

Summary
Human trisomies can alter cellular phenotypes and produce congenital abnormalities such as Down syndrome (DS). Here we have generated induced pluripotent stem cells (iPSCs) from DS fibroblasts and introduced a TKNEO transgene into one copy of chromosome 21 by gene targeting. When selecting against TKNEO, spontaneous chromosome loss was the most common cause for survival, with a frequency of ∼10−4, while point mutations, epigenetic silencing, and TKNEO deletions occurred at lower frequencies in this unbiased comparison of inactivating mutations. Mitotic recombination events resulting in extended loss of heterozygosity were not observed in DS iPSCs. The derived, disomic cells proliferated faster and produced more endothelia in vivo than their otherwise isogenic trisomic counterparts, but in vitro hematopoietic differentiation was not consistently altered. Our study describes a targeted removal of a human trisomy, which could prove useful in both clinical and research applications.
Results and Discussion
Human trisomies are a major health problem, accounting for 23% of spontaneous abortions (Hassold et al., 1980). Down syndrome (DS) is the most frequent autosomal trisomy (chromosome 21), with numerous clinical manifestations including heart defects, impaired cognition, premature aging, and a high frequency of specific leukemias (Hasle et al., 2000). Trisomies produce diverse effects on cultured cells. Fibroblasts from trisomic mice proliferate slowly and have increased sensitivity to specific drugs (Tang et al., 2011; Williams et al., 2008), while human pluripotent stem cells with trisomy 12 or 17 have an apparent growth advantage in vitro (Baker et al., 2007). In DS, the effects of an extra copy of chromosome 21 have been studied by comparing normal and trisomic cells from distinct individuals (Chou et al., 2008; Costa et al., 2010; Crossen and Morgan, 1980; Segal and McCoy, 1974; Sikora et al., 1993), where differences in age, medical history, genetic polymorphisms, and cell passaging may have influenced results. Ideally, the effects of human trisomy should be compared in otherwise isogenic, disomic cell lines, as done previously in yeast (Torres et al., 2010) and mice (Tang et al., 2011; Williams et al., 2008).
Induced pluripotent stem cells (iPSCs) offer a promising way to study human trisomy because they can be derived from the somatic cells of trisomic individuals or fetuses, cultured indefinitely, and differentiated into diverse cell types. We generated six independent iPSC lines from DS fibroblasts as described (Yu et al., 2007) and validated their trisomy 21 karyotypes (example in Figure S1A available online). These iPSCs were used to derive disomic subclones as outlined in Figure 1A. A TKNEO fusion gene encoding both thymidine kinase and neomycin resistance was introduced into chromosome 21 of DS iPSCs for positive-negative selection. The Amyloid Precursor Protein (APP) gene was chosen as the target locus due to its location on the long arm of chromosome 21 and high expression in iPSCs. The adeno-associated virus vector AAV-APPe3ITKNA was used to introduce an internal ribosome entry site (IRES)-TKNEO-polyadenylation site cassette into exon 3 of the APP gene by homologous recombination (Figure 1A). Five transduced, G418-resistant clones were isolated from each of three DS iPSC lines and analyzed by Southern blots. Twelve of these clones were targeted correctly at one APP allele and lacked random integrants (Figure 1B). Based on these results, 0.14% of all iPSC colony-forming units (CFUs) underwent gene targeting (Table S1 available online), comparable to previous reports (Chamberlain et al., 2004; Khan et al., 2010).
Figure 1. Targeting and Removal of Chromosome 21 from DS iPSCs.

(A) Schematic overview of experimental approach.
(B) Identification of chromosome 21 targeted clones. Maps of untargeted and targeted APP loci are shown. X, Xba I sites. The Southern blots show parental DS iPSC clones C1, C2, and C3 and their derived G418-resistant subclones after digestion with Xba I and being probed for APP or TKNEO.
(C) Copy numbers of three chromosome 21 loci and a chromosome 6 control locus were determined by quantitative Southern blots in 33 GCV-resistant subclones. Probe loci are shown in matched colors.
