

Abstract
Substantia nigra pars reticulata (SNr) GABAergic neurons are projection neurons that convey output from the basal ganglia to target structures. These neurons exhibit spontaneous regular firing, but also exhibit burst firing in the presence of NMDA or when excitatory glutamatergic input to the SNr is activated. Notably, an increase in burst firing is also seen in Parkinson's disease. Therefore, elucidating conductances that mediate spontaneous activity and changes of firing pattern in these neurons is essential for understanding how the basal ganglia control movement. Using ex vivo slices of guinea pig midbrain, we show that SNr GABAergic neurons express transient receptor potential melastatin 2 (TRPM2) channels that underlie NMDA-induced burst firing. Furthermore, we show that spontaneous firing rate and burst activity are modulated by the reactive oxygen species H2O2 acting via TRPM2 channels. Thus, our results indicate that activation of TRPM2 channels is necessary for burst firing in SNr GABAergic neurons and their responsiveness to modulatory H2O2. These findings have implications not only for normal regulation, but also for Parkinson's disease, which involves excitotoxicity and oxidative stress.
Introduction
The substantia nigra pars reticulata (SNr) is one of two major output nuclei of the basal ganglia network. This nucleus contains GABAergic neurons that are spontaneously active and provide tonic inhibition of neurons in target structures, which is essential for motor control. These SNr neurons typically exhibit regular spontaneous activity; however, stimulation of glutamatergic input from the subthalamic nucleus (STN) to the SNr can induce burst firing (Shen and Johnson, 2006). Moreover, in Parkinson's disease the regular discharge of SNr neurons is replaced by rhythmic burst firing, consistent with observations in other basal ganglia nuclei in Parkinson's and animal models that a change to burst firing contributes to the pathophysiology of the disease (Rivlin-Etzion et al., 2008; Weinberger and Dostrovsky, 2011).
The importance of both regular spontaneous activity and burst firing in SNr GABAergic neurons to basal ganglia function has spurred investigations to identify intrinsic mechanisms that modulate the firing rate and pattern of these neurons. It is recognized that tonic activity in SNr GABAergic neurons persists in the absence of synaptic input and that the depolarizing drive necessary for spontaneous activity is mediated in part by tetrodotoxin (TTX)-sensitive and TTX-insensitive Na+ channels, some of which convey a persistent Na+ current (Atherton and Bevan, 2005). Additional depolarizing drive from transient receptor potential (TRP) canonical 3 (TRPC3) channels (Zhou et al., 2008) is enhanced by dopamine, leading to an increase in firing rate (Zhou et al., 2009). Moreover, SNr GABAergic neurons exhibit burst firing in response to NMDA when recorded ex vivo (Ibáñez-Sandoval et al., 2007). NMDA-induced bursting persists as membrane potential oscillations when voltage-gated Na+ channels are blocked with TTX and is dependent on Ca2+ (Ibáñez-Sandoval et al., 2007). However, a more complete understanding of the ionic and molecular basis of NMDA-induced burst firing has remained elusive.
Among the intrinsic conductances that might contribute to burst firing in SNr neurons is a Ca2+-activated nonselective-cation conductance that underlies a plateau potential observed in response to depolarizing current pulses (Lee and Tepper, 2007b). Additionally, SNr GABAergic neurons exhibit an increase in firing rate in response to elevation of the reactive oxygen species, H2O2 (Lee et al., 2011). Both the plateau potential and H2O2-induced increases in firing rate are blocked by a nonselective TRP channel blocker (Lee and Tepper, 2007b; Lee et al., 2011), implicating TRP channels in these effects. Notably, although a variety of TRP channels are expressed in the brain, only the TRP melastatin 2 (TRPM2) channel is uniquely sensitive to activation by intracellular Ca2+ and by H2O2 (Fleig and Penner, 2004).
Here we demonstrate the presence of functional TRPM2 channels in guinea pig SNr GABAergic neurons and show that TRPM2 channel activity affects the firing rate of these neurons. Moreover, we show for the first time in any neuron that activation of TRPM2 channels is necessary for NMDA-induced burst firing and underlies SNr neuron responsiveness to H2O2.
Materials and Methods
Ex vivo slice preparation for electrophysiology.
Adult male guinea pigs (Hartley, 150–250 g) were anesthetized with 50 mg/kg pentobarbital, then perfused transcardially with ice-cold modified artificial CSF (ACSF) containing the following (in mm): 225 sucrose; 2.5 KCl 2.5; 7 MgCl2; 28 NaHCO3 28; 1.25 NaH2PO4; 7 glucose; 1 ascorbate; 3 pyruvate; and 0.5 CaCl2, equilibrated with 95% O2/5% CO2 (Avshalumov et al., 2005; Lee and Tepper, 2007a,b). The midbrain was sectioned into 300-μm-thick coronal slices containing the substantia nigra in the same medium using a Leica VT1200S vibrating blade microtome (Leica Microsystems). Slices were then transferred to another ACSF solution at 34°C, which contained the following (in mm): 125 NaCl; 2.5 KCl; 1.25 NaH2PO4; 25 NaHCO3; 1 MgCl2; 25 glucose; 1 ascorbate; 3 pyruvate; 0.4 myo-inositol; and 2 CaCl2, equilibrated with 95% O2/5% CO2 and allowed to cool to room temperature over the next hour (Avshalumov et al., 2005; Lee and Tepper, 2007a,b). Slices were maintained in this solution until use.
Whole-cell electrophysiology and drugs.
Slices were placed in a recording chamber and superfused at 1.4 ml/min with ACSF (at 32°C) containing the following (in mm): 124 NaCl; 3.7 KCl; 26 NaHCO3; 1.3 MgSO4; 1.3 KH2PO4; 10 glucose; and 2.4 CaCl2, equilibrated with 95% O2/5% CO2 (Avshalumov et al., 2005). In some experiments, NaCl was replaced with an equimolar concentration of choline chloride or meglumine and NaHCO3 was replaced with choline bicarbonate (Johnson et al., 1992; Zhu et al., 2004; Lee and Tepper, 2007b; Khaliq and Bean, 2010). Neurons in the SNr were visualized using an Olympus BX51WI microscope equipped with infrared differential interference contrast optics (Olympus America). Patch pipettes for current-clamp recordings were filled with a solution containing the following (in mm): 129 potassium gluconate; 11 KCl; 10 HEPES; 2 MgCl2; 10 EGTA; 3 Na2-ATP; and Na3-GTP 0.3, adjusted to a pH of 7.2–7.3 with KOH (Lee and Tepper, 2007 a,b). Pipettes had resistances of 3–5 MΩ. In some experiments an antibody directed against a TRP channel (1:100) was added to the intracellular solution, or the solution supplemented with the Ca2+ chelator 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetra-acetic acid (BAPTA; 10 mm) (Zhu et al., 2004). In some cases in which BAPTA was added, EGTA concentration was decreased to 1 mm. Pipettes for voltage-clamp recordings were filled with a solution containing (in mm): 115 potassium methyl sulfate, 20 NaCl, 1.5 MgCl2, 5 HEPES, 10 BAPTA, 2 Na2-ATP, and 0.3 Na3-GTP (Ford et al., 2010). The ACSF used in voltage-clamp experiments did not include MgCl2. In addition, the following blockers were added to the ACSF for voltage-clamp recordings (in μm): 2 CGP 55845, 20 6,7-dinitroquinoxaline-2,3-dione (DNQX), 100 picrotoxin, and 2 TTX. Whole-cell current and voltage-clamp recordings were obtained with an Axopatch 200B amplifier, lowpass filtered at 2 kHz, and digitized by a Digidata 1322A for acquisition using Clampex 9 (Molecular Devices).
GABAergic neurons in the SNr were distinguished from dopaminergic neurons using electrophysiological characteristics. These included little or no sag caused by Ih current activation in response to hyperpolarizing current pulses, a faster spontaneous firing rate when compared with dopaminergic neurons, and narrow action potentials with short latency afterhyperpolarizations, all of which have been attributed to GABAergic neurons in the substantia nigra (Nakanishi et al., 1987; Grace and Onn, 1989; Lacey et al., 1989; Hainsworth et al., 1991; Yung et al., 1991; Hajós and Greenfield, 1994; Richards et al., 1997; Lee and Tepper, 2007a). Drugs were applied via the superfusing ACSF or intracellularly through the recording electrode. NMDA, BAPTA, picrotoxin, flufenamic acid (FFA), H2O2, adenosine 5′-diphosphoribose sodium salt (ADPR), dl-2-amino-phosphonopentanoic acid (AP5), choline chloride, choline bicarbonate, and meglumine were purchased from Sigma-Aldrich. TTX (as tetrodotoxin citrate), CGP 55845, DNQX, Pyr3, and (±)-Bay K 8644 were from Tocris Bioscience. Catalase (bovine liver) was from Calbiochem. CGP 55845, picrotoxin, DNQX, FFA, Bay K 8644, and Pyr3 were dissolved in dimethylsulfoxide (DMSO; Sigma-Aldrich) before addition to the ACSF. Final concentrations of DMSO did not exceed 0.06% in current-clamp experiments and 0.12% in voltage-clamp experiments, and DMSO added to ACSF at equivalent concentrations had no effect on electrophysiology.
