

Abstract
Background: Avibacterium paragallinarum, the causative agent of infectious coryza, is a highly contagious respiratory acute disease of poultry, which affects commercial chickens, laying hens and broilers worldwide. Methodology: In this study, we performed the whole genome sequencing, assembly and annotation of a Peruvian isolate of A. paragallinarum. Genome was sequenced in a 454 GS FLX Titanium system. De novo assembly was performed and annotation was completed with GS De Novo Assembler 2.6 using the H. influenzae str. F3031 gene model. Manual curation of the genome was performed with Artemis. Putative function of genes was predicted with Blast2GO. Virulence factors were identified by comparison with the Virulence Factor Database. Results: The genome obtained has a length of 2.47 Mb with 40.66% of GC content. Seventy five large contigs (>500 nt) were obtained, which comprised 1,204 predicted genes. All the contigs are available in Genbank [GenBank: PRJNA64665]. A total of 103 virulence factors, reported in the Virulence Factor Database, were found in A. paragallinarum. Forty four of them are present in 7 species of Haemophilus, which are related with pathogenesis, virulence and host immune system evasion. A tetracycline-resistance associated transposon (Tn10), was found in A. paragallinarum, possibly acting as a defense mechanism. Discussion and conclusion: The availability of A. paragallinarum genome represents an important source of information for the development of diagnostic tests, genotyping, and novel antigens for potential vaccines against infectious coryza. Identification of virulence factors contributes to better understanding the pathogenesis, and planning efforts for prevention and control of the disease.
Keywords: Infectious coryza, genome sequencing, virulence factors, syntenic homology, Tn10 transposon
Background
Infectious coryza is a highly contagious acute respiratory disease of layers and chickens, caused by the gram negative bacterium Avibacterium paragallinarum. This organism was initially classified as Haemophilus paragallinarum, but later re-classified as A. paragallinarum [1]. The disease has a worldwide distribution, being endemic in commercial laying hens. Broilers may also be severely affected with depression, decreased feed and water consumption that negatively affect their growth. This lead to important economic losses associated to poor productivity, increased mortality up to 48% and a reduction up to 75% of egg production. Although this disease is rarely seen in broilers, an outbreak in Panama caused mortality and 45% of production losses [2].
A. paragallinarum produces an acute catarrhal inflammation of mucous membranes and sinus passages, as well as catarrhal and subcutaneous edema of face and wattles. It is characterized by nasal discharge, watery eyes, facial swelling, anorexia, diarrhea and swelling of wattles. A. paragallinarum infection was also reported in non-respiratory organs such as liver, kidney and tarsus [3].
Nowadays, the use of inactivated A. paragallinarum vaccines against infectious coryza formulated from local strains is the best way to control the disease [4, 5]. However, these vaccines have the disadvantage of inducing protection only against the serotypes included in the vaccine, but not to other strains [6]. The lack of effective vaccines for A. paragallinarum requires new efforts and the use of novel approaches. The A. paragallinarum genome sequence is an important source of information for a better understanding of the biology of this pathogen, in particular in the development of vaccines and more accurate methods of genotyping.
In this study we present the genome sequence and its annotation of a circulating pathogenic strain of A. paragallinarum isolated from a broiler outbreak in Ica, a city in the central coast of Peru. This strain was previously identified as serovar C. This genome was compared with related organisms, focusing on virulence factors.
Methodology
Microbiological culture:
A Peruvian local isolate of A. paragallinarum was obtained by direct culture of the infraorbital sinuses of a broiler from an infectious coryza outbreak in a local farm (Ica, Peru). The isolate was cultured in chocolate agar [7] with factor X-V and incubated in a microenvironment with 5% CO2 at 40°C for 48 hours. After three passages, the colonies were collected in a modified BHI culture with 50% glycerol and stored at -80°C.
DNA library and sequencing:
Genomic DNA was extracted from reactivated bacteria using DNeasy Blood & Tissue Kit (Qiagen, Valencia, CA, USA) with slightly modification that includes a lysozyme treatment at 37°C for 1h followed by incubation with Proteinase K at 56°C overnight. The quantity and quality of the eluted DNA was tested with picogreen kit (Invitrogen, Carlsbad, CA, USA) and Biophotometer Plus (Eppendorf, Hamburg, Germany) respectively.