(D) TKNEO transgene presence in a subset of GCVR subclones as shown by Southern blot using a TKNEO probe. Parental trisomic iPSCs (C2) and targeted trisomic iPSCs (C2-4) were included as controls.
(E) Northern blot demonstrating TKNEO gene expression in a subset of the GCVR subclones containing TKNEO transgenes. Parental trisomic iPSCs (C2) and targeted trisomic iPSCs (C2-4) were included as controls.
(F) TKNEO coding region mutations found in the five GCVR subclones with expressed TKNEO transgenes. Wild-type sequences are shown above in red and mutant sequences are shown below in blue.
(G) Total frequency of GCV resistance and the percentage of GCVR subclones due to each type of event. See also Figure S1 and Tables S1 and S2.
APP-targeted clones were cultured in the absence of G418 selection for three passages to allow chromosome loss to take place, and then were cultured in the presence of gancyclovir (GCV) to select against TKNEO expression. A total of 33 GCV-resistant clones derived from four different DS iPSCs were screened for disomy by quantitative Southern blot analysis using three probes from the long arm of chromosome 21 (centromeric, APP target locus, and telomeric) and a reference probe on chromosome 6 (Figure 1C). By comparison to trisomic and disomic controls, we determined that 23 of the 33 GCV-resistant clones had lost one copy of chromosome 21. Seven of these clones were karyotyped (Table S2) and all had two copies of chromosome 21 (example in Figure S1A), validating the Southern blot method and demonstrating that disomic iPSCs had been derived from trisomic DS iPSCs. We also showed that different copies of chromosome 21 had been targeted and lost, based on SNP analysis of the disomic clones (Figure S1B).
Because the targeted copy of chromosome 21 was not required for cell growth, we were able to measure the frequencies of all causes of GCV resistance without bias. Overall, loss of an entire copy of chromosome 21 was the most common cause in five out of five independent experiments with an average frequency of 1.4 × 10−4. The remaining 10 clones were characterized further to determine the basis for their GCV resistance. Southern blots showed that eight clones were still trisomic at chromosome 21 (Figure 1C and Table S2). Six of these contained the TKNEO gene, which was either mutated in the coding region (five clones) or silenced based on the lack of mRNA expression (one clone) (Figures 1D–1F). The frequencies of TKNEO coding mutations and silencing in DS iPSCs were similar to those observed previously in mouse ESCs (Chan et al., 2001). Two other trisomy 21 clones (C1-5-13 and C3-5-10) had lost the TKNEO gene. These TKNEO deletions were analyzed further to see if loss of heterozygosity (LOH) extending from a recombination crossover point to the telomere had occurred. However, three distinct chromosome 21 haplotypes were present based on SNP analysis, suggesting that more localized gene conversion events were responsible (Figure S1C). The absence of clones with extended LOH was unexpected because this is a major cause of gene inactivation/loss in mouse ESCs and human lymphocytes (Chan et al., 2001; Gupta et al., 1997; Lefebvre et al., 2001). The two remaining GCV-resistant clones contained three copies of a portion of chromosome 21 (Figure 1C), but only two copies of more telomeric sequences. One of these clones contained a silenced TKNEO gene (C1-5-12), while the other lacked TKNEO and was disomic at chromosome 21 by karyotyping (C3-1-2), suggesting that there may have been a localized duplication of pericentromeric sequences not detected by cytogenetic analysis. Based on this classification, the different causes of GCV resistance were chromosome loss > TKNEO coding mutation > TKNEO transgene deletion > TKNEO transgene silencing (Figure 1G).