Antibodies.
Rabbit polyclonal anti-TRPM2 antibodies from Abcam were used for Western blots and for immunohistochemical and electrophysiological experiments. The TRPM2-N antibody Abcam ab11168 was raised against the peptide ILKELSKEEDTDSSEEMLA, which represents residues 658–677 in the N-terminal region of the TRPM2 channel. The TRPM2-C antibody Abcam ab72802 was raised against residues 1430–1508 in the C-terminal region of the TRPM2 protein (Perraud et al., 2001; Maruyama et al., 2007). In some electrophysiological experiments, a rabbit polyclonal anti-TRPC3 antibody (ACC-016, Alomone Labs) raised against residues 822–835 in the C-terminal region of mouse TRPC3 was used. A mouse monoclonal antibody to parvalbumin (PV) (P3088; Sigma-Aldrich) was also used for immunohistochemical experiments. Additionally, a sheep polyclonal antibody to tyrosine hydroxylase (TH) (ab113; Abcam), for which the immunogen was the full-length native protein purified from rat pheochromocytoma, was used to identify dopaminergic neurons in some immunohistochemical studies. Secondary antibodies were donkey anti-sheep Cy5, donkey anti-rabbit Cy3, and donkey anti-mouse Cy3 (Jackson ImmunoResearch), and Alexa 488 donkey anti-rabbit and Alexa 488 donkey anti-mouse (Invitrogen).
In situ hybridization.
In situ hybridization was performed as described previously (Batista-Brito et al., 2008) using a digoxigenin-labeled cRNA probe directed against exon 13 of the guinea pig TRPM2 gene on coronal sections (4–6 sections spanning the SNr) from two brains. Following hybridization, the signal for TRPM2 mRNA was visualized using alkaline phosphatase histochemistry. PCR primers used to generate the cDNA fragment from guinea pig genomic DNA for antisense probe generation were as follows: 5′-CTCAGATCTGCACCCCACGAT-3′ and 5′-ATTAATACGACTCACTATAGTGAGTTTGACGTGGGGCACAG-3′. For combined immunofluorescence and in situ hybridization, the anti-PV immunofluorescence staining was performed as described above before in situ hybridization using Cy3-tyramide to visualize the presence of TRPM2 mRNA.
Protein extraction and Western blotting.
Guinea pig midbrain was sonicated in lysis buffer and the protein concentration determined using a bicinchoninic acid protein assay kit (Sigma-Aldrich) with bovine serum albumin (BSA) as a reference standard. After dilution 1:1 with Laemmle's buffer containing β-mercaptoethanol, the protein was resolved on a 6% polyacrylamide gel and transferred to nitrocellulose, using standard procedures. After blotting in 3% defatted milk, the membrane was reacted with the TRPM2-N antibody (1:500) for 1.5 h, then washed and reacted with donkey anti-rabbit biotinylated horseradish peroxidase secondary (Promega). Finally the nitrocellulose membrane was exposed to enhanced chemiluminescence reagent (Western Lightning) and developed with x-ray film.
Immunohistochemistry.
Guinea pigs (n = 6) were deeply anesthetized with 50 mg/kg pentobarbital administered intraperitoneally, then transcardially perfused with 0.1 m PBS, pH 7.3, at room temperature, followed by freshly prepared 4% paraformaldehyde in 0.1 m PBS. The brain was then removed and postfixed for 1 h at room temperature, washed 3 × 15 min in PBS, and transferred to ice-cold 30% sucrose in PBS for 16–20 h. A block containing the midbrain was cut in 20–25 μm sections on a cryostat, the sections mounted on Superfrost plus slides (Fisher Scientific), dried for 1–2 h at 37°C, and stored frozen. Frozen sections were thawed, washed in PBS, then in blocking solution (10 ml PBS, 100 mg BSA, 30 μl Triton X-100, 100 μl 10% (w/v) sodium azide) before exposure to a primary antibody or a mixture of primaries in blocking solution for 2 h at room temperature. After further washes in PBS, the sections were exposed to secondary antibodies for 2 h, washed in PBS, coverslipped in Vectashield (Vector Laboratories), and examined with a Nikon PM 800 confocal microscope equipped with a digital camera controlled by the Spot software program. Digital images were acquired separately from each laser channel. Digital files were processed with deconvolution software (AutoQuant Imaging). Final images were created in Photoshop 7.0 (Adobe). Images were adjusted for brightness and contrast with all adjustments made uniformly to all parts of the image.
Data analysis.
Electrophysiological data were analyzed in Clampfit 9.2 (Molecular Devices). Bursts were identified visually and start and end times set manually. Action potentials were detected by threshold search. Neurons that required a change in bias current of >25% during recording in NMDA to prevent excessive depolarization and loss of burst firing were excluded from quantitative analysis. Burst parameters reported were measured from four consecutive bursts from each neuron under each condition and averaged. Interburst intervals were averaged from three consecutive intervals. Burst frequency was calculated using four consecutive bursts. Firing rate data were acquired from 60 s of spontaneous activity with zero holding current. Data from some of the neurons used to calculate the control firing rate of SNr GABAergic neurons were also reported previously (Lee et al., 2011). Data are presented as mean ± SEM. Voltages in current-clamp recordings were corrected for a liquid junction potential, which was estimated to be 13 mV using JPCalc (Barry, 1994). Statistical tests were performed in SAS 9.3 and data were compared using paired or unpaired t tests as appropriate. Significance was defined as p < 0.05.
Results
NMDA induces burst firing in SNr GABAergic neurons
All SNr GABAergic neurons recorded exhibited regular, single spike firing at rest (Fig. 1A). Bath application of NMDA (15–30 μm) and injection of a hyperpolarizing current caused these neurons to shift from a regular firing pattern to an oscillatory bursty firing pattern (Fig. 1B). Hyperpolarizing current was gradually injected during NMDA application until stable burst firing emerged. Current was maintained at or near this threshold level to avoid alteration in burst characteristics in a given cell. Final averaged current was −184 ± 14 pA (n = 29). Bursts occurred at a frequency of 0.31 ± 0.02 Hz and had an average interburst interval of 2.5 ± 0.3 s (n = 29). The average duration of a burst was 1.9 ± 0.2 s (n = 29). Bursts consisted of 61 ± 8 spikes, with an average firing rate of 31.5 ± 2.3 Hz within the burst (n = 29).
Figure 1.

NMDA induces burst firing in SNr GABAergic neurons. A, Spontaneous, regular spiking activity of an SNr GABAergic neuron recorded under control conditions, with no current injected through the recording pipette. B, NMDA (30 μm) caused the same neuron to exhibit rhythmic burst firing when a tonic hyperpolarizing current of −150 pA was applied.
Burst firing is dependent on intracellular Ca2+
To identify the mechanisms underlying burst firing in SNr GABAergic neurons, we first examined the ionic dependence of this behavior. Addition of the Ca2+ chelator BAPTA (10 mm) to the intracellular solution abolished NMDA-induced burst firing. All of the neurons recorded under these conditions (n = 11) initially exhibited a shift to burst firing in response to NMDA (Fig. 2A), but transitioned to an irregular firing pattern during recording (Fig. 2B) as BAPTA diffused from the pipette into the neuron during whole-cell recording; NMDA-induced bursting was lost after an average total recording time of 7.7 ± 0.6 min (n = 11). Notably, voltage-clamp recordings (−70 mV holding potential) confirmed that inclusion of BAPTA (10 mm) in the intracellular solution did not abolish an NMDA-dependent inward current. Under these conditions, NMDA evoked an inward current with a peak amplitude of 104 ± 16 pA (n = 7) (Fig. 2C), when applied after an average recording time of 8.7 ± 0.6 min to ensure diffusion of BAPTA from the pipette; peak NMDA currents were measured at 11.6 ± 0.3 min. These currents were completely prevented by AP5 (100 μm) (Fig. 2D). Together, these data demonstrate the Ca2+ dependence of NMDA-induced burst firing in SNr neurons.
Figure 2.

NMDA-induced burst firing is dependent on intracellular Ca2+. A, Burst firing induced by NMDA (30 μm) in an SNr GABAergic neuron after 5 min of recording with the intracellular solution supplemented with the Ca2+ chelator BAPTA (10 mm). B, Four minutes later, burst firing was lost and the neuron exhibited irregular, single spike activity. A tonic hyperpolarizing current of −75 pA was applied in both A and B. C, Voltage-clamp recording of an SNr GABAergic neuron showing a reversible inward current caused by NMDA (30 μm) with BAPTA (10 mm) in the intracellular solution. D, The NMDA-induced current was completely prevented by AP5 (100 μm).
