

Abstract
Simple Sequence Repeats (SSR), also called microsatellite, is very useful for genetic marker development and genome application. The increasing whole sequences of more and more large genomes provide sources for SSR mining in silico. However currently existing SSR mining tools can’t process large genomes efficiently and generate no or poor statistics. Genome-wide Microsatellite Analyzing Tool (GMATo) is a novel tool for SSR mining and statistics at genome aspects. It is faster and more accurate than existed tools SSR Locator and MISA. If a DNA sequence was too long, it was chunked to short segments at several Mb followed by motifs generation and searching using Perl powerful pattern match function. Matched loci data from each chunk were then merged to produce final SSR loci information. Only one input file is required which contains raw fasta DNA sequences and output files in tabular format list all SSR loci information and statistical distribution at four classifications. GMATo was programmed in Java and Perl with both graphic and command line interface, either executable alone in platform independent manner with full parameters control. Software GMATo is a powerful tool for complete SSR characterization in genomes at any size.
Availability
The soft GMATo is freely available at http://sourceforge.net/projects/gmato/files/?source=navbar or on contact
Keywords: Genome, Microsatellite, SSR, Marker development, Software
Background
Simple Sequence Repeats (SSR) or microsatellite is a relative short tandem repeats of DNA [1, 2]. Its length polymorphism is specie specific and inheritable, which makes SSR very useful for developing genetic SSR marker widely used in linking genome sequence with traits, diversity investigation, map-base cloning and molecular breeding [2]. There are some useful software and tools developed for SSRs discovery and marker development in silico. However, they were designed before large genome era and have two major limitations: i) too low sequence processing capability and slow speed as pointed by Sharma [2] et al to deal with large genomes while more large genomes i.e. those from crops become important sources for SSR characterization with the benefit from the advanced next generation sequencing technology, ii) no or simple statistical function provided such as TROLL [3]. In addition, some tools have platform dependence i.e. SSR Locator [4] and SciRoko [5]. Most command tools have no graphic interface and very limited other functions, i.e. tool MISA [6, 7]. In order to overcome those limitations mentioned above, novel software named GMATo was developed for faster and accurate SSR discovery and comprehensively statistical analyzing especially for large genomes running at multiple platforms with both graphic and command interface.
Methodology
The soft GMATo was written in Perl and Java language. Java was used for developing graphic interface. Perl was used to discover the microsatellite and perform statistical analyzing. In GMATo DNA sequences are formatted first and the long DNA sequence is chunked to small segments at several Mb for easy processing. All microsatellite motifs consisting of A, T, G and C nucleotide of DNA at user controlled length are generated using Perl meta-characters and regular expression pattern. All motifs are searched greedily through each DNA chunk using Perl powerful pattern matching function. The returned values are used to generate SSR loci information at each chunk and the final SSR loci data at a chromosome after merging data from chunks. This method allows microsatellite discover efficiently in any genome with any size theoretically. Statistical classification and summarization were performed at four levels i.e. motif length, motif composition, grouped complementary motifs and chromosome/scaffold. A flowchart was shown in (Figure 1).
Figure 1.
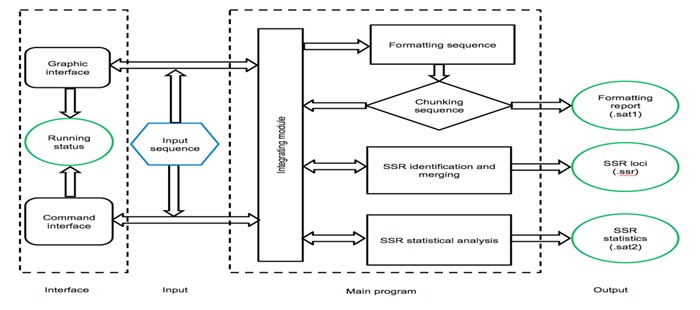
A flowchart of software GMATo.
Validation
The performance of microsatellite identification in recent published Setaria Italica entire genome[8] showed GMATo ran faster than either of most widely used tools, SSR Locator and MISA, in all three platforms Table 1 (see supplementary material). It was also easily to mine SSR in the genome in a normal computer because processing one chunked segment at a time in GMATo required less computing memory. A total number of 46,739, 46,625 and 46,782 microsatellite loci were identified by GMATo, SSR Locator and MISA respectively (Table 1), suggesting more accurate SSR mining than SSR Locator. Manually comparison of these loci revealed that the extra loci from MISA are mined redundantly in the overlapped microsatellites.
