

Summary
Age-related macular degeneration causes irreversible central blindness in people over the age of 50 and is increasing in prevalence among elderly populations. There are currently limited treatment options available for the exudative form of the disease and no formal treatments for the geographic atrophy form aside from lifestyle change and incorporation of antioxidant supplements in the diet. As such, it is important to be able to assess high-risk AMD patients as early as possible in order to prescribe preventative measures. Carboxyethylpyrrole (CEP) is a promising plasma biomarker suited to this purpose. Both CEP immunoreactivity levels as well as anti-CEP autoantibody titers are significantly elevated in AMD patients and thus provide the potential to assess AMD susceptibility with approximately 80% accuracy when evaluated alongside genomic AMD markers. Moreover, strong evidence implicates CEP as functionally related to AMD pathogenesis, a role which must be explored further. This avenue of research will foster improved understanding of the disease itself and perhaps reveal better therapeutic targets and options. Further research into the role of CEP in AMD pathogenesis and the application of CEP as an AMD biomarker is merited.
Introduction
Age-related macular degeneration (AMD) is the world's leading cause of irreversible central blindness in people over the age of 50 (1–3). Since the prevalence of AMD is expected to increase by 50% to approximately 3 million people in the United States alone by the year 2020 (4), the disease constitutes a major public health concern. To date, there is no effective cure for the disease and the only available treatments include either diet and lifestyle change for early-stage patients and anti-vascular endothelial growth factor (anti-VEGF) antibody injection to prevent angiogenesis in exudative AMD cases (5).
Early AMD is characterized by extracellular deposition of debris (drusen) on Bruch's membrane, which separates the retinal pigment epithelium (RPE) from the choriocapillaris (5). The disease may result in geographic atrophy, in which case areolar loss of the photoreceptor and RPE cells in the macula occurs, or exudative/neovascular AMD, which is characterized by choroidal neovascularization (CNV), retinal exudates, and hemorrhages. CNV is only present in 10–15% of total AMD cases, and yet it accounts for severe vision loss and blindness in more than 80% of all AMD patients seen by ophthalmologists (5). Although the disease pathogenesis is not fully understood, it is widely agreed that AMD is an inflammatory disease resulting from different immuno/inflammatory pathways such as dysregulation of the complement system (6–7). In humans, several complement pathway genes have been associated with AMD, including CFH, C2, and C3 (8). Strong genetic associations have also been made with human HTRA1 (serine protease) and ARMS2 (mitochondrial protein of unknown function) (8–9). Nonetheless, AMD is a multifactorial disease with environmental components as well. Specifically, oxidative damage is a significant risk factor, as it has been shown that smoking increases the chance of developing AMD (10) and incorporating antioxidants and zinc into the diet reduces this risk (11).
In the last 10 years significant progress has been made into establishing plasma biomarkers for use alongside genomic markers to characterize AMD susceptibility (7, 12–14). One promising biomarker, carboxyethylpyrrole (CEP), belongs to the family of 2-(omega-carboxyalkyl)pyrrole adducts. Protein adducts form with reactive fragments generated from oxidation of cellular polyunsaturated fatty acids such as docasehexaenoic acid (Figure 1) (12, 15). Studies have shown that the use of anti-CEP immunoreactivity measurements in plasma and titers of anti-CEP autoantibody levels provide good means to assess AMD susceptibility (14), particularly when used alongside genomic markers (13).
Figure 1.
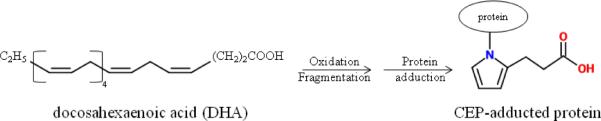
CEP structure and formation. Oxidation of DHA creates CEP fragments that react with cellular protein to form CEP adducts.
CEP as an AMD biomarker
One of the earliest hallmarks of AMD susceptibility is the accumulation of drusen on Bruch's membrane. Small hard drusen are found in people over the age of 50 and are considered in most cases to be a part of normal aging (5). However, excess and large soft drusen may cause lesions in the RPE and chronic inflammation and are thus thought to play a role in AMD pathogenesis (6). Proteomic analysis revealed there to be significantly larger quantities of CEP protein adducts in drusen obtained from AMD donors as opposed to those extracted from age-matched normal donors (12). CEP had been previously identified as being uniquely generated from free-radical induced oxidation of docosahexaenoic acid (DHA), the most oxidizable polyunsaturated fatty acid comprising ~80 mol % of all lipids in photoreceptor outer segments (16). Considering the great potential for photooxidative stress in ocular tissues, CEP may be an ideal marker for oxidative damage in RPE cells. This stands in direct contrast to other carboxylalkylpyrroles, such as omega-(2-carboxyheptyl)pyrrole (CHP, oxidized from linoleic acid) and omega-(2-carboxypropyl)pyrrole (CPP, oxidized from arachidonic acid), which may be generated due to various other causes (14).