We next addressed whether increasing intracellular Ca2+ through another mechanism could similarly induce burst firing in SNr neurons. The L-type Ca2+ channel agonist Bay K 8644 (5 μm) has been reported to induce burst-like firing in rat SNr GABAergic neurons (Lee and Tepper, 2007b). We found that Bay K 8644 (5 μm) also induced burst firing in guinea pig SNr GABAergic neurons (Fig. 3) (n = 7). Bursts induced by Bay K 8644 had an average frequency of 0.15 ± 0.04 Hz and an average duration of 3.5 ± 1.0 s. Bursts consisted of an average of 76 ± 20 spikes with an intraburst firing rate of 25.0 ± 7.5 Hz. The average interburst interval was 8.2 ± 2.8 s. To eliminate involvement of NMDA receptors, AP5 (50 μm) was included in the superfusing ACSF for five of seven recordings with no effect on burst firing. These results indicate that Ca2+ entry through either NMDA or L-type Ca2+ channels is sufficient to trigger burst firing in SNr GABAergic neurons.
Figure 3.

Burst firing can be induced by activation of L-type Ca2+ channels. A, Spontaneous activity of an SNr GABAergic neuron under control conditions. B, The same neuron exhibited burst firing in the presence of the L-type Ca2+ channel agonist Bay K 8644 (5 μm) and injection of a tonic hyperpolarizing current of −110 pA. AP5 (50 μm) was applied with Bay K 8644 to prevent activation of NMDA receptors.
Burst firing is dependent on Na+
We then shifted our focus to the role of Na+ in burst firing in SNr GABAergic neurons. Blockade of voltage-gated Na+ channels with TTX (1–2 μm) revealed an underlying oscillation of the membrane potential induced by NMDA (Fig. 4A,B). The oscillation induced by NMDA in the presence of TTX had an average frequency of 0.27 ± 0.03 Hz and duration of 1.72 ± 0.23 s (n = 12). The average interval between oscillations was 3.3 ± 0.6 s (n = 12). In most cases (n = 10) burst firing was induced before exposure to TTX, allowing the comparison of slow membrane potential oscillations before and after blockade of fast Na+ channels. Most neurons did not require adjustment of the tonic hyperpolarizing current required to maintain burst firing during NMDA exposure when switching to TTX; indeed, averaged pre- and post-TTX currents did not differ significantly (−194 ± 21 vs −200 ± 21 pA; n = 10; p > 0.05). The frequency of NMDA-induced bursts did not differ significantly from that of the membrane potential oscillation in the presence of TTX (NMDA alone 0.29 ± 0.04 Hz; NMDA and TTX 0.30 ± 0.03 Hz; n = 10; p > 0.05). The interval between bursts or oscillations was similarly unaffected (NMDA alone 2.81 ± 0.61 s; NMDA and TTX 2.91 ± 0.60 s; n = 10; p > 0.05) and there was no change in the duration of the oscillation observed in TTX when compared with bursting observed with spiking intact (NMDA 1.88 ± 0.22 s; NMDA and TTX 1.61 ± 0.19 s; n = 10; p > 0.05). Overall, the NMDA-induced membrane potential oscillations had nearly identical properties to those observed during NMDA-induced burst firing and were insensitive to blockade by TTX.
Figure 4.

NMDA induces a Na+-dependent membrane potential oscillation. A, Recording from an SNr GABAergic neuron exhibiting burst firing in the presence of NMDA (30 μm) and a tonic hyperpolarizing current injection of −220 pA. B, Blockade of voltage-gated Na+ channels with TTX (1 μm) revealed an underlying membrane potential oscillation. C, The membrane potential oscillation was abolished in Na+-free ACSF. D, The oscillation resumed following return of Na+ to the ACSF. A tonic hyperpolarizing current of −235 pA was applied in B–D.
We next tested whether NMDA-induced oscillations in the presence of TTX were mediated by Na+. In all neurons tested, a switch to Na+-free ACSF completely abolished the membrane potential oscillation caused by NMDA in the presence of TTX (Fig. 4C) (n = 7); this oscillation could not be restored by depolarizing or hyperpolarizing the neuron through current injection (data not shown). The oscillation returned in most neurons following superfusion of standard ACSF with NMDA and TTX (Fig. 4D) (n = 5).
NMDA-induced burst firing is abolished by blocking TRP channels
The results from the previous sets of experiments revealed that the conductance underlying NMDA-induced burst firing is dependent on Ca2+ and uses Na+ as a charge carrier. A conductance with these characteristics could be mediated by one or more TRP channels. To test the hypothesis that TRP channels are involved in NMDA-induced burst firing in SNr GABAergic neurons, we first tested whether FFA, which has been shown to block multiple TRP channel subtypes (Hill et al., 2004; Clapham, 2007; Zhou et al., 2008; Chung et al., 2011) and has been used to implicate TRP channels in NMDA-induced burst firing in other neurons (Zhu et al., 2004; Mrejeru et al., 2011), could affect burst firing in SNr neurons. Indeed, FFA (40–100 μm) completely abolished NMDA-induced burst firing and returned the neuron to a more regular firing pattern (Fig. 5A–C) (n = 6). Additionally, FFA (40–60 μm) abolished the membrane potential oscillation induced by NMDA in the presence of TTX (Fig. 5D–F) (n = 3). In the presence of FFA, neither burst firing nor the membrane potential oscillation induced by NMDA in TTX could be restored by either depolarizing or hyperpolarizing current injection (data not shown). To address the possible confounding factor that FFA might have a nonspecific effect on the inward current mediated by NMDA channels, we compared the amplitude of the NMDA-induced current in the presence and absence of FFA. As with voltage-clamp recordings made without FFA (Fig. 2), NMDA was applied after a sufficient time for BAPTA to infiltrate the neuron and prevent recruitment of other Ca2+-dependent currents (9.4 ± 0.3 min; n = 6). FFA was present throughout. Notably, when NMDA was then applied in the continued presence of FFA (60 μm), the inward current induced by NMDA (30 μm) did not differ from that measured in the absence of FFA (NMDA 104 ± 16 pA; n = 7; NMDA and FFA 104 ± 17 pA; n = 6; p > 0.05). Peak NMDA currents in the presence of FFA were measured at 12.4 ± 0.6 min of total recording time. These results strongly indicate the participation of one or more TRP channel subtypes in NMDA-induced burst firing in SNr neurons.
Figure 5.

NMDA-induced burst firing is abolished by FFA. A, Spontaneous regular-spiking activity recorded under control conditions. B, Burst firing induced by NMDA (30 μm). C, Burst firing is abolished by FFA (60 μm). A tonic hyperpolarizing current of −170 pA was applied in B and C. D, Recording from another SNr GABAergic neuron exhibiting NMDA-induced burst firing. A tonic hyperpolarizing current of −140 pA was applied. E, Blockade of voltage-gated Na+ channels with TTX reveals a membrane potential oscillation induced by NMDA. F, The NMDA-induced membrane potential oscillation is blocked by FFA (60 μm). A tonic hyperpolarizing current of −160 pA was applied in E and F.
SNr GABAergic neurons express TRPM2 channels
One TRP channel subtype, TRPM2, meets the criteria we have identified, including activation by Ca2+ and the use of Na+ as a charge carrier (Fleig and Penner, 2004). However, the presence of TRPM2 channels on SNr GABAergic neurons has not been established. We therefore first examined whether guinea pig SNr GABAergic neurons express TRPM2 channels by performing in situ hybridization for TRPM2 mRNA on tissue sections containing the SNr. By itself, in situ hybridization revealed diffuse staining for TRPM2 mRNA within the SNr (Fig. 6A), and in combination with immunohistochemistry for PV, a Ca2+ binding protein found exclusively in GABAergic neurons in the substantia nigra (González-Hernández and Rodríguez, 2000), in situ hybridization revealed the presence of TRPM2 mRNA in SNr GABAergic neuron cell bodies (Fig. 6B,C).
Figure 6.

PV-containing SNr neurons express TRPM2 channel mRNA. A, Coronal section of guinea pig midbrain reveals diffuse staining for TRPM2 channel mRNA in the SNr. Scale bar, 900 μm. Dotted box indicates the approximate location of the fields shown in B and C. B, Immunohistochemistry for PV (green) before in situ hybridization (ISH) for TRPM2. A nuclear stain (DAPI) is shown in blue. Scale bar, 50 μm. C, Immunohistochemical staining for PV was combined with fluorescent in situ hybridization for TRPM2 (red) revealing that PV-containing neurons colocalize TRPM2 mRNA. Scale bar, 50 μm.
To determine whether TRPM2 channel protein is expressed in guinea pig midbrain, we used an antibody directed against the N-terminal region of the TRPM2 channel protein (TRPM2-N), on Western blots (n = 3), each prepared from homogenized midbrain containing the substantia nigra from a single guinea pig. This antibody revealed a single band (Fig. 7A, right column) near 180 kDa, a molecular mass corresponding to that of the full-length TRPM2 channel (Zhang et al., 2003). Pre-adsorption of the antibody by its immunogenic peptide resulted in a complete loss of immunostaining (Fig. 7A, left column). These results confirm the presence of TRPM2 channel protein in guinea pig midbrain.
Figure 7.