A 454-FLX shotgun library was prepared with 500 ng of genomic DNA using a GS FLX Titanium Rapid Library Preparation Kit (Roche, Branford, CT, USA). Quality assessment was performed with the Agilent Bioanalyzer using High Sensitivity DNA Kit (Agilent Technologies, Santa Clara, CA, USA). The obtained library was clonally amplified within a water-in-oil emulsion (EmPCR). EmPCR and titration by enrichment were made using GS FLX Titanium SV emPCR Kit Lib-L (Roche, Branford, CT, USA). Then, the DNA beads were sequenced in a GS FLX Titanium PicoTiterPlate 70×75 (Roche, Branford, CT, USA) on the GS FLX+ Sequencing System.
Data processing and assembly:
The raw signal data were processed with the software GS Run Processor to obtain the reads. Given the lack of a reference genome for mapping assembly, de novo assembly was conducted using GS De Novo Assembler 2.6. The chicken genome [GenBank: PRJNA10808] was filtered out of the assembly. Quality control was performed using GS De Novo Assembler and CLC Main Workbench 6.7. We discarded chimeric sequences and homopolymeric errors originated by the pyrosequencing process itself.
Genome annotation:
A pseudomolecule was built by arbitrarily joining the 75 largest contigs, adding 100 ambiguous nucleotides (represented with N) between each pair of contigs. The pseudomolecule was tested for presence of coding DNA sequences using Glimmer v3.02 [8] SNAP [9], and Augustus [10] with a gene model trained with the annotated genome of H. influenzae str. F3031 [GenBank:PRJNA62123]. Genome annotation was performed using the semi-automatic annotation pipeline Maker-GMOD [11]. Specific analysis included ab-initio gene prediction from SNAP, Augustus, Fgenesh [12] and GeneMark [13]; protein alignments from EXONERATE [14] and BLASTx [15], and repetitive regions identified by RepeatMasker [16]. Gene function and metabolic pathways predictions were obtained with the Blast2GO annotation pipeline [17]. A manual curation of the genome annotation was performed using Artemis [18]. This procedure included the verification of Open Reading Frames, stop/start codon of coding sequences and indels.
Virulence factors analysis:
Local BLAST [15] was performed between the A. paragallinarum genome and the Virulence Factor Database (VFDB) [19], using e-value 1.10-3 and 60% of identity as cutoff. The predicted virulence factors in A. paragallinarum were compared with the virulence factors compiled in a comparative table indexed in VFDB for all Haemophilus species available in this database, which are: H. ducreyi 35000HP [GenBank:NC_002940], H. influenzae Rd KW20 [GenBank:NC_000907], H. influenzae 86-028NP [GenBank:NC_007146], H. influenzae PittEE [GenBank:NC_009566], H. influenzae PittGG [GenBank:NC_009567], H. somnus 129PT [GenBank:NC_008309] and H. somnus 2336 [GenBank:NC_010519]. We compare with Haemophilus because it was the closest organism with information available in VFDB.
Results
Sequencing, assembly and annotation:
The average fragment length was 600-900 nt. The whole shotgun sequencing reached a mean depth of 23X, producing 183,434 reads (62'190,061 nt). 98.12% of the total reads formed contigs, obtaining 93 contigs (2'465,440 nt) with a N50 of 113,569 nt. The 75 largest contigs (>500 nt) comprised 2'459,730 nt. 99.70% of these showed a quality greater than Q40. The largest contig size was 439,531 nt and the average contig size was 32,796 nt. This assembly produced an estimated genome size of 2.47 Mb with 40.66% of GC content (Figure 1). All the contigs are available in Genbank [GenBank: PRJNA64665].
Figure 1.
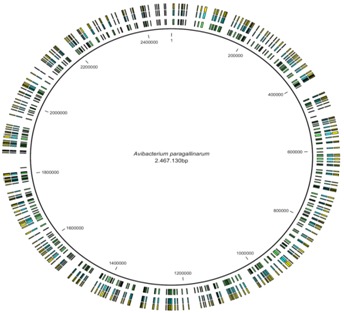
Circular representation of Avibacterium paragallinarum genome. Contigs obtained from whole genome sequencing was joined in a contiguous pseudo molecule, which was used to predict genes and estimate the GC content. This circular plot shows the distribution of coding DNA sequences, genes and mRNAs obtained in the A. paragallinarum genome. Each bar in the internal circle represents the mRNAs, in the middle circle represents the genes, and in the outer circle represents the coding DNA sequences, respectively.