DS fibroblasts and lymphocytes have been reported to have either increased or decreased cell growth in comparison to nonisogenic, disomic cells (Crossen and Morgan, 1980; Segal and McCoy, 1974; Sikora et al., 1993). We measured the proliferation of four passage-matched, isogenic pairs of disomic and trisomic iPSCs and two control pairs with GCV-resistant trisomic clones that had TKNEO mutations, using mixed cultures in which we tracked the percentage of trisomic cells by Southern blots for the TKNEO gene (Figure 2A). In all cases the disomic cells expanded preferentially during coculture, with an average population doubling time of 37 ± 0.7 hr as compared to 45 ± 0.9 hr for trisomic cells. Pure cultures of disomic clones also expanded more quickly than their trisomic counterparts, with 3–7 hr shorter doubling times during the proliferative period (Figure 2B). Our results are consistent with those obtained from isogenic mouse cell lines showing that trisomies inhibit proliferation (Williams et al., 2008) and with the observation that disomy 21 mosaicism increases over time in DS (Jenkins et al., 1997). Based on the profiling of cell cycle phases with bromodeoxyuridine (BrdU) and propidium iodide (PI), a greater percentage of trisomic cells were in the G1 phase (Figure 2C), which was not found in mouse trisomy models (Williams et al., 2008). These results were not due to altered differentiation kinetics or apoptosis, since expression of the pluripotency surface marker SSEA3 and the apoptosis marker Annexin V were similar in disomic and trisomic cells (Figures 2D and 2E).
Figure 2. Proliferation and Gene Expression of Trisomic and Disomic iPSCs.
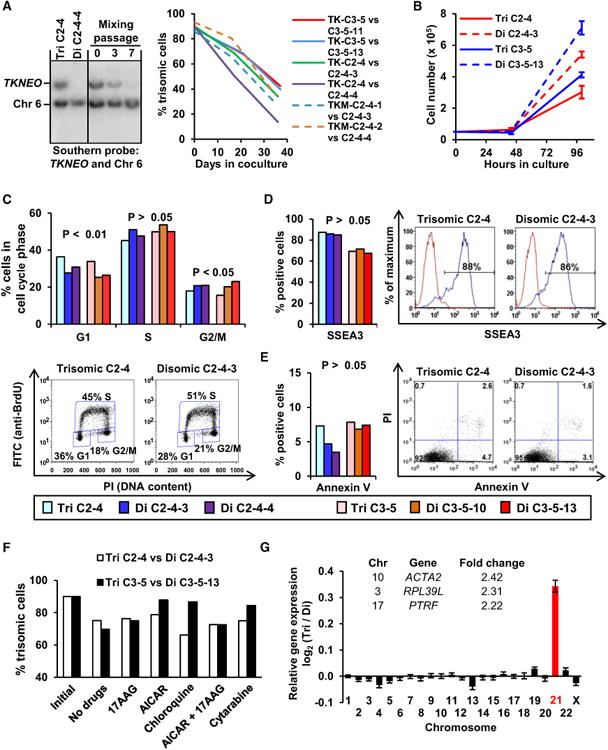
(A) Mixed culture measurements of trisomic and disomic iPSCs (initial seeding with 90% trisomic cells) by Southern blot analysis with TKNEO and chromosome 6 probes at 0, 3, and 7 passages. Representative blots with trisomic clone C2-4, disomic subclone C2-4-4, and pure cell population controls are shown. At right, the trisomic cell percentages of four mixed cultures are shown (solid lines). Controls (dashed lines) included cultures with trisomic cells containing mutated TKNEO transgenes (TKM C2-4-1 and C2-4-2).
(B) Growth curves of two isogenic pairs of iPSCs.
(C) The percents of G1, S, and G2/M phase cells as determined by BrdU-PI labeling in trisomic and disomic cells with representative flow cytometry examples shown below. Bars are color-coded as indicated below for (C)–(E).
(D and E) The percentages of SSEA3+ pluripotent cells (D) and Annexin V+ apoptotic cells(E) as determined by flow cytometry of trisomic and disomic clones, with examples shown at the right.
(F) Percentage of trisomic iPSCs present in mixed cultures performed in the absence of drugs or in the presence of 200 nM 17-AAG, 1 mM AICAR, 10 mM chloroquine, 200 nM 17-AAG plus 1 mM AICAR, or 20 ng/ml cytarabine. DNA samples were obtained at the initial plating and after 15 days in culture.
(G) Average gene expression changes for each chromosome in trisomic iPSCs. Genes with >2-fold induction and statistical significance (p < 0.05) are listed.
p values in (C)–(E) and (G) were determined by paired t tests. Data in (B and G) are represented as mean ± SEM.
See also Figure S2.