As a consequence of the link between oxidative damage and AMD (10), CEP was tested as a potential marker for AMD susceptibility. Immunocytochemical analysis showed that CEP localizes to photoreceptor rod outer segments and RPE cells in both mouse and human retinas (12, 14). Additionally, anti-CEP immunoreactivity is significantly higher in ocular tissue from human AMD retinas than from healthy donor retinas (12, 14). Gu et al.(2003) reported (14) that anti-CEP immunoreactivity in AMD human plasma (n=19, average age 82) was 1.5-fold higher (p = 0.004) than in both age-matched controls (n=19, average age 83) and in young controls (n=9, average age 27, p = 0.05). Additionally, they found that sera from AMD patients demonstrated mean titers of anti-CEP autoantibody 2.3-fold higher than controls (p=0.02). The autoantibody results were confirmed using a dot-blot assay, which revealed 2.5-fold higher anti-CEP autoantibody levels in AMD eyes than in age-matched controls (14). Of the patients in this study who had both elevated anti-CEP immunoreactivity and autoantibody measurements above the non-AMD mean, 92% had AMD. The authors concluded that using both anti-CEP immunoreactivity and autoantibody titer measurements together resulted in the best predictive value of AMD susceptibility. Though statistically significant, the data was nonetheless weak due to a very small sample size.
A later study used a significantly larger sample size (916 AMD and 488 control donors) and determined a 1.6-fold increase in mean plasma anti-CEP immunoreactivity (p < 0.0001) and a 1.3-fold increase in mean plasma anti-CEP autoantibody (p < 0.0001) titer in AMD donors with respect to control donors (13). This study found similar c-statistic values to those presented by the earlier study (14) when using both anti-CEP plasma immunoreactivity levels and autoantibody titers to predict AMD likelihood (13). Interestingly, proteomic CEP markers alone can distinguish between AMD and normal donors with approximately 76% accuracy and when analyzed together with genomic markers, the discriminatory accuracy increased to about 80% (13). Donors with ARMS2, HRTA1, CFH, and C3 AMD risk alleles were found to be approximately 5–10 times more likely to have AMD, whereas when coupled with elevated CEP markers the odds ratios increased an additional 2-to-3-fold (13). Furthermore, donors with AMD risk genotypes at the ARMS2/HRTA1 loci were about 2 times more likely to have elevated CEP markers in their plasma whereas donors with CFH/C3 risk alleles showed an inappreciable increase (13).
Both above studies present compelling evidence for the use of CEP plasma biomarkers to predict AMD susceptibility (13–14), particularly when analyzed alongside genomic markers that have been previously linked to AMD (8, 17–21). However, work remains to be done to define confounding factors surrounding the use of these biomarkers (13). For instance, the authors indicate that smoking seemed to have a relatively low impact on plasma CEP marker levels within both AMD and control groups (13), which contrasts with the well-established link between smoking and AMD (10, 22–23). Additionally, the study by Gu et al. (2009) determined that both hypertensive and hyperlipidemic patients had slightly increased levels of CEP markers (13), and by and large it seems there is a link between cardiovascular disease and AMD in many population studies (24–28). It is, however, uncertain as to whether or not CEP markers may be affected in this case and further analysis is required in this area. Furthermore, diabetics with AMD exhibited no increase in plasma CEP adducts whereas those without AMD appeared to have elevated CEP autoantibody levels (13). Again, further analyses are warranted in these cases, especially due to the small sample size of the study. Aside from biological confounding factors, there is importantly as of yet no standard method for analyzing patient samples, such as whether the or not patients be required to fast, how samples are stored, and how they are assayed (13). These varying experimental conditions across studies may have a role in differing and confusing results.