PV-containing SNr neurons express TRPM2 channels. A, Western blots performed on tissue extracts of guinea pig midbrain using an antibody directed against the N-terminal region of the TRPM2 protein (TRPM2-N) (right column) revealed a band at 180 kDa (arrow). Staining was eliminated by pre-adsorption (pre-ad) of the TRPM2-N antibody with its immunogenic peptide (left column). B, Immunohistochemical localization of TRPM2 in SNr GABAergic neurons. Top, SNr GABAergic neuron stained for TRPM2 channels using an antibody directed against the N-terminal region of the channel protein (TRPM2-N) as well as the same neuron stained for PV. Staining for TRPM2 was absent following pre-adsorption of the primary antibody with its immunogenic peptide. Bottom, SNr neuron stained for TRPM2 using an antibody directed against the C-terminal region of the protein (TRPM2-C) and the same neuron stained for PV. Merged images demonstrate colocalization of PV and TRPM2. Scale bar, 10 μm. C, Section of SNc immunostained for TH, TRPM2-C, and PV. This neuron exhibited immunostaining for TH but not PV identifying it as a dopaminergic neuron. Merged image demonstrates colocalization of TH and TRPM2-C immunostaining. Scale bar, 20 μm.
Next, we used immunohistochemistry to determine whether TRPM2 channels are expressed on SNr GABAergic neurons. Two antibodies directed against the TRPM2 channel protein were used for this purpose. The first was the TRPM2-N antibody described above, and the second was an antibody directed against the C-terminal region of the TRPM2 protein (TRPM2-C). GABAergic neurons were identified by immunostaining for PV. Both TRPM2 antibodies produced robust immunostaining in PV-containing GABAergic SNr neurons (Fig. 7B). Immunoreactivity (ir) to TRPM2 was punctate and tended to cluster around the perikaryal plasma membrane, whereas PV-ir was smooth and evenly distributed throughout the perikaryal cytoplasm. TRPM2-ir and PV-ir were colocalized in cell bodies, as well as dendrites. Pre-adsorption of the TPRM2-N antibody with its immunogenic peptide resulted in a complete loss of immunostaining (Fig. 7B). The degree of colocalization between PV and TRPM2 staining was assessed using the TRPM2-N antibody on a series of five coronal sections spanning the entire rostral to caudal extent of the nucleus, with each section containing the substantia nigra in both hemispheres. Examination of 200 perikarya with evident processes and staining for PV revealed colocalization with TRPM2 in 197/200. Consistent with immunohistochemical results from the rat (Chung et al., 2011), we also identified TRPM2 channels using the TRPM2-C antibody in dopaminergic neurons of guinea pig substantia nigra pars compacta (SNc), identified by immunostaining for TH (Fig. 7C). These data demonstrate TRPM2 channel expression in SNr GABAergic neurons.
TRPM2 channels are required for burst firing in SNr GABAergic neurons
We next examined whether activation of TRPM2 channels is necessary for NMDA-induced burst firing in SNr GABAergic neurons. Given the limited specificity of pharmacological agents to study TRP channels, we used an anti-TRPM2 antibody to block TRPM2 channels selectively in these neurons. Anti-TRP channel antibodies have been used successfully to block specific TRP channels in previous work (Amaral and Pozzo-Miller, 2007; Hecquet et al., 2008; Zhou et al., 2008; Cvetkovic-Lopes et al., 2010).
When the TRPM2-C antibody was included in the intracellular solution (1:100) and the neurons exposed to NMDA (30 μm), burst firing was blocked (n = 7) (Fig. 8), indicating the presence of functional TRPM2 channels in SNr neurons. In 3/7 neurons burst firing was absent at all times during recording. The remaining neurons (4/7) initially exhibited weak oscillatory burst firing in response to NMDA that was lost as the antibody diffused into the cell; in those neurons bursting was abolished after an average total recording time of 9.6 ± 1.2 min. A constant hyperpolarizing current was applied during NMDA application as usual; however, once blocked, burst firing was not restored by varying the injected current.
Figure 8.

NMDA-induced burst firing is occluded by a TRPM2-specific antibody. A, Spontaneous activity of an SNr GABAergic neuron with the TRPM2-C antibody included in the intracellular solution (1:100). B, With the continued presence of the TRPM2-C antibody in the pipette, the neuron exhibited irregular single spike firing but no burst firing with application of NMDA (30 μm), regardless of the level of tonic current injection (hyperpolarizing current of −145 pA was used in the record illustrated).
To test whether the presence of the TRPM2-C antibody in the intracellular solution might act through a direct effect on the inward current mediated by NMDA channels, we compared the amplitude of the current in the presence and absence of intracellular application of the antibody. Again, NMDA was applied after a delay, in this case of 9.5 ± 0.3 min, to allow infiltration the TRPM2-C antibody into the recorded neuron. Under these conditions, NMDA continued to evoke an inward current that reached a peak at 12.7 ± 1.0 min of total recording time with peak amplitude that was not affected by the presence of the antibody (NMDA 104 ± 16 pA; n = 7; NMDA with TRPM2-C antibody 98 ± 20 pA; n = 4; p > 0.05), confirming a lack of effect on NMDA-induced currents.
Previous studies have shown that an antibody directed against TRPC3 channels blocks activity of that channel in SNr GABAergic neurons from juvenile mice (Zhou et al., 2008). However, inclusion of a TRPC3 channel antibody did not occlude NMDA-induced burst firing in guinea pig SNr neurons (data not shown; n = 4). Additionally, Pyr3 (6–60 μm), a pharmacological blocker of TRPC3 channels (Kiyonaka et al., 2009), had no effect on NMDA-induced burst firing (data not shown; n = 9). We conclude that TRPC3 channels do not play a significant role in NMDA-induced burst firing in SNr GABAergic neurons. Instead, our data indicate that TRPM2 channels are an essential participant in NMDA-induced burst firing in SNr GABAergic neurons.
TRPM2 channel activity contributes to the tonic firing rate of SNr GABAergic neurons and underlies their responsiveness to H2O2
We and others have previously reported that the spontaneous firing rate of SNr GABAergic neurons decreases when TRP channels are broadly blocked by FFA (Zhou et al., 2008; Lee et al., 2011). Conversely, H2O2 induces an increase in the firing rate of SNr GABAergic neurons of ∼40% that is reversed by FFA, suggesting that H2O2 activates one or more TRP channel subtypes in these cells leading to an increase in their excitability (Lee et al., 2011). We therefore determined whether TRPM2 channel activation affects the tonic firing rate of SNr GABAergic neurons and whether these channels convey sensitivity to H2O2.
We again used antibodies to block TRPM2 channels. Inclusion of the TRPM2-C antibody in the intracellular solution (1:100) caused a decrease in spontaneous firing rate in SNr GABAergic neurons, indicating a key role in basal cell activity. The spontaneous firing rate was 15.3 ± 0.4 Hz (n = 129) under control conditions and 11.0 ± 1.2 Hz with the antibody in the intracellular solution (Fig. 9A,B) (n = 6; p < 0.05). To determine whether H2O2 modulates the firing rate of SNr GABAergic neurons via TRPM2 channels, exogenous H2O2 (1.5 mm) (Avshalumov et al., 2005; Lee et al., 2011) was applied in the presence of intracellular antibodies. Under these conditions, H2O2 caused a decrease in firing rate to 4.4 ± 1.5 Hz (n = 6; p < 0.05) (Fig. 9A,B). We have previously reported a decrease in spontaneous firing rate in response to H2O2 in the presence of TRP channel blockade with FFA, and have shown this decrease in firing rate to be caused by the unopposed activation of ATP-sensitive potassium (KATP) channels by H2O2 (Lee et al., 2011).
Figure 9.