A total of 1,204 genes were predicted from the pseudomolecule. All of these were assigned with a putative function using Blast2GO Table 1 (see supplementary material). Eight major categories and 49 sub-categories from gene ontology were identified Table 2 (see supplementary material)). The distribution of orthologous genes clusters is presented in (Figure 2). H. influenzae, followed by Aggregatibacter actinomycetemcomitans and P. multocida were the organisms that showed the highest number of homologue genes with A. paragallinarum (Figure 3).
Figure 2.
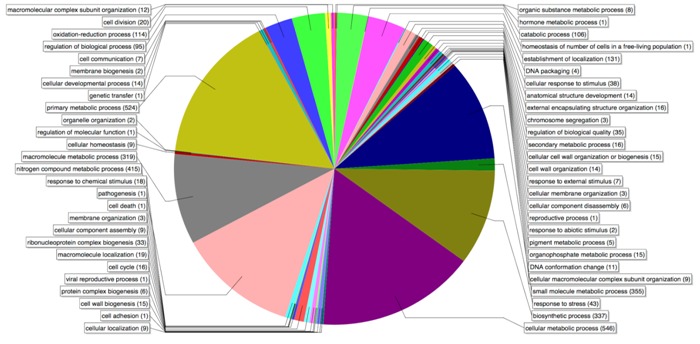
Functional classification of genes. Predicted genes from annotation process were processed in Blast2GO for functional annotation. This graphics shows the relative abundance of biological functions assigned.
Figure 3.
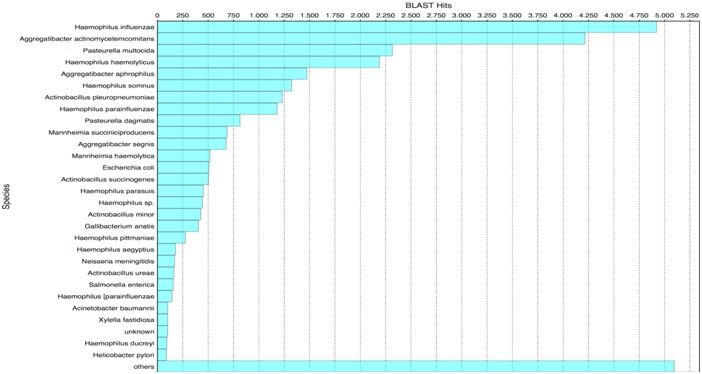
Amount of sequences matching with Avibacterium paragallinarum, by organism. Distribution of the number sequences which matches with the predicted ORFs of A. paragallinarum, by organism, in the Blast2GO functional annotation. Horizontal bars corresponds to the number of significant BLAST hits between A. paragallinarum predicted ORFs and the non-redundant database from GenBank, for each organism listed. H. influenzae, A. actinomycetemcomitans, P. multocida, and H. haemolyticus were the mayor contributors for A. paragallinarum genome annotation.
Virulence factors analysis:
One hundred and three virulence factors from the VFDB were found in A. paragallinarum Table 3 (see supplementary material), and 44 of them were found in common with the 7 Haemophilus compared in the database [19] Table 4 (Available with authors). From these results, we found an IgA protease, adherence-related factors (ompP5 and type IV pili proteins); and a region of 6,488 nt highly identical (>99%) to the transposon Tn10, containing four tetracycline resistance genes Table 4 (Available with authors) with (Figure 4).
Figure 4.
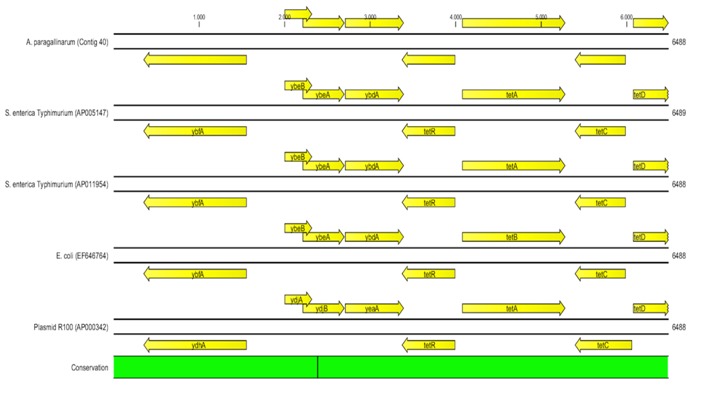
Tn10 transposon and tetracycline-resistance genes from Avibacterium paragallinarum. Sequence comparison and gene annotations (arrows) in a region of Tn10 transposon found in A. paragallinarum genome sequencing (Contig40). This transposon was reported in plasmid sequences of chicken pathogens: Salmonella enterica subsp. enterica serovar Typhimurium plasmid R64 [GenBank: AP005147], Salmonella enterica subsp. enterica serovar Typhimurium plasmid R621a [GenBank:AP011954], Plasmid R100 [GenBank:AP000342] and Escherichia coli transposon Tn10 [GenBank:EF646764]. Conservation bar (in green) shows there are few variations (black lines inside) between the compared sequences. This region was found with 99.9% of identity with the organisms listed above.