CEP is not the only biomarker available for AMD. Various studies have implicated A2E, carboxymethllysine (CML), and pentosidine as potential biomarkers for AMD. A2E is found primarily in lipofuscin granules in the retinal pigment epithelium (RPE) cells and lipofuscin deposits on Bruch's membrane (29–30). A2E accumulates progressively with age (31), induces VEGF expression in ARPE-19 cells (29), and has pro-angiogenic properties in vivo (32), which constitutes it as a possible contributor to wet AMD. However, no studies have yet been performed which attempt to associate A2E levels with humans afflicted with AMD. Plasma protein pentosidine and CML, two advanced glycation end products, have been examined alongside CEP (7). Both CML and pentosidine were found in significantly higher levels in AMD serum (p<0.0001) in a cohort of 58 AMD and 32 control donors; using CEP adducts in combination with CML and pentosidine increased the discriminatory accuracy of both (7). Being able to use these biomarkers and any others that may be found in the future will prove useful in taking preventative measures for people with a high-risk of developing AMD.
A functional role for CEP in AMD
It is not surprising that CEP biomarkers are useful in assessing AMD susceptibility, because CEP seems to have a functional role in AMD pathogenesis, specifically in the neovascularization process observed in neovascular AMD (Figure 2) (33). Ebrahem et al. provided convincing evidence that CEP-modified peptides or proteins induce neovascularization in animal models (34). First, in vivo implantation of CEP-adduct-containing methylcellulose discs in chick embryos induced angiogenesis similarly to exogenous VEGF implantation (34). Additionally, rat corneas which received CEP-adduct-containing pellets underwent neovascularization in a pattern similar to that induced by VEGF positive controls (34). Neovascularization was also observed in mouse corneas implanted with CEP pellets, but a differential response was observed in eyes which received pellets containing mixtures of either CEP and anti-CEP or CEP and anti-VEGF antibody. Neovascularization was only attenuated when anti-CEP antibodies were included in the CEP pellet, whereas anti-VEGF antibody addition failed to eliminate new blood vessel growth (34). Moreover, subretinal injection of CEP-modified proteins into laser-induced CNV mouse models exacerbated the extent of CNV (34). Based on these data alone, it seems that CEP may independently induce neovascularization, and when considering that ARPE-19 cell cultures treated with CEP do not secrete VEGF (34), it seems likely that an intercellular signaling cascade involving multiple cell types or pathways could be involved. Subsequently, CEP was shown to induce angiogenesis via toll-like receptor 2 (TLR2) in a VEGF-independent mechanism in endothelial cells from the human umbilical vein, mouse lung, and mouse aorta (35). No studies with respect to AMD have been performed.
Figure 2.
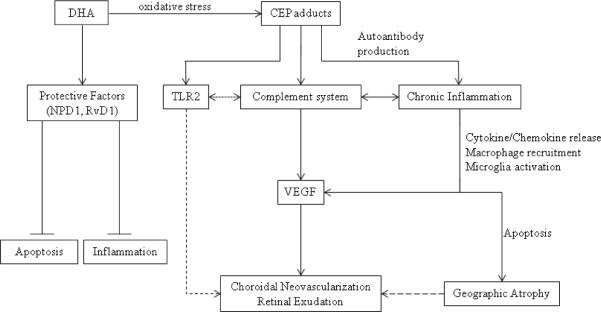
CEP functionality in AMD pathogenesis. CEP adducts generated from oxidation of DHA likely activate inflammatory and complement pathways that result in neovascularization and/or geographic atrophy. Neovascularization occurs via VEGF and another, VEGF-independent pathway involing TLR2. Chronic inflammation may also result in geographic atrophy via a macrophage-regulated process, and may even lead to neovascularization over time. DHA normally regulates protective mechanisms in the cell.
It must be noted, however, that not all data suggests CEP plays a role in exudative AMD, as two studies by the same group failed to detect CNV in CEP-immunized mice (36–37). Mice immunized with CEP-adducted serum albumin develop high autoantibody levels and characteristic dry AMD features including drusen formation and RPE lesions that mimic geographic atrophy (36). The long-term response to CEP immunization includes thick swelling in retinal tissues in addition to RPE cell lysis, pyknosis, and infiltration of macrophages and lymphocytes (37). Importantly, the invading cells were reported to not play a role in lesion formation due to the fact that they contained melanin pigment, indicating they were recruited post-lesion by cytokine secretion in order to clear debris (37). Additionally, not all lesions were found to have been infiltrated by inflammatory cells (37).
