsen O, Schuler M, Newmeyer DD, Green DR. Cytochrome c maintains mitochondrial transmembrane potential and ATP generation after outer mitochondrial membrane permeabilization during the apoptotic process. J Cell Biol. 2001;153:319–328. doi: 10.1083/jcb.153.2.319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lamkanfi M, Kanneganti TD. Nlrp3: an immune sensor of cellular stress and infection. Int J Biochem Cell Biol. 2010;42:792–795. doi: 10.1016/j.biocel.2010.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Embry CA, Franchi L, Nunez G, Mitchell TC. Mechanism of impaired NLRP3 inflammasome priming by monophosphoryl lipid A. Sci Signal. 2011;4:ra28. doi: 10.1126/scisignal.2001486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hirschfeld M, Ma Y, Weis JH, Vogel SN, Weis JJ. Cutting edge: repurification of lipopolysaccharide eliminates signaling through both human and murine toll-like receptor 2. J Immunol. 2000;165:618–622. doi: 10.4049/jimmunol.165.2.618. [DOI] [PubMed] [Google Scholar]
- 41.Yang CS, Shin DM, Kim KH, Lee ZW, Lee CH, Park SG, Bae YS, Jo EK. NADPH oxidase 2 interaction with TLR2 is required for efficient innate immune responses to mycobacteria via cathelicidin expression. J Immunol. 2009;182:3696–3705. doi: 10.4049/jimmunol.0802217. [DOI] [PubMed] [Google Scholar]
- 42.Konorev EA, Kennedy MC, Kalyanaraman B. Cell-permeable superoxide dismutase and glutathione peroxidase mimetics afford superior protection against doxorubicin-induced cardiotoxicity: the role of reactive oxygen and nitrogen intermediates. Arch Biochem Biophys. 1999;368:421–428. doi: 10.1006/abbi.1999.1337. [DOI] [PubMed] [Google Scholar]
- 43.Nakamura Y, Feng Q, Kumagai T, Torikai K, Ohigashi H, Osawa T, Noguchi N, Niki E, Uchida K. Ebselen a glutathione peroxidase mimetic seleno-organic compound as a multifunctional antioxidant Implication for inflammation-associated carcinogenesis. J Biol Chem. 2002;277:2687–2694. doi: 10.1074/jbc.M109641200. [DOI] [PubMed] [Google Scholar]
- 44.Kwon MJ, Han J, Kim BH, Lee YS, Kim TY. Superoxide dismutase 3 suppresses hyaluronic Acid fragments mediated skin inflammation by inhibition of toll-like receptor 4 signaling pathway: superoxide dismutase 3 inhibits reactive oxygen species-induced trafficking of toll-like receptor 4 to lipid rafts. Antioxid Redox Signal. 2012;16:297–313. doi: 10.1089/ars.2011.4066. [DOI] [PubMed] [Google Scholar]
- 45.Seror C, Melki MT, Subra F, Raza SQ, Bras M, Saidi H, Nardacci R, Voisin L, Paoletti A, Law F, Martins I, Amendola A, Abdul-Sater AA, Ciccosanti F, Delelis O, Niedergang F, Thierry S, Said Sadier N, Lamaze C, Metivier D, Estaquier J, Fimia GM, Falasca L, Casetti R, Modjtahedi N, Kanellopoulos J, Mouscadet JF, Ojcius DM, Piacentini M, Gougeon ML, Kroemer G, Perfettini JL. Extracellular ATP acts on P2Y2 purinergic receptors to facilitate HIV-1 infection. J Exp Med. 2011;208:1823–1834. doi: 10.1084/jem.20101805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shi CS, Shenderov K, Huang NN, Kabat J, Abu Asab M, Fitzgerald KA, Sher A, Kehrl JH. Activation of autophagy by inflammatory signals limits IL-1beta production by targeting ubiquitinated inflammasomes for destruction. Nat Immunol. 2012;13:255–263. doi: 10.1038/ni.2215. [DOI] [PMC free article] [PubMed] [Google Scholar]