Discussion & Conclusion
The present study presents for first time a draft genome sequence of A. paragallinarum, its annotation and comparison with Haemophilus, identifying potential virulence factors. Interestingly, Tn10 transposon was found partially in A. paragallinarum sequencing data. This transposon was found in plasmids from several chicken pathogens, including Escherichia coli, and Salmonella enterica serovar Typhimurium. Tn10 has been used to induce mutagenesis to study the effect of mutations in the fitness [20], and for the construction of a tagged mini-Tn10 plasmid bank to attenuate the pathogen virulence, which could be used as live attenuated vaccine [21]. Tn10 is a transposon of 9,147 nt, comprising four genes associated to tetracycline resistance (tetR/A/C/D) [22]. These genes may potentially cause tetracycline resistance in A. paragallinarum, which needs to be further studied. Tn10 typically has two insertion sequences (IS10-L and IS10-R) and two transposases (ydgA and yedA) flanking them. However, these sequences were not found in the A. paragallinarum corresponding contig (6,488 nt), probably due to lack of coverage. IgA protease was found in the A. paragallinarum genome, suggesting that this specie may be able to hydrolyze chicken IgA-like inmunoglobulins. IgA proteases were reported in related species as H. influenzae [23], Neisseria meningitidis [24]. This protease is known to cleave host secreted IgA immunoglobulin enabling to circumvent host mucosal defense mechanisms; enhancing the ability to infect respiratory tract [25].
Studies in bacterial pathogens have shown that the profile of virulence genes are associated with disease [26]. Therefore, genomic comparison analysis provides the basis for understanding pathogenicity and for rational vaccine design and immunoassays development. It was interesting to find OmpP5 and Type IV pili virulence factors. It is known that OmpP5 is an outer membrane protein homologue to E. coli OmpA [27], the major protective antigen responsible for the integrity of the outer membrane, which induces strong antibody response in chickens [28]. Type IV pili are involved in a variety of bacterial functions, including cell adhesion [29], bacteriophage adsorption, plasmid transfer [30], and twitching motility, a form of flagellum-independent locomotion [31]. Contamination with chicken DNA was reduced from the assembly by filtering the chicken genome. Assembly errors, contig ordering and genome closure were not made since no paired-end library was produced and no reference genome was available yet. Therefore, it is important to perform complementary studies to build the complete chromosome in order to define the genetic structure and perform more accurate comparisons with related organisms.
The availability of A. paragallinarum genome is an important achievement for poultry industry, which would facilitate the development of useful tools against infectious coryza. Furthermore, the identification of virulence factors and immunogenic and antibiotic-resistant factors contributes to understanding the pathogenesis, and contribute to efforts for prevention and control of the disease.
Authors' contributions
Conception and design: DR, AC and MFD. Acquisition, analysis and interpretation of data: DR, AC, MT, OA, MR, MZi. Drafting of the manuscript and revising it critically: DR, AC, MT, OA, MR, HV, AHG, RIL, LES, MZa, LTL, IB, MFS, EI, MZi and MFD. Final approval of the version to be published: DR, MZi, MFD.
Supplementary material
Acknowledgments
To Rocío Quispe, Isabel García, David Tarazona and Lenin Maturrano for their participation in the course of this project.