There is no consensus on how CEP works within the AMD disease state. Looking at AMD without considering CEP, one leading hypothesis regarding disease pathogenesis purports that chronic inflammation mediated by dysregulation of complement proteins, M1 and M2 macrophages, and microglia activation leads to geographic atrophy (Figure 2). CNV development in later AMD stages is explained by observations implicating M2 macrophages to have pro-angiogenic effects (See Patel and Chan (38) and Ding et al. (39) for a thorough review). Because mice immunized with CEP-adducts produce anti-CEP antibody and fix complement component-3 in Bruch's membrane (36), the process seems to be complement-regulated. Evidence exists linking the complement system to a variety of developmental processes including regulation of angiogenesis (40–42). Complement activation leads to recruitment of the membrane-attack complex which mediates targeted-cell lysis. Within the activation cascade, binding of anaphylatoxins to blood basophils and mast cells induces degranulation, smooth muscle contraction, and increased vascular permeability. It has been previously shown that the anaphylatoxins C3a and C5a, the downstream effectors of the complement system, are present in AMD drusen and stimulate choroidal neovascularization via VEGF in vivo and in vitro, and that anti-C3a and anti-C5a antibody treatment prevents angiogenesis (43). In contrast, immunocompromised mice (Rag-deficient mice that lack B and T cells) treated with CEP-adducts mount no immune response and develop no signs of AMD (36). It is therefore possible that complement dysregulation, together with processes involved in antibody production, causes a prolonged inflammatory/immunologic response that contributes to the AMD pathology.
However, complement activation has also been convincingly shown to inhibit neovascularization via macrophage-regulated mechanisms in mouse models of retinopathy of prematurity (ROP) (41). AMD and ROP pathogenesis differ in that different tissues are involved, namely that the RPE is an important factor in AMD but not in ROP models. Thus, the overall message is that the complement system is intricately involved in neovascularization and further study is required to evaluate its role in AMD pathogenesis. It could very well be that CEP triggers an autoimmune response mediated by the complement system, and that immune cell recruitment by proinflammatory cytokines worsens the condition and results in RPE damage. The finding that CEP induces VEGF-independent neovascularization via TLR2 (35) suggests that the link between increased CEP adducts levels in AMD patients and activation of the complement system could be TLR2 (Figure 2), which would subsequently implicate a role for innate immunity in regulating angiogenesis. Consequently, manipulation of the TLR2 signaling pathway in the retina may be a viable therapy for AMD patients. At least one other AMD marker, A2E, has been shown to activate the complement system via the alternative pathway and contribute to the chronic inflammation observed in AMD pathologies (44–45), and is also pro-angiogenic in vivo (32). Research into the largely unknown interplay between these various factors will undoubtedly be the key to fully understanding AMD pathogenesis.
CEP and Omega-3 Fatty Acids
The negative effects caused by CEP accumulation question the treatment value of omega-3 fatty acid supplements recommended for AMD patients (46–47). DHA, one of the omega-3 fatty acids and the precursor of CEP, has been shown to inhibit apoptosis in RPE cells (48–49), protect cells from oxidative-stress and promote survival via two of its metabolic products, neuroprotectin D1 (NPD1) and resolvin D1 (RvD1) (50–51), and suppress retinal inflammation in an AMD mouse model (47). One hypothesis is that omega-3 fatty acids repress NF-kappaB signaling in RPE cells and thus prevent an inflammatory response (52). If CEP accumulation is responsible for at least a subset of all AMD cases, then increasing DHA consumption could potentially worsen the AMD condition in those patients who are more susceptible to oxidizing DHA into CEP. It is also important to consider omega-3 fatty acids along with omega-6 fatty acids. Studies have shown that more important than absolute quantities of the fatty acids is their relative proportions (53). For example, in animal models, a high omega-6/omega-3 ratio results in abnormal electroretinograms (54–55), and there are suggestions that omega-6 fatty acids have pro-inflammatory properties (56). Thus further advances into understanding the biochemistry between DHA and CEP, as well how the effects are mediated based on the omega-6/omega-3 fatty acid ratio, are required to validate the efficacy of this treatment option. In particular, the controversy could be resolved if we knew whether the formation of CEP is substrate (DHA)-dominant or shunted, with conversion to CEP favored in individuals with at-risk genetic or epigenetic profiles, as well as a macular microenvironment.
Conclusions
AMD poses a tangible threat to public health in the future (1–2, 5). It is of vital importance to be able to identify at-risk individuals for AMD as early as possible so that proper preventative measures can be taken prior to disease progression. Towards early detection, plasma biomarkers such as CEP provide a good measurement for assessing risk (14), particularly when they are analyzed along with genetic predisposition (13) and other potential disease biomarkers such as CML and pentosidine (7).