The impaired proliferation of trisomic cells has been attributed to gene dosage effects and compensatory changes in protein degradation and energy consumption that result in increased sensitivity to compounds affecting these processes (Tang et al., 2011). We performed mixed culture proliferation experiments in the presence of three of these compounds (17-allylamino-17-demethoxygeldanamycin [17-AAG], 5-aminoimidazole-4-carboxamide riboside [AICAR], and chloroquine), but even at the highest tolerated doses, none of these drugs selectively inhibited the proliferation of trisomy 21 iPSCs (Figure 2F). Thus, the impaired proliferation of trisomic DS iPSCs is not due to the same alterations in protein degradation and energy consumption observed in trisomic mouse fibroblasts (Tang et al., 2011). We also measured proliferation in the presence of cytarabine (Figure 2F) because DS leukemia cells are particularly sensitive to this drug (Zwaan et al., 2002); however, no differences were observed.
We compared the global gene expression profiles of two parental trisomic iPSCs, four derived disomic iPSCs, and human embryonic stem cells (H1 ESCs). Pearson correlation analysis showed that both trisomic and disomic iPSCs clustered closely together (coefficients >0.98), similar to replicate samples of H1 ESCs. A direct comparison of isogenic trisomic and disomic pairs showed the expected upregulation of chromosome 21 genes with trisomy, and remarkably little additional variation. Only three other genes were >2-fold dysregulated in trisomic cells: ACTA2, PTRF, and RPL39L were overexpressed (Figure 2G). It is tempting to speculate that altered expression of ACTA2 (Alpha 2 actin) may play a role in DS cardiovascular malformations because ACTA2 mutations are known to cause vascular abnormalities (Guo et al., 2007).
Trisomy 21 has been proposed to influence hematopoietic and endothelial differentiation (Baek et al., 2009; Chou et al., 2008; Reynolds et al., 2010). We analyzed cells from day 14 embryoid bodies (EBs) for the presence of hematopoietic and endothelial surface markers and progenitor cells, but did not detect consistent differences between two isogenic pairs of trisomic and disomic iPSCs (Figures S2A–S2C), arguing that this in vitro model poorly recapitulates the known effects of trisomy 21 on hematopoiesis during human development (Fong and Brodeur, 1987). In vivo, all four clones formed trilineage teratomas after implantation into immunodeficient mice (example in Figure S2D), and the trisomic lines produced fewer endothelia within these teratomas (Figure S2E). Impaired endothelia formation is consistent with prior observations of DS cells (Baek et al., 2009; Reynolds et al., 2010), suggesting that in vivo modeling may be more accurate.
In summary, we have generated DS iPSCs and removed the extra copy of chromosome 21 by a combination of gene targeting and negative selection. Our study provides a measurement of spontaneous mutation frequencies in human pluripotent stem cells, with chromosome loss being the most common cause of GCV resistance. Previously, an alternative strategy for chromosome loss was described in mouse cells that consisted of Cre-mediated recombination at engineered, inverted loxP sites (Lewandoski and Martin, 1997). The advantages of our approach are that recombinase protein is not required, abnormal chromosomal forms are not produced, and the frequency of spontaneous chromosome loss can be measured.
Trisomy removal has practical applications. DS patients have a 10- to 20-fold increased risk of leukemia (Fong and Brodeur, 1987), so trisomy removal might eventually be used to treat DS leukemia by autologous transplantation of patient-derived, disomic cells that lack the proleukemic effects of the extra copy of chromosome 21. Our approach could also be used to eliminate the unwanted trisomies that frequently arise during stem cell culture (Draper et al., 2004; Mayshar et al., 2010), thereby allowing one to revert a valuable, trisomic clone to a normal karyotype.
Supplementary Material
Acknowledgments
This work was supported by a grant to D.R. from Horizon Discovery and NIH grants DK55759, HL53750, and GM086497 to D.R.; DK077864 to K.C.; and HL46557 to T.P. The authors thank Yi Jiang, Gaoying Ren, Raisa Stolitenko, Andrew Herman, and Sam Lam for technical assistance.