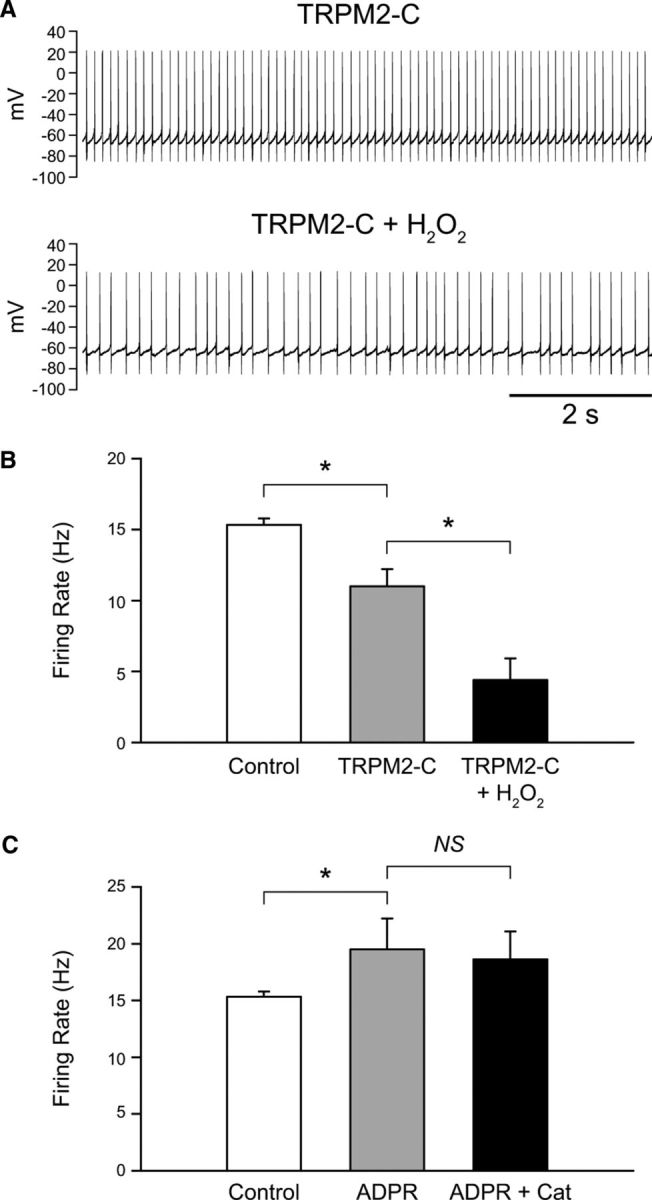
H2O2 increases the firing rate of SNr GABAergic neurons by activating TRPM2 channels. A, Spontaneous activity of an SNr GABAergic neuron before and after exogenous H2O2 (1.5 mm) application with the TRPM2-C antibody (1:100) in the intracellular solution. B, Blockade of TRPM2 channels by the TRPM2-C antibody caused a decrease in firing rate of SNr GABAergic neurons indicating tonic activation of TRPM2 channels. H2O2 caused a further decrease in firing rate when TRPM2 channels were blocked. C, Intracellular application of the TRPM2 channel activator ADPR (500 μm or 1 mm) increased the spontaneous firing rate of SNr GABAergic neurons. Under these conditions basal H2O2 depletion by catalase (Cat; 500 U/ml) had no effect on firing rate (n = 9) (*p < 0.05). Data are presented as mean ± SEM.
In contrast to the TRPM2-C antibody, the TRPM2-N antibody did not have a significant effect on SNr GABAergic neuron firing rate, which was 15.3 ± 0.4 Hz under control conditions (n = 129) and 13.6 ± 1.6 Hz with the TRPM2-N antibody in the intracellular solution (n = 4; p > 0.05). Additionally, this antibody failed to block the usual H2O2-induced increase in firing rate (control 13.6 ± 1.6 Hz; H2O2 18.5 ± 2.2 Hz; n = 4; p < 0.05). Our finding that the TRPM2-C antibody was efficacious in blocking TRPM2 channel activation by H2O2 but the TRPM2-N antibody was not is consistent with previous studies showing that the C-terminal region of the TRPM2 channel contains a Nudix domain that is the site of action of ADPR, an endogenous TRPM2 channel activator (Perraud et al., 2001; Kolisek et al., 2005; Lange et al., 2008) that can be formed in response to H2O2 (Perraud et al., 2001, 2005).
We next examined whether endogenous H2O2 also modulates SNr GABAergic neuron activity through TRPM2 channel activation. Like blocking TRPM2 channels, depleting endogenous H2O2 with catalase causes a decrease in the spontaneous firing rate of SNr GABAergic neurons (Lee et al., 2011). We therefore hypothesized that the suppressive effect of H2O2 depletion on firing rate would be occluded when TRPM2 channels were activated by elevating intracellular ADPR (Perraud et al., 2001; Kolisek et al., 2005; Lange et al., 2008). Inclusion of ADPR (500 μm or 1 mm) in the intracellular solution resulted in a significant increase in SNr GABAergic neuron firing rate from 15.3 ± 0.4 Hz (n = 129) under control conditions to 19.5 ± 2.7 Hz (Fig. 9C) (n = 9; p < 0.05). This sensitivity to ADPR provided further evidence for the presence of functional TRPM2 channels in SNr GABAergic neurons. In the presence of ADPR, modulatory regulation by endogenous H2O2 was abolished, as seen in loss of the usual suppressive effect of catalase (500 U/ml) on firing rate (n = 9; p > 0.05 ADPR vs ADPR + catalase) (Fig. 9C). Thus, these data indicate that H2O2 exerts a tonic excitatory effect on these cells via TRPM2 channel activation.
H2O2 modulates NMDA-induced burst firing
Based on our findings that both NMDA-induced burst firing and responsiveness to H2O2 in SNr GABAergic neurons are dependent on activation of TRPM2 channels, we then tested whether H2O2 can modulate NMDA-induced burst firing. H2O2 was increased by either applying exogenous H2O2 (1.5 mm) or by amplifying endogenous H2O2 through inhibition of glutathione peroxidase with mercaptosuccinate (1 mm; Avshalumov et al., 2005; Lee et al., 2011). These manipulations produced comparable results so the data were pooled. Increasing H2O2 caused NMDA-induced bursts to become longer and resulted in a decrease in burst frequency (Fig. 10). Burst duration increased from 2.3 ± 0.4 s to 4.2 ± 1.2 s in the presence of elevated H2O2 (n = 9; p < 0.05) while burst frequency decreased from 0.29 ± 0.05 to 0.21 ± 0.03 Hz (n = 9; p < 0.05) under the same conditions. There was an increase in the average number of spikes per burst from 87.1 ± 20.3 to 158.4 ± 50.4, which did not reach significance in this sample (n = 9; p = 0.07). The firing rate within bursts as well as the interburst interval were unaffected by H2O2. There was no difference in the average hyperpolarizing current needed to maintain burst firing in the presence of NMDA under control conditions or in the presence of increased H2O2 (−178 ± 24 vs −181 ± 24 pA); indeed, only one neuron required slightly more hyperpolarizing current to maintain burst firing. As a control for recording time, we found no increase in burst duration or decrease in burst frequency in NMDA alone (n = 2) when measured at latencies of NMDA exposure approximating those used for baseline burst duration and burst duration in the presence of H2O2 (baseline duration measured after ∼7 min of NMDA exposure and H2O2 duration after ∼11 min of NMDA exposure). H2O2 also caused an increase in the duration of the oscillation in membrane potential induced by NMDA in the presence of TTX. The oscillation had a duration of 2.1 ± 0.4 s under control conditions and increased to 3.5 ± 0.6 s in the presence of exogenous H2O2 (n = 5; p < 0.05). No change in hyperpolarizing current was required in these experiments. These results indicate that H2O2 can modulate NMDA-induced burst firing in SNr GABAergic neurons.
Figure 10.

H2O2 modulates NMDA-induced burst firing. A, Whole-cell recording illustrating the effect of H2O2 (1.5 mm) on NMDA-induced (30 μm) burst firing in an SNr GABAergic neuron. A tonic hyperpolarizing current of −170 pA was applied throughout. B, Summary data indicate that H2O2 caused an increase in burst duration and a decrease in burst frequency (*p < 0.05). Data are presented as mean ± SEM.
Discussion
The mechanisms underlying NMDA-induced burst firing in SNr GABAergic neurons have remained incompletely understood. Here we show that such bursts are mediated by TRPM2 channels that convey a Na+ conductance activated by Ca2+ entry. Additionally, we show that H2O2 causes an increase in the firing rate of SNr GABAergic neurons by activating TRPM2 channels, and also increases the duration of NMDA-induced bursts, while decreasing burst frequency.
TRPM2 channels in SNr GABAergic neurons
The presence of TRPM2 channels on guinea pig SNr GABAergic neurons was demonstrated using several approaches, including in situ hybridization showing TRPM2 mRNA in these cells, Western blot to confirm TRPM2 channel protein in midbrain, and immunohistochemistry localizing TRPM2 protein in SNr GABAergic neurons. Finally, the presence of functional TRPM2 channels was demonstrated by the sensitivity of SNr neuron firing rate to modulation by the known TRPM2 activators, H2O2 and ADPR, and by the efficacy of a specific TRPM2 antibody to block NMDA-induced burst firing and H2O2-induced modulation.
Previous single-cell RT-PCR data from SNr GABAergic neurons in young mice identified TRPC3 channels as the primary subtype expressed (Zhou et al., 2008; Zhou and Lee, 2011). However, the absence of evidence for TRPM2 mRNA transcript does not rule out TRPM2 channel expression in those cells. Indeed, in situ hybridization data from mouse SNr shows neuronal-like staining for TRPM2 mRNA (Allen Institute for Brain Science; Lein et al., 2007). Immunohistochemical data also indicate that rat nigral GABAergic neurons express TRPM2 channels, with expression of TRPM2 immunoreactivity in TH-positive dopaminergic neurons, as well as in TH-negative SNr neurons (Chung et al., 2011). Other studies have shown TRPM2 mRNA in mouse SNc tissue containing TH mRNA, as well (Mrejeru et al., 2011). Consistent with these observations, we found TRPM2 immunoreactivity in TH-positive SNc neurons, as well as in SNr GABAergic neurons in guinea pig midbrain (Fig. 7B,C).