Footnotes
Citation:Requena et al, Bioinformation 9(10): 528-536 (2013)
References
- 1.Blackall PJ, et al. Int J Syst Evol Microbiol. 2005;55:353. doi: 10.1099/ijs.0.63357-0. [DOI] [PubMed] [Google Scholar]
- 2.Calderon EN, et al. Avian Dis. 2010;54:1095. doi: 10.1637/9123-110409-Case.1. [DOI] [PubMed] [Google Scholar]
- 3.Droual R, et al. Avian Dis. 1990;34:1009. [PubMed] [Google Scholar]
- 4.Wambura PN, et al. Trop Anim Health Prod. 2010;42:483. doi: 10.1007/s11250-009-9447-z. [DOI] [PubMed] [Google Scholar]
- 5.Chukiatsiri K, et al. Avian Dis. 2009;53:382. doi: 10.1637/8563-121908-Reg.1. [DOI] [PubMed] [Google Scholar]
- 6.Wu JR, et al. Vaccine. 2011;29:660. doi: 10.1016/j.vaccine.2010.11.040. [DOI] [PubMed] [Google Scholar]
- 7.Gunn BA. J Clin Microbiol. 1984;20:822. doi: 10.1128/jcm.20.4.822-823.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Salzberg SL, et al. Nucleic Acids Res. 1998;26:544. doi: 10.1093/nar/26.2.544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Korf I. BMC Bioinformatics. 2004;5:59. doi: 10.1186/1471-2105-5-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Keller O, et al. Bioinformatics. 2011;27:757. doi: 10.1093/bioinformatics/btr010. [DOI] [PubMed] [Google Scholar]
- 11.Cantarel BL, et al. Genome Res. 2008;18:188. doi: 10.1101/gr.6743907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tyson GW, et al. Nature. 2004;428:37. doi: 10.1038/nature02340. [DOI] [PubMed] [Google Scholar]
- 13.Besemer J, et al. Nucleic Acids Res. 2001;29:2607. doi: 10.1093/nar/29.12.2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Slater GS, Birney E. BMC Bioinformatics. 2005;6:31. doi: 10.1186/1471-2105-6-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Altschul SF, et al. Nucleic Acids Res. 1997;25:3389. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bao Z, Eddy SR. Genome Res. 2002;12:1269. doi: 10.1101/gr.88502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Conesa A, et al. Bioinformatics. 2005;21:3674. doi: 10.1093/bioinformatics/bti610. [DOI] [PubMed] [Google Scholar]
- 18.Rutherford K, et al. Bioinformatics. 2000;16:944. doi: 10.1093/bioinformatics/16.10.944. [DOI] [PubMed] [Google Scholar]
- 19.Chen L, et al. Nucleic Acids Res. 2012;40:D641. doi: 10.1093/nar/gkr989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tamura M, et al. J Bacteriol. 2012;194:1919. doi: 10.1128/JB.06652-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fuller TE, et al. Microb Pathog. 2000;29:25. doi: 10.1006/mpat.2000.0365. [DOI] [PubMed] [Google Scholar]
- 22.Haniford DB. Crit Rev Biochem Mol Biol. 2006;41:407. doi: 10.1080/10409230600987415. [DOI] [PubMed] [Google Scholar]
- 23.Mistry DV, Stockley RA. Virulence. 2011;2:103. doi: 10.4161/viru.2.2.15401. [DOI] [PubMed] [Google Scholar]
- 24.Vitovski S, Sayers JR. Infect Immun. 2007;75:2875. doi: 10.1128/IAI.01671-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mistry D, Stockley RA. Int J Biochem Cell Biol. 2006;38:1244. doi: 10.1016/j.biocel.2005.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sassetti C, Rubin EJ. Curr Opin Microbiol. 2002;5:27. doi: 10.1016/s1369-5274(02)00281-3. [DOI] [PubMed] [Google Scholar]
- 27.Sugawara E, Nikaido H. J Biol Chem. 1992;267:2507. [PubMed] [Google Scholar]
- 28.Fadl AA, et al. J Appl Microbiol. 2002;92:180. doi: 10.1046/j.1365-2672.2002.01511.x. [DOI] [PubMed] [Google Scholar]
- 29.Manning PA, Meyer TF. Gene. 1997;192:1. doi: 10.1016/s0378-1119(97)00036-x. [DOI] [PubMed] [Google Scholar]
- 30.Kim SR, Komano T. J Bacteriol. 1997;179:3594. doi: 10.1128/jb.179.11.3594-3603.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Merz AJ, et al. Nature. 2000;407:98. doi: 10.1038/35024105. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.