Currently, the biggest drawbacks include a limited number of studies and no clear progress towards using preliminary data to create a practical clinical assay. Moreover, strong evidence implicating the aforementioned biomarkers as functionally related to AMD pathogenesis must be further explored, particularly the finding that CNV may result from at least one other VEGF-independent mechanism (34) which may be induced via TLR2 (35). Exploring these avenues of research will provide a better understanding of the disease itself and perhaps reveal better therapeutic targets and options. It may potentially divulge other biomarkers which may aid in predicting not only AMD susceptibility, but also in indicating to what stage the disease has progressed and how the disease has responded to therapy. For instance, Hollyfield et al. reported that CEP is sufficient to induce AMD in a mouse model that causes geographic atrophy of the macula (36–37), whereas Ebrahem et al. reported that CEP plays a role in exudative AMD (34). Per these studies, it could be the case that CEP accumulation initiates geographic atrophy and may potentially induce CNV in later stages after sufficient time is allowed or some other pathogenic pathway is activated. Further research into the role of CEP in AMD pathogenesis and the application of CEP as an AMD biomarker is merited.
Disclosures and Acknowledgments
This review was funded by the NEI Intramural Research Program. Only those individuals listed as authors contributed to writing this article.
Footnotes
The authors state no conflict of interests.
References
- 1.Pascolini D, Mariotti SP, Pokharel GP, Pararajasegaram R, Etya'ale D, Negrel AD, Resnikoff S. 2002 global update of available data on visual impairment: a compilation of population-based prevalence studies. Ophthalmic Epidemiol. 2004;11(2):67–115. doi: 10.1076/opep.11.2.67.28158. [DOI] [PubMed] [Google Scholar]
- 2.Congdon N, O'Colmain B, Klaver CC, et al. Causes and prevalence of visual impairment among adults in the United States. Arch Ophthalmol. 2004;122(4):477–85. doi: 10.1001/archopht.122.4.477. [DOI] [PubMed] [Google Scholar]
- 3.Visual impairment and blindness. [Accessed June 6, 2011];World Health Organization. 2011 Apr; http://www.who.int/mediacentre/factsheets/fs282/en/index.html.
- 4.Friedman DS, O'Colmain BJ, Munoz B, Tomany SC, McCarthy C, de Jong PT, Nemesure B, Mitchell P, Kempen J. Prevalence of age-related macular degeneration in the United States. Arch Ophthalmol. 2004;122:564–72. doi: 10.1001/archopht.122.4.564. [DOI] [PubMed] [Google Scholar]
- 5.Jager RD, Mieler WF, Miller JW. Age-related Macular Degeneration. N Engl J Med. 2008;358(24):2606–17. doi: 10.1056/NEJMra0801537. [DOI] [PubMed] [Google Scholar]
- 6.de Jong PT. Age-related macular degeneration. N Engl J Med. 2006;355:1474–85. doi: 10.1056/NEJMra062326. [DOI] [PubMed] [Google Scholar]
- 7.Ni J, Yuan X, Gu J, Yue X, Gu X, Nagaraj RH, Crabb JW. Plasma Protein Pentosidine and Carboxymethyllysine, Biomarkers for Age-related Macular Degeneration. Mol Cell Proteomics. 2009;8(8):1921–33. doi: 10.1074/mcp.M900127-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen Y, Bedell M, Zhang K. Age-related Macular Degeneration: Genetic and Environmental Factors of Disease. Mol Interv. 2010;10(5):271–81. doi: 10.1124/mi.10.5.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yu Y, Bhangale TR, Fagerness J, et al. Common Variants near FRK/COL10A1 and VEGFA are Associated with Advanced Age-related Macular Degeneration. Hum Mol Genet. 2011 doi: 10.1093/hmg/ddr270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Seddon JM, Willett WC, Speizer FE, Hankinson SE. A prospective study of cigarette smoking and age-related macular degeneration in women. JAMA. 1996;276(14):1141–6. [PubMed] [Google Scholar]