In contrast to the prevention of NMDA-induced bursting in guinea pig SNr neurons by intracellular TRPM2 antibodies, TRPC3 antibodies had no effect, indicating that they do not contribute to burst firing. This is not surprising, given previous evidence that TRPC3 is an effector channel for metabotropic G-protein-coupled receptors (Zhou and Lee, 2011). Specifically, TRPC3 channels mediate an increase in the firing rate of mouse SNr GABAergic neurons by dopamine (Zhou et al., 2009) or serotonin (Zhou and Lee, 2011), with an unidentified TRPC channel mediating the effects of muscarinic acetylcholine receptor activation in these cells (Michel et al., 2004, 2005). Instead, TRPM2 channels are activated by other mechanisms including increases in intracellular Ca2+ (Lee and Tepper, 2007b; present results), H2O2, or ADPR (Perraud et al., 2001; Hara et al., 2002; Wehage et al., 2002; Fleig and Penner, 2004; Lange et al., 2008; Tóth and Csanády, 2010; present results).
Burst firing in SNr GABAergic neurons
The present findings show that NMDA receptor activation can lead to TRPM2 channel-dependent burst firing. Previous studies have shown that stimulation of glutamatergic input from the STN can induce burst firing in SNr GABAergic neurons (Shen and Johnson, 2006). The mechanisms underlying STN-induced burst firing are not fully known, but could involve prolonged excitation of SNr neurons by repetitive excitatory input from recurrent activation of STN neurons (Shen and Johnson, 2006, 2012; Ammari et al., 2010). In addition, the occurrence of prolonged plateau depolarizations in individual SNr GABAergic neurons in response to brief depolarizing current pulses (Nakanishi et al., 1987; Lee and Tepper, 2007b) and burst firing in response to NMDA ex vivo (Ibáñez-Sandoval et al., 2007; present results) indicate that SNr GABAergic neurons have intrinsic mechanisms that amplify depolarizing input and enable burst firing. Here we demonstrate that activation of TRPM2 channels is a key intrinsic mechanism in SNr GABAergic neuron burst firing.
Interestingly, neither H2O2 nor ADPR alone induced burst firing in SNr GABAergic neurons, despite the ability of each to activate TRPM2 channels. These data imply an essential, synergistic role for both Ca2+ and TRPM2 channels in the rhythmicity of burst firing, possibly through intracellular Ca2+ dynamics and rhythmic activation of TRPM2 channels or from the activation of other Ca2+-activated channels including Ca2+-activated potassium channels (Atherton and Bevan, 2005; Yanovsky et al., 2005, 2006; Ibáñez-Sandoval et al., 2007; Zhou and Lee, 2011). Although TRPM2 channels can be permeable to Ca2+ (Sumoza-Toledo and Penner, 2011), our results suggest that TRPM2 channels in SNr GABAergic neurons predominantly use Na+ as a charge carrier, as the NMDA-induced oscillation was abolished in Na+-free ACSF. It seems unlikely, therefore, that activation of TRPM2 channels alone would directly cause a significant increase in intracellular Ca2+, though an increase in Ca2+ secondary to depolarization induced by TRPM2 channel activation and further activation of voltage-gated Ca2+ channels is possible.
Our data further indicate that activation of any of several Ca2+ conductances, not only NMDA receptors, can induce burst firing through TRPM2 channels. Indeed, activation of L-type Ca2+ channels with Bay K 8644 was sufficient to induce burst firing in SNr GABAergic neurons. This is consistent with previous work that showed the Ca2+ dependence of NMDA-induced burst firing in these cells and identified activation of T-type (CaV3.2) Ca2+ channels as an initiating event in burst generation (Ibáñez-Sandoval et al., 2007).
Involvement of TRP channels in burst firing in other basal ganglia nuclei
Burst firing plays particularly important roles for neurons in the basal ganglia, given established links between bursting and neuronal function (e.g., SNc dopaminergic neurons; Morikawa and Paladini, 2011), as well as dysfunction (e.g., SNr neurons). Notably, characteristics of NMDA-induced burst firing in other basal ganglia neurons mirror those reported here for SNr GABAergic neurons. Specifically, in SNc dopaminergic neurons, NMDA induces a Na+-dependent membrane oscillation that persists in TTX (Johnson et al., 1992). Moreover, prolonged application of intracellular BAPTA can block this effect, indicating that bursting in SNc dopaminergic neurons also depends on Ca2+ (Mrejeru et al., 2011). Interestingly, however, based on pharmacological studies with FFA and other TRP channel blockers, TRPM4 channels were implicated in mouse SNc neurons (Mrejeru et al., 2011). STN neurons also exhibit NMDA-induced burst firing that is abolished by FFA (Zhu et al., 2004), a Ca2+-dependent plateau potential in response to depolarizing current pulses (Beurrier et al., 1999; Otsuka et al., 2001), like that seen in SNr GABAergic neurons (Lee and Tepper, 2007b), as well as FFA-sensitive Ca2+-activated inward currents (Zhu et al., 2005). These properties implicate TRP channels, and in particular TRPM2, which exhibits robust expression in the mouse STN (Allen Institute for Brain Science; Lein et al., 2007). Overall, these data suggest that burst firing mediated by TRPM2 channels could be an important neuronal mechanism in the basal ganglia, as well as in other regions of the CNS.
H2O2 modulation of firing rate and NMDA-induced burst firing
We reported previously that elevation of endogenous H2O2 increases the spontaneous firing rate of SNr GABAergic neurons (Lee et al., 2011). Here we identified TRPM2 channels as the target of modulatory H2O2 in these cells. A growing body of evidence points to H2O2 as a neuromodulator under physiological conditions, in which it is produced tonically by mitochondria and elevated in response to neuronal activity (Lee et al., 2011; Rice, 2011). Identification of TRPM2 channels as mediators of H2O2 modulation in SNr neurons implies a mechanism for positive feedback in these cells, thus revealing a new link between neuronal activity and metabolism.
Demonstration that both NMDA and H2O2 lead to activation of TRPM2 channels in SNr GABAergic neurons led to the question of whether H2O2 might modulate NMDA-induced burst firing via this common target. Indeed, H2O2 increased the duration of NMDA-induced bursts, and as a result decreased burst frequency, although firing rate within bursts and interburst interval were unchanged. We reported previously that H2O2 also activates KATP channels in SNr GABAergic neurons, which decrease firing rate (Lee et al., 2011). It is possible that concurrent activation of KATP channels by H2O2 might mask more dramatic enhancement of burst parameters. Nevertheless, the net effect of H2O2 in guinea pig SNr GABAergic neurons is to increase excitability, reflected in an increase in NMDA-induced burst duration, as well as in the increase in spontaneous firing rate reported previously (Lee et al., 2011).
Relevance of TRPM2 channels for Parkinson's disease
Understanding regulation of burst firing in SNr GABAergic neurons has particular implications for Parkinson's disease and associated animal models, given the increase in burst firing of SNr neurons seen with dopamine depletion (Filion and Tremblay, 1991; Hutchison et al., 1994; Wichmann et al., 1999; Wichmann and Dostrovsky, 2011). Contributing factors could include an increase in glutamatergic input to the SNr from the STN (Bergman et al., 1994; Wilson and Bevan, 2011). Furthermore, in Parkinson's disease, there is evidence for increased production of reactive oxygen species, including H2O2, from mitochondrial dysfunction and from altered α-synuclein (Turnbull et al., 2001; Dauer and Przedborski, 2003). Evidence for H2O2 modulation of NMDA-induced burst dynamics suggests that increased glutamatergic input from the STN might interact with elevated H2O2 to promote burst firing in Parkinson's disease via TRPM2, further suggesting a novel therapeutic target.
Footnotes
This work was supported by National Institutes of Health–National Institute of Neurological Disorders and Stroke Grants NS036362 and NS057458 (M.E.R.) and in part by F32 NS063656 (C.R.L.), the Richard H. Chartrand Foundation (P.W.), and the Attilio and Olympia Ricciardi Research Fund (M.E.R.).We thank Professor Yasuo Mori for generously providing TRPM2 antibodies and Pyr3 used in preliminary experiments for this work. We thank Nicole Zeak for excellent technical assistance.