Footnotes
Supplemental Information: Supplemental Information for this article includes two figures, two tables, and Supplemental Experimental Procedures and can be found with this article online at http://dx.doi.org/10.1016/j.stem.2012.08.004.
The authors declare no competing financial interests.
References
- Baek KH, Zaslavsky A, Lynch RC, Britt C, Okada Y, Siarey RJ, Lensch MW, Park IH, Yoon SS, Minami T, et al. Down's syndrome suppression of tumour growth and the role of the calcineurin inhibitor DSCR1. Nature. 2009;459:1126–1130. doi: 10.1038/nature08062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker DE, Harrison NJ, Maltby E, Smith K, Moore HD, Shaw PJ, Heath PR, Holden H, Andrews PW. Adaptation to culture of human embryonic stem cells and oncogenesis in vivo. Nat Biotechnol. 2007;25:207–215. doi: 10.1038/nbt1285. [DOI] [PubMed] [Google Scholar]
- Chamberlain JR, Schwarze U, Wang PR, Hirata RK, Hankenson KD, Pace JM, Underwood RA, Song KM, Sussman M, Byers PH, Russell DW. Gene targeting in stem cells from individuals with osteogenesis imperfecta. Science. 2004;303:1198–1201. doi: 10.1126/science.1088757. [DOI] [PubMed] [Google Scholar]
- Chan MF, van Amerongen R, Nijjar T, Cuppen E, Jones PA, Laird PW. Reduced rates of gene loss, gene silencing, and gene mutation in Dnmt1-deficient embryonic stem cells. Mol Cell Biol. 2001;21:7587–7600. doi: 10.1128/MCB.21.22.7587-7600.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou ST, Opalinska JB, Yao Y, Fernandes MA, Kalota A, Brooks JS, Choi JK, Gewirtz AM, Danet-Desnoyers GA, Nemiroff RL, Weiss MJ. Trisomy 21 enhances human fetal erythro-megakaryocytic development. Blood. 2008;112:4503–4506. doi: 10.1182/blood-2008-05-157859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa V, Sommese L, Casamassimi A, Colicchio R, Angelini C, Marchesano V, Milone L, Farzati B, Giovane A, Fiorito C, et al. Impairment of circulating endothelial progenitors in Down syndrome. BMC Med Genomics. 2010;3:40. doi: 10.1186/1755-8794-3-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crossen PE, Morgan WF. Lymphocyte proliferation in Down's syndrome measured by sister chromatid differential staining. Hum Genet. 1980;53:311–313. doi: 10.1007/BF00287048. [DOI] [PubMed] [Google Scholar]
- Draper JS, Smith K, Gokhale P, Moore HD, Maltby E, Johnson J, Meisner L, Zwaka TP, Thomson JA, Andrews PW. Recurrent gain of chromosomes 17q and 12 in cultured human embryonic stem cells. Nat Biotechnol. 2004;22:53–54. doi: 10.1038/nbt922. [DOI] [PubMed] [Google Scholar]
- Fong CT, Brodeur GM. Down's syndrome and leukemia: epidemiology, genetics, cytogenetics and mechanisms of leukemogenesis. Cancer Genet Cytogenet. 1987;28:55–76. doi: 10.1016/0165-4608(87)90354-2. [DOI] [PubMed] [Google Scholar]
- Guo DC, Pannu H, Tran-Fadulu V, Papke CL, Yu RK, Avidan N, Bourgeois S, Estrera AL, Safi HJ, Sparks E, et al. Mutations in smooth muscle alpha-actin (ACTA2) lead to thoracic aortic aneurysms and dissections. Nat Genet. 2007;39:1488–1493. doi: 10.1038/ng.2007.6. [DOI] [PubMed] [Google Scholar]
- Gupta PK, Sahota A, Boyadjiev SA, Bye S, Shao C, O'Neill JP, Hunter TC, Albertini RJ, Stambrook PJ, Tischfield JA. High frequency in vivo loss of heterozygosity is primarily a consequence of mitotic recombination. Cancer Res. 1997;57:1188–1193. [PubMed] [Google Scholar]