- 11.Age-related Eye Disease Study Research Group A randomized, placebo-controlled, clinical trial of high-dose supplementation with vitamins C and E, beta carotene, and zinc for age-related macular degeneration and vision loss: AREDS report no. 8. Arch Ophthalmol. 2001;119:1417–36. doi: 10.1001/archopht.119.10.1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Crabb JW, Miyagi M, Gu X, et al. Drusen proteome analysis: an approach to the etiology of age-related macular degeneration. Proc Natl Acad Sci U S A. 2002;99(23):14682–7. doi: 10.1073/pnas.222551899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gu J, Pauer GJ, Yue X, Narendra U, Sturgill GM, Bena J, Gu X, Peachey NS, Salomon RG, Hagstrom SA, Crabb JW, the Clinical Genomic and Proteomic AMD Study Group Assessing susceptibility to age-related macular degeneration with proteomic and genomic biomarkers. Mol Cell Proteomics. 2009;8(6):1338–49. doi: 10.1074/mcp.M800453-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gu X,M,SG, Miyagi M, Rayborn ME, Hollyfield JG, Crabb JW, Salomon RG. Carboxyethylpyrrole protein adducts and autoantibodies, biomarkers for age-related macular degeneration. J Biol Chem. 2003;278(43):42027–35. doi: 10.1074/jbc.M305460200. [DOI] [PubMed] [Google Scholar]
- 15.Gu X, Sun M, Gugiu B, Hazen S, Crabb JW, Salomon RG. Oxidatively truncated docosahexaenoate phospholipids: total synthesis, generation, and Peptide adduction chemistry. J Org Chem. 2003;68(10):3749–61. doi: 10.1021/jo026721t. [DOI] [PubMed] [Google Scholar]
- 16.Fliesler SJ, Anderson RE. Chemistry and metabolism of lipids in the vertebrate retina. Prog Lipid Res. 1983;22(2):79–131. doi: 10.1016/0163-7827(83)90004-8. [DOI] [PubMed] [Google Scholar]
- 17.Edwards AO, Ritter R, Abel KJ, Manning A, Panhuysen C, Farrer LA. Complement factor H polymorphism and age-related macular degeneration. Science. 2005;308(5720):421–4. doi: 10.1126/science.1110189. [DOI] [PubMed] [Google Scholar]
- 18.Katta S, Kaur I, Chakrabarti S. The molecular genetic basis of age-related macular degeneration: an overview. J Genet. 2009;88(4):425–49. doi: 10.1007/s12041-009-0064-4. [DOI] [PubMed] [Google Scholar]
- 19.Dewan A, Liu MG, Hartman S, et al. HTRA1 promoter polymorphism in wet age-related macular degeneration. Science. 2006;314(5801):989–92. doi: 10.1126/science.1133807. [DOI] [PubMed] [Google Scholar]
- 20.Sobrin L, Reynolds R, Yu Y, et al. ARMS2/HTRA1 Locus Can Confer Differential Susceptibility to the Advanced Subtypes of Age-Related Macular Degeneration. Am J Ophthalmol. 2011;151(2):345–52. doi: 10.1016/j.ajo.2010.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ryu E, Fridley BL, Tosakulwong N, Bailey KR, Edwards AO. Genome-wide association analyses of genetic, phenotypic, and environmental risks in the age-related eye disease study. Mol Vis. 2010;16(301–02):2811–21. [PMC free article] [PubMed] [Google Scholar]
- 22.Kabasawa S, Mori K, Horie-Inoue K, et al. Associations of Cigarette Smoking But Not Serum Fatty Acids with Age-related Macular Degeneration in a Japanese Population. Ophthalmology. 2011;118(6):1082–8. doi: 10.1016/j.ophtha.2010.10.012. [DOI] [PubMed] [Google Scholar]
- 23.Cackett P, Yeo I, Cheung CMG, et al. Relationship of Smoking and Cardiovascular Risk Factors with Polypoidal Choroidal Vasculopathy and Age-related Macular Degeneration in Chinese Persons. Ophthalmology. 2011;118(5):846–52. doi: 10.1016/j.ophtha.2010.09.026. [DOI] [PubMed] [Google Scholar]
- 24.Fraser-Bell S, Wu J, Klein R, Azen SP, Hooper C, Foong AWP, Varma R. Cardiovascular risk factors and age-related macular degeneration: The Los Angeles Latino Eye Study. American Journal of Ophthalmology. 2008;145(2):308–16. doi: 10.1016/j.ajo.2007.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Haas P, Aggermann T, Steindl K, et al. Genetic cardiovascular risk factors and age-related macular degeneration. Acta Ophthalmol. 2011;89(4):335–8. doi: 10.1111/j.1755-3768.2009.01697.x. [DOI] [PubMed] [Google Scholar]