Software input
Both graphic user and command line interface were provided in GMATo, either executable independently in Windows, Linux or Mac OS system. Only one input file containing DNA sequence(s) in (raw) (multi-) FASTA format is required to be chosen in graphic mode or typed in command mode if taken the default parameters. The parameters are the motif length range, the minimum repeated times and an option for highlighting microsatellite (Figure 2A). The motif length can be set to any range instead of 1-10 bp given in most SSR mining tools.
Figure 2.
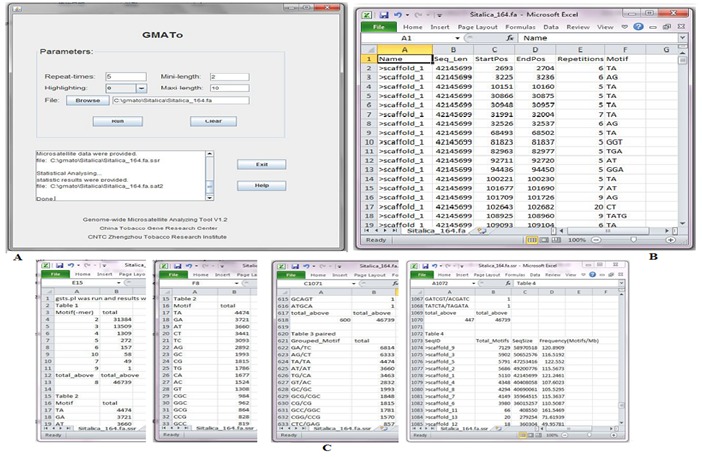
Image showing input and output of soft GMATo; (A) graphic input interface; (B) SSR loci information produced by GMATo; (C) SSR statistical data produced by GMATo.
Software output
The output files generated by GMATo are one formatting report, one file containing SSR loci information and another file containing statistical distribution of SSR. All output files are in a tab delimited plain text format for easily importing to other applications i.e. spread sheet for viewing or other manipulation (Figure 2 B, C).The formatting report summarizes the input sequence(s). The SSR loci file lists the input sequence ID and its length, starting and ending position of a microsatellite, the repeated times and the motif sequence.
The statistical distribution file provides statistical data at four different classifications at genome aspect. A summary of total is generated in the end of each classification. Classification I is the motif length statistics, providing overview information for the type, abundance in rank order. Classification II is the motif statistics based on sequence composition, i.e. motif composition, occurrence in ranked order. Classification III is the statistics of grouped complementary motifs, providing distribution data for complementary motifs such as TC/GA in a group and their occurrence in ranked order. Classification IV is the statistics of chromosome level distribution, providing the total occurrence of motif(s) and SSR frequency (loci/Mb) at each chromosome or super-scaffold.
Utility
GMATo can be used for efficient and faster microsatellite sequence identification from any given DNA sequences or genomes at any size. Detailed statistic distribution of microsatellites can be used for genome analysis.
Caveat and future development
Current version provides each perfect SSR loci information. The compound and long imperfect microsatellites can be calculated from the SSR loci output using additional script. For a future development, more functions including displaying statistical data graphically, primer designing, marker generation and electronic mapping markers into a genome will be added. The final goal is to develop an integrated powerful toolkit facilitating microsatellite characterization and marker development in large genomes.
Supplementary material
Footnotes
Citation:Wang et al, Bioinformation 9(10): 541-544 (2013)
References
- 1.Ellegren H. Nat Rev Genet. 2004;5:435. doi: 10.1038/nrg1348. [DOI] [PubMed] [Google Scholar]
- 2.Sharma P. Trends Biotechnology. 2007;25:490. doi: 10.1016/j.tibtech.2007.07.013. [DOI] [PubMed] [Google Scholar]
- 3.Castelo AT, et al. Bioinformatics. 2002;18:634. doi: 10.1093/bioinformatics/18.4.634. [DOI] [PubMed] [Google Scholar]
- 4.da Maia LC, et al. Int J Plant Genomics. 2008;412:696. doi: 10.1155/2008/412696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kofler R, et al. Bioinformatics. 2007;23:1683. doi: 10.1093/bioinformatics/btm157. [DOI] [PubMed] [Google Scholar]
- 6.Thiel TT, et al. Theor Appl Genet. 2003;106:411. doi: 10.1007/s00122-002-1031-0. [DOI] [PubMed] [Google Scholar]
- 7.Sonah H, et al. PLoS ONE. 2011;6:e21298. doi: 10.1371/journal.pone.0021298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bennetzen JL, et al. Nat Biotechnol. 2012;30:555. doi: 10.1038/nbt.2196. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.