References
- Allen Institute for Brain Science. Allen Mouse Brain Atlas Internet. Seattle, WA: Allen Institute for Brain Science; 2009. Available from: http://mouse.brain-map.org. [Google Scholar]
- Amaral MD, Pozzo-Miller L. TRPC3 channels are necessary for brain-derived neurotrophic factor to activate a nonselective cationic current and to induce dendritic spine formation. J Neurosci. 2007;27:5179–5189. doi: 10.1523/JNEUROSCI.5499-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ammari R, Lopez C, Bioulac B, Garcia L, Hammond C. Subthalamic nucleus evokes similar long lasting glutamatergic excitations in pallidal, entopeduncular and nigral neurons in the basal ganglia slice. Neuroscience. 2010;166:808–818. doi: 10.1016/j.neuroscience.2010.01.011. [DOI] [PubMed] [Google Scholar]
- Atherton JF, Bevan MD. Ionic mechanisms underlying autonomous action potential generation in the somata and dendrites of GABAergic substantia nigra pars reticulata neurons in vitro. J Neurosci. 2005;25:8272–8281. doi: 10.1523/JNEUROSCI.1475-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avshalumov MV, Chen BT, Koós T, Tepper JM, Rice ME. Endogenous hydrogen peroxide regulates the excitability of midbrain dopamine neurons via ATP-sensitive potassium channels. J Neurosci. 2005;25:4222–4231. doi: 10.1523/JNEUROSCI.4701-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barry PH. JPCalc, a software package for calculating liquid junction potential corrections in patch-clamp, intracellular, epithelial and bilayer measurements and for correcting junction potential measurements. J Neurosci Methods. 1994;51:107–116. doi: 10.1016/0165-0270(94)90031-0. [DOI] [PubMed] [Google Scholar]
- Batista-Brito R, Machold R, Klein C, Fishell G. Gene expression in cortical interneuron precursors is prescient of their mature function. Cereb Cortex. 2008;18:2306–2317. doi: 10.1093/cercor/bhm258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergman H, Wichmann T, Karmon B, DeLong MR. The primate subthalamic nucleus. II. Neuronal activity in the MPTP model of parkinsonism. J Neurophysiol. 1994;72:507–520. doi: 10.1152/jn.1994.72.2.507. [DOI] [PubMed] [Google Scholar]
- Beurrier C, Congar P, Bioulac B, Hammond C. Subthalamic nucleus neurons switch from single-spike activity to burst-firing mode. J Neurosci. 1999;19:599–609. doi: 10.1523/JNEUROSCI.19-02-00599.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung KK, Freestone PS, Lipski J. Expression and functional properties of TRPM2 channels in dopaminergic neurons of the substantia nigra of the rat. J Neurophysiol. 2011;106:2865–2875. doi: 10.1152/jn.00994.2010. [DOI] [PubMed] [Google Scholar]
- Clapham DE. SnapShot: mammalian TRP channels. Cell. 2007;129:220. doi: 10.1016/j.cell.2007.03.034. [DOI] [PubMed] [Google Scholar]
- Cvetkovic-Lopes V, Eggermann E, Uschakov A, Grivel J, Bayer L, Jones BE, Serafin M, Mühlethaler M. Rat hypocretin/orexin neurons are maintained in a depolarized state by TRPC channels. PLoS One. 2010;5:e15673. doi: 10.1371/journal.pone.0015673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dauer W, Przedborski S. Parkinson's disease: mechanisms and models. Neuron. 2003;39:889–909. doi: 10.1016/s0896-6273(03)00568-3. [DOI] [PubMed] [Google Scholar]
- Filion M, Tremblay L. Abnormal spontaneous activity of globus pallidus neurons in monkeys with MPTP-induced parkinsonism. Brain Res. 1991;547:142–151. [PubMed] [Google Scholar]
- Fleig A, Penner R. The TRPM ion channel subfamily: molecular, biophysical and functional features. Trends Pharmacol Sci. 2004;25:633–639. doi: 10.1016/j.tips.2004.10.004. [DOI] [PubMed] [Google Scholar]
- Ford CP, Gantz SC, Phillips PE, Williams JT. Control of extracellular dopamine at dendrite and axon terminals. J Neurosci. 2010;30:6975–6983. doi: 10.1523/JNEUROSCI.1020-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González-Hernández T, Rodríguez M. Compartmental organization and chemical profile of dopaminergic and GABAergic neurons in the substantia nigra of the rat. J Comp Neurol. 2000;421:107–135. doi: 10.1002/(sici)1096-9861(20000522)421:1<107::aid-cne7>3.3.co;2-6. [DOI] [PubMed] [Google Scholar]
- Grace AA, Onn SP. Morphology and electrophysiological properties of immunocytochemically identified rat dopamine neurons recorded in vitro. J Neurosci. 1989;9:3463–3481. doi: 10.1523/JNEUROSCI.09-10-03463.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hainsworth AH, Röper J, Kapoor R, Ashcroft FM. Identification and electrophysiology of isolated pars compacta neurons from guinea-pig substantia nigra. Neuroscience. 1991;43:81–93. doi: 10.1016/0306-4522(91)90419-o. [DOI] [PubMed] [Google Scholar]
- Hajós M, Greenfield SA. Synaptic connections between pars compacta and pars reticulata neurones: electrophysiological evidence for functional modules within the substantia nigra. Brain Res. 1994;660:216–224. doi: 10.1016/0006-8993(94)91292-0. [DOI] [PubMed] [Google Scholar]
- Hara Y, Wakamori M, Ishii M, Maeno E, Nishida M, Yoshida T, Yamada H, Shimizu S, Mori E, Kudoh J, Shimizu N, Kurose H, Okada Y, Imoto K, Mori Y. LTRPC2 Ca2+-permeable channel activated by changes in redox status confers susceptibility to cell death. Mol Cell. 2002;9:163–173. doi: 10.1016/s1097-2765(01)00438-5. [DOI] [PubMed] [Google Scholar]
- Hecquet CM, Ahmmed GU, Vogel SM, Malik AB. Role of TRPM2 channel in mediating H2O2-induced Ca2+ entry and endothelial hyperpermeability. Circ Res. 2008;102:347–355. doi: 10.1161/CIRCRESAHA.107.160176. [DOI] [PubMed] [Google Scholar]
- Hill K, Benham CD, McNulty S, Randall AD. Flufenamic acid is a pH-dependent antagonist of TRPM2 channels. Neuropharmacology. 2004;47:450–460. doi: 10.1016/j.neuropharm.2004.04.014. [DOI] [PubMed] [Google Scholar]
- Hutchison WD, Lozano AM, Davis KD, Saint-Cyr JA, Lang AE, Dostrovsky JO. Differential neuronal activity in segments of globus pallidus in Parkinson's disease patients. Neuroreport. 1994;5:1533–1537. doi: 10.1097/00001756-199407000-00031. [DOI] [PubMed] [Google Scholar]
- Ibáñez-Sandoval O, Carrillo-Reid L, Galarraga E, Tapia D, Mendoza E, Gomora JC, Aceves J, Bargas J. Bursting in substantia nigra pars reticulata neurons in vitro: possible relevance for Parkinson disease. J Neurophysiol. 2007;98:2311–2323. doi: 10.1152/jn.00620.2007. [DOI] [PubMed] [Google Scholar]
- Khaliq ZM, Bean BP. Pacemaking in dopaminergic ventral tegmental area neurons: depolarizing drive from background and voltage-dependent sodium conductances. J Neurosci. 2010;30:7401–7413. doi: 10.1523/JNEUROSCI.0143-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiyonaka S, Kato K, Nishida M, Mio K., Numaga T, Sawaguchi Y, Yoshida T, Wakamori M, Mori E, Numata T, Ishii M, Takemoto H, Ojida A, Watanabe K, Uemura A, Kurose H, Morii T, Kobayashi T, Sato Y, Sato C, et al. Selective and direct inhibition of TRPC3 channels underlies biological activities of a pyrazole compound. Proc Natl Acad Sci U S A. 2009;106:5400–5405. doi: 10.1073/pnas.0808793106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolisek M, Beck A, Fleig A, Penner R. Cyclic ADP-ribose and hydrogen peroxide synergize with ADP-ribose in the activation of TRPM2 channels. Mol Cell. 2005;18:61–69. doi: 10.1016/j.molcel.2005.02.033. [DOI] [PubMed] [Google Scholar]
- Lacey MG, Mercuri NB, North RA. Two cell types in rat substantia nigra zona compacta distinguished by membrane properties and the actions of dopamine and opioids. J Neurosci. 1989;9:1233–1241. doi: 10.1523/JNEUROSCI.09-04-01233.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange I, Penner R, Fleig A, Beck A. Synergistic regulation of endogenous TRPM2 channels by adenine dinucleotides in primary human neutrophils. Cell Calcium. 2008;44:604–615. doi: 10.1016/j.ceca.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CR, Tepper JM. Morphological and physiological properties of parvalbumin- and calretinin-containing gamma-aminobutyric acidergic neurons in the substantia nigra. J Comp Neurol. 2007a;500:958–972. doi: 10.1002/cne.21220. [DOI] [PubMed] [Google Scholar]
- Lee CR, Tepper JM. A calcium-activated nonselective cation conductance underlies the plateau potential in rat substantia nigra GABAergic neurons. J Neurosci. 2007b;27:6531–6541. doi: 10.1523/JNEUROSCI.1678-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CR, Witkovsky P, Rice ME. Regulation of substantia nigra pars reticulata GABAergic neuron activity by H2O2 via flufenamic acid-sensitive channels and KATP channels. Front Syst Neurosci. 2011;5:14. doi: 10.3389/fnsys.2011.00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lein ES, Hawrylycz MJ, Ao N, Ayres M, Bensinger A, Bernard A, Boe AF, Boguski MS, Brockway KS, Byrnes EJ, Chen L, Chen L, Chen TM, Chin MC, Chong J, Crook BE, Czaplinska A, Dang CN, Datta S, Dee NR, et al. Genome-wide atlas of gene expression in the adult mouse brain. Nature. 2007;445:168–176. doi: 10.1038/nature05453. [DOI] [PubMed] [Google Scholar]