- 26.Klein R, Klein BEK, Tomany SC, Cruickshanks KJ. The association of cardiovascular disease with the long-term incidence of age-related maculopathy: the Beaver Dam Eye Study. Ophthalmology. 2003;110(6):1273–80. doi: 10.1016/S0161-6420(03)00599-2. [DOI] [PubMed] [Google Scholar]
- 27.van Leeuwen R, Ikram MK, Vingerling JR, Witteman JCM, Hofman A, de Jong P. Blood pressure, atherosclerosis, and the incidence of age-related maculopathy: The Rotterdam Study. Invest Ophthalmol Vis Sci. 2003;44(9):3771–7. doi: 10.1167/iovs.03-0121. [DOI] [PubMed] [Google Scholar]
- 28.Hogg RE, Woodside JV, Gilchrist S, et al. Cardiovascular disease and hypertension are strong risk factors for choroidal neovascularization. Ophthalmology. 2008;115(6):1046–52. doi: 10.1016/j.ophtha.2007.07.031. [DOI] [PubMed] [Google Scholar]
- 29.Iriyama A, Fujiki R, Inoue Y, et al. A2E, a pigment of the lipofuscin of retinal pigment epithelial cells, is an endogenous ligand for retinoic acid receptor. J Biol Chem. 2008;283(18):11947–53. doi: 10.1074/jbc.M708989200. [DOI] [PubMed] [Google Scholar]
- 30.Ng KP, Gugiu B, Renganathan K, Davies MW, Gu X, Crabb JS, Kim SR, Rózanowska MB, Bonilha VL, Rayborn ME, Salomon RG, Sparrow JR, Boulton ME, Hollyfield JG, Crabb JW. Retinal pigment epithelium lipofuscin proteomics. Mol Cell Proteomics. 2008;7(7):1397–405. doi: 10.1074/mcp.M700525-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Murdaugh LS, Wang Z, Del Priore LV, Dillon J, Gaillard ER. Age-related accumulation of 3-nitrotyrosine and nitro-A2E in human Bruch's membrane. Exp Eye Res. 2010;90(5):564–71. doi: 10.1016/j.exer.2010.01.014. [DOI] [PubMed] [Google Scholar]
- 32.Iriyama A, Inoue Y, Takahashi H, Tamaki Y, Jang WD, Yanagi Y. A2E, a component of lipofuscin, is pro-angiogenic in vivo. J Cell Physiol. 2009;220(2):469–75. doi: 10.1002/jcp.21792. [DOI] [PubMed] [Google Scholar]
- 33.Ramkumar HL, Zhang J, Chan CC. Retinal ultrastructure of murine models of dry age-related macular degeneration (AMD) Prog Retin Eye Res. 2010;29(3):169–90. doi: 10.1016/j.preteyeres.2010.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ebrahem Q, Renganathan K, Sears J, Vasanji A, Gu X, Lu L, Salomon RG, Crabb JW, Anand-Apte B. Carboxyethylpyrrole oxidative protein modifications stimulate neovascularization: Implications for age-related macular degeneration. Proc Natl Acad Sci U S A. 2006;103(36):13480–4. doi: 10.1073/pnas.0601552103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.West XZ, Malinin NL, Merkulova AA, et al. Oxidative stress induces angiogenesis by activating TLR2 with novel endogenous ligands. Nature. 2010;467(7318):972–6. doi: 10.1038/nature09421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hollyfield JG, Bonilha VL, Rayborn ME, Yang X, Shadrach KG, Lu L, Ufret RL, Salomon RG, Perez VL. Oxidative damage-induced inflammation initiates age-related macular degeneration. Nat Med. 2008;14(2):194–8. doi: 10.1038/nm1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hollyfield JG, Perez VL, Salomon RG. A hapten generated from an oxidation fragment of docosahexaenoic acid is sufficient to initiate age-related macular degeneration. Mol Neurobiol. 2010;41(2–3):290–8. doi: 10.1007/s12035-010-8110-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Patel M, Chan CC. Immunopathological aspects of age-related macular degeneration. Semin Immunopathol. 2008;30(2):97–110. doi: 10.1007/s00281-008-0112-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ding X, Patel M, Chan CC. Molecular pathology of age-related macular degeneration. Prog Retin Eye Res. 2009;28(1):1–18. doi: 10.1016/j.preteyeres.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rutkowski MJ, Sughrue ME, Kane AJ, Ahn BJ, Fang S, Parsa AT. The complement cascade as a mediator of tissue growth and regeneration. Inflamm Res. 2010;59(11):897–905. doi: 10.1007/s00011-010-0220-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Langer HF, Chung KJ, Orlova VV, et al. Complement-mediated inhibition of neovascularization reveals a point of convergence between innate immunity and angiogenesis. Blood. 2010;116(22):4395–403. doi: 10.1182/blood-2010-01-261503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kahr WH. Complement halts angiogenesis gone wild. Blood. 2010;116(22):4393–4. doi: 10.1182/blood-2010-08-297648. [DOI] [PubMed] [Google Scholar]
- 43.Nozaki M, Raisler BJ, Sakurai E, et al. Drusen complement components C3a and C5a promote choroidal neovascularization. Proc Natl Acad Sci U S A. 2006;103(7):2328–33. doi: 10.1073/pnas.0408835103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhou J, Kim SR, Westlund BS, Sparrow JR. Complement activation by bisretinoid constituents of RPE lipofuscin. Invest Ophthalmol Vis Sci. 2009;50(3):1392–9. doi: 10.1167/iovs.08-2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sparrow JR. Bisretinoids of RPE lipofuscin: trigger for complement activation in age-related macular degeneration. Adv Exp Med Biol. 2010;703:63–74. doi: 10.1007/978-1-4419-5635-4_5. [DOI] [PubMed] [Google Scholar]
- 46.Ho L, van Leeuwen R, Witteman JC, et al. Reducing the Genetic Risk of Age-Related Macular Degeneration With Dietary Antioxidants, Zinc, and {omega}-3 Fatty Acids: The Rotterdam Study. Arch Ophthalmol. 2011;129(6):758–66. doi: 10.1001/archophthalmol.2011.141. [DOI] [PubMed] [Google Scholar]
- 47.Tuo J, Ross RJ, Herzlich AA, et al. A high omega-3 fatty acid diet reduces retinal lesions in a murine model of macular degeneration. Am J Pathol. 2009;175(2):799–807. doi: 10.2353/ajpath.2009.090089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.German OL, Insua MF, Gentili C, Rotstein NP, Politi LE. Docosahexaenoic acid prevents apoptosis of retina photoreceptors by activating the ERK/MAPK pathway. J Neurochem. 2006;98(5):1507–20. doi: 10.1111/j.1471-4159.2006.04061.x. [DOI] [PubMed] [Google Scholar]
- 49.Rotstein NP, Politi LE, German OL, Girotti R. Protective effect of docosahexaenoic acid on oxidative stress-induced apoptosis of retina photoreceptors. Invest Ophthalmol Vis Sci. 2003;44(5):2252–9. doi: 10.1167/iovs.02-0901. [DOI] [PubMed] [Google Scholar]
- 50.Bazan NG. Neuroprotecting D1 (NPD1): a DHA-derived mediator that protects brain and retina against cell injury-induced oxidative stress. Brain Pathol. 2005;15:159–66. doi: 10.1111/j.1750-3639.2005.tb00513.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mukherjee P, Marcheselli V, de Rivero Vacari J, Gordon W, Jackson F, Bazan N. Photoreceptor outer segment phagocytosis attenuates oxidative-stress induced apoptosis with concomitant neuroprotecting D1 synthesis. Proc Natl Acad Sci U S A. 2007;104:13158–63. doi: 10.1073/pnas.0705963104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kaarniranta K, Salminen A. NF-kappaB signaling as a putative target for omega-3 metabolites in the prevention of age-related macular degeneration (AMD) Exp Gerontol. 2009;44(11):685–8. doi: 10.1016/j.exger.2009.09.002. [DOI] [PubMed] [Google Scholar]
- 53.Schweigert FJ, Reimann J. Micronutrients and their Relevance for the Eye - Function of Lutein, Zeaxanthin and Omega-3 Fatty Acids. Klin Monbl Augenheilkd. 2011;228(6):537–43. doi: 10.1055/s-0029-1245527. [DOI] [PubMed] [Google Scholar]
- 54.Pawlosky RJ, Denkins Y, Ward G, Salem N., Jr Retinal and brain accretion of long-chain polyunsaturated fatty acids in developing felines: the effects of corn oil-based maternal diets. Am J Clin Nutr. 1997;65(2):465–72. doi: 10.1093/ajcn/65.2.465. [DOI] [PubMed] [Google Scholar]
- 55.Neuringer M, Connor WE, Van Petten C, Barstad L. Dietary omega-3 fatty acid deficiency and visual loss in infant rhesus monkeys. J Clin Invest. 1984;73(1):272–6. doi: 10.1172/JCI111202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kishan AU, Modjtahedi BS, Martins EN, Modjtahedi SP, Morse LS. Lipids and age-related macular degeneration. Surv Ophthalmol. 56(3):195–213. doi: 10.1016/j.survophthal.2010.08.008. [DOI] [PubMed] [Google Scholar]