- Maruyama Y, Ogura T, Mio K, Kiyonaka S, Kato K, Mori Y, Sato C. Three-dimensional reconstruction using transmission electron microscopy reveals a swollen, bell-shaped structure of transient receptor potential melastatin type 2 cation channel. J Biol Chem. 2007;282:36961–36970. doi: 10.1074/jbc.M705694200. [DOI] [PubMed] [Google Scholar]
- Michel FJ, Robillard JM, Trudeau LE. Regulation of rat mesencephalic GABAergic neurones through muscarinic receptors. J Physiol. 2004;556:429–445. doi: 10.1113/jphysiol.2003.057737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel FJ, Fortin GD, Martel P, Yeomans J, Trudeau LE. M3-like muscarinic receptors mediate Ca2+ influx in rat mesencephalic GABAergic neurones through a protein kinase C-dependent mechanism. Neuropharmacology. 2005;48:796–809. doi: 10.1016/j.neuropharm.2005.01.006. [DOI] [PubMed] [Google Scholar]
- Morikawa H, Paladini CA. Dynamic regulation of midbrain dopamine neuron activity: intrinsic, synaptic, and plasticity mechanisms. Neuroscience. 2011;198:95–111. doi: 10.1016/j.neuroscience.2011.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mrejeru A, Wei A, Ramirez JM. Calcium-activated non-selective cation currents are involved in generation of tonic and bursting activity in dopamine neurons of the substantia nigra pars compacta. J Physiol. 2011;589:2497–2514. doi: 10.1113/jphysiol.2011.206631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakanishi H, Kita H, Kitai ST. Intracellular study of rat substantia nigra pars reticulata neurons in an in vitro slice preparation: electrical membrane properties and response characteristics to subthalamic stimulation. Brain Res. 1987;437:45–55. doi: 10.1016/0006-8993(87)91525-3. [DOI] [PubMed] [Google Scholar]
- Otsuka T, Murakami F, Song WJ. Excitatory postsynaptic potentials trigger a plateau potential in rat subthalamic neurons at hyperpolarized states. J Neurophysiol. 2001;86:1816–1825. doi: 10.1152/jn.2001.86.4.1816. [DOI] [PubMed] [Google Scholar]
- Perraud AL, Takanishi CL, Shen B, Kang S, Smith MK, Schmitz C, Knowles HM, Ferraris D, Li W, Zhang J, Stoddard BL, Scharenberg AM. Accumulation of free ADP-ribose from mitochondria mediates oxidative stress-induced gating of TRPM2 cation channels. J Biol Chem. 2005;280:6138–6148. doi: 10.1074/jbc.M411446200. [DOI] [PubMed] [Google Scholar]
- Perraud AL, Fleig A, Dunn CA, Bagley LA, Launay P, Schmitz C, Stokes AJ, Zhu Q, Bessman MJ, Penner R, Kinet JP, Scharenberg AM. ADP-ribose gating of the calcium-permeable LTRPC2 channel revealed by Nudix motif homology. Nature. 2001;411:595–599. doi: 10.1038/35079100. [DOI] [PubMed] [Google Scholar]
- Rice ME. H2O2: a dynamic neuromodulator. Neuroscientist. 2011;17:389–406. doi: 10.1177/1073858411404531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards CD, Shiroyama T, Kitai ST. Electrophysiological and immunocytochemical characterization of GABA and dopamine neurons in the substantia nigra of the rat. Neuroscience. 1997;80:545–557. doi: 10.1016/s0306-4522(97)00093-6. [DOI] [PubMed] [Google Scholar]
- Rivlin-Etzion M, Marmor O, Saban G, Rosin B, Haber SN, Vaadia E, Prut Y, Bergman H. Low-pass filter properties of basal ganglia cortical muscle loops in the normal and MPTP primate model of parkinsonism. J Neurosci. 2008;28:633–649. doi: 10.1523/JNEUROSCI.3388-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen KZ, Johnson SW. Subthalamic stimulation evokes complex EPSCs in the rat substantia nigra pars reticulata in vitro. J Physiol. 2006;573:697–709. doi: 10.1113/jphysiol.2006.110031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen KZ, Johnson SW. Regulation of polysynaptic subthalamonigral transmission by D2, D3 and D4 dopamine receptors in rat brain slices. J Physiol. 2012;590:2273–2284. doi: 10.1113/jphysiol.2011.225672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumoza-Toledo A, Penner R. TRPM2: a multifunctional ion channel for calcium signalling. J Physiol. 2011;589:1515–1525. doi: 10.1113/jphysiol.2010.201855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tóth B, Csanády L. Identification of direct and indirect effectors of the transient receptor potential melastatin 2 (TRPM2) cation channel. J Biol Chem. 2010;285:30091–30102. doi: 10.1074/jbc.M109.066464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbull S, Tabner BJ, El-Agnaf OM, Moore S, Davies Y, Allsop D. alpha-Synuclein implicated in Parkinson's disease catalyses the formation of hydrogen peroxide in vitro. Free Radic Biol Med. 2001;30:1163–1170. doi: 10.1016/s0891-5849(01)00513-5. [DOI] [PubMed] [Google Scholar]
- Wehage E, Eisfeld J, Heiner I, Jüngling E, Zitt C, Lückhoff A. Activation of the cation channel long transient receptor potential channel 2 (LTRPC2) by hydrogen peroxide. A splice variant reveals a mode of activation independent of ADP-ribose. J Biol Chem. 2002;277:23150–23156. doi: 10.1074/jbc.M112096200. [DOI] [PubMed] [Google Scholar]
- Weinberger M, Dostrovsky JO. A basis for the pathological oscillations in basal ganglia: the crucial role of dopamine. Neuroreport. 2011;22:151–156. doi: 10.1097/WNR.0b013e328342ba50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wichmann T, Dostrovsky JO. Pathological basal ganglia activity in movement disorders. Neuroscience. 2011;198:232–244. doi: 10.1016/j.neuroscience.2011.06.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wichmann T, Bergman H, Starr PA, Subramanian T, Watts RL, DeLong MR. Comparison of MPTP-induced changes in spontaneous neuronal discharge in the internal pallidal segment and in the substantia nigra pars reticulata in primates. Exp Brain Res. 1999;125:397–409. doi: 10.1007/s002210050696. [DOI] [PubMed] [Google Scholar]
- Wilson CJ, Bevan MD. Intrinsic dynamics and synaptic inputs control the activity patterns of subthalamic nucleus neurons in health and in Parkinson's disease. Neuroscience. 2011;198:54–68. doi: 10.1016/j.neuroscience.2011.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanovsky Y, Zhang W, Misgeld U. Two pathways for the activation of small-conductance potassium channels in neurons of substantia nigra pars reticulata. Neuroscience. 2005;136:1027–1036. doi: 10.1016/j.neuroscience.2005.08.026. [DOI] [PubMed] [Google Scholar]
- Yanovsky Y, Velte S, Misgeld U. Ca2+ release-dependent hyperpolarizations modulate the firing pattern of juvenile GABA neurons in mouse substantia nigra pars reticulata in vitro. J Physiol. 2006;577:879–890. doi: 10.1113/jphysiol.2006.117622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yung WH, Häusser MA, Jack JJ. Electrophysiology of dopaminergic and non-dopaminergic neurones of the guinea-pig substantia nigra pars compacta in vitro. J Physiol. 1991;436:643–667. doi: 10.1113/jphysiol.1991.sp018571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Chu X, Tong Q, Cheung JY, Conrad K, Masker K, Miller BA. A novel TRPM2 isoform inhibits calcium influx and susceptibility to cell death. J Biol Chem. 2003;278:16222–16229. doi: 10.1074/jbc.M300298200. [DOI] [PubMed] [Google Scholar]
- Zhou FM, Lee CR. Intrinsic and integrative properties of substantia nigra pars reticulata neurons. Neuroscience. 2011;198:69–94. doi: 10.1016/j.neuroscience.2011.07.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou FW, Matta SG, Zhou FM. Constitutively active TRPC3 channels regulate basal ganglia output neurons. J Neurosci. 2008;28:473–482. doi: 10.1523/JNEUROSCI.3978-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou FW, Jin Y, Matta SG, Xu M, Zhou FM. An ultra-short dopamine pathway regulates basal ganglia output. J Neurosci. 2009;29:10424–10435. doi: 10.1523/JNEUROSCI.4402-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu ZT, Munhall A, Shen KZ, Johnson SW. Calcium-dependent subthreshold oscillations determine bursting activity induced by N-methyl-D-aspartate in rat subthalamic neurons in vitro. Eur J Neurosci. 2004;19:1296–1304. doi: 10.1111/j.1460-9568.2004.03240.x. [DOI] [PubMed] [Google Scholar]
- Zhu ZT, Munhall A, Shen KZ, Johnson SW. NMDA enhances a depolarization-activated inward current in subthalamic neurons. Neuropharmacology. 2005;49:317–327. doi: 10.1016/j.neuropharm.2005.03.018. [DOI] [PubMed] [Google Scholar]