- Hasle H, Clemmensen IH, Mikkelsen M. Risks of leukaemia and solid tumours in individuals with Down's syndrome. Lancet. 2000;355:165–169. doi: 10.1016/S0140-6736(99)05264-2. [DOI] [PubMed] [Google Scholar]
- Hassold T, Chen N, Funkhouser J, Jooss T, Manuel B, Matsuura J, Matsuyama A, Wilson C, Yamane JA, Jacobs PA. A cytogenetic study of 1000 spontaneous abortions. Ann Hum Genet. 1980;44:151–178. doi: 10.1111/j.1469-1809.1980.tb00955.x. [DOI] [PubMed] [Google Scholar]
- Jenkins EC, Schupf N, Genovese M, Ye LL, Kapell D, Canto B, Harris M, Devenny D, Lee JH, Brown WT. Increased low-level chromosome 21 mosaicism in older individuals with Down syndrome. Am J Med Genet. 1997;68:147–151. doi: 10.1002/(sici)1096-8628(19970120)68:2<147::aid-ajmg5>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- Khan IF, Hirata RK, Wang PR, Li Y, Kho J, Nelson A, Huo Y, Zavaljevski M, Ware C, Russell DW. Engineering of human pluripotent stem cells by AAV-mediated gene targeting. Mol Ther. 2010;18:1192–1199. doi: 10.1038/mt.2010.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefebvre L, Dionne N, Karaskova J, Squire JA, Nagy A. Selection for transgene homozygosity in embryonic stem cells results in extensive loss of heterozygosity. Nat Genet. 2001;27:257–258. doi: 10.1038/85808. [DOI] [PubMed] [Google Scholar]
- Lewandoski M, Martin GR. Cre-mediated chromosome loss in mice. Nat Genet. 1997;17:223–225. doi: 10.1038/ng1097-223. [DOI] [PubMed] [Google Scholar]
- Mayshar Y, Ben-David U, Lavon N, Biancotti JC, Yakir B, Clark AT, Plath K, Lowry WE, Benvenisty N. Identification and classification of chromosomal aberrations in human induced pluripotent stem cells. Cell Stem Cell. 2010;7:521–531. doi: 10.1016/j.stem.2010.07.017. [DOI] [PubMed] [Google Scholar]
- Reynolds LE, Watson AR, Baker M, Jones TA, D'Amico G, Robinson SD, Joffre C, Garrido-Urbani S, Rodriguez-Manzaneque JC, MartinoEcharri E, et al. Tumour angiogenesis is reduced in the Tc1 mouse model of Down's syndrome. Nature. 2010;465:813–817. doi: 10.1038/nature09106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal DJ, McCoy EE. Studies on Down's syndrome in tissue culture I Growth rates and protein contents of fibroblast cultures. J Cell Physiol. 1974;83:85–90. doi: 10.1002/jcp.1040830112. [DOI] [PubMed] [Google Scholar]
- Sikora E, Radziszewska E, Kmiec T, Maslinska D. The impaired transcription factor AP-1 DNA binding activity in lymphocytes derived from subjects with some symptoms of premature aging. Acta Biochim Pol. 1993;40:269–272. [PubMed] [Google Scholar]
- Tang YC, Williams BR, Siegel JJ, Amon A. Identification of aneuploidy-selective antiproliferation compounds. Cell. 2011;144:499–512. doi: 10.1016/j.cell.2011.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres EM, Dephoure N, Panneerselvam A, Tucker CM, Whittaker CA, Gygi SP, Dunham MJ, Amon A. Identification of aneuploidy-tolerating mutations. Cell. 2010;143:71–83. doi: 10.1016/j.cell.2010.08.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams BR, Prabhu VR, Hunter KE, Glazier CM, Whittaker CA, Housman DE, Amon A. Aneuploidy affects proliferation and spontaneous immortalization in mammalian cells. Science. 2008;322:703–709. doi: 10.1126/science.1160058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J, Vodyanik MA, Smuga-Otto K, Antosiewicz-Bourget J, Frane JL, Tian S, Nie J, Jonsdottir GA, Ruotti V, Stewart R, et al. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007;318:1917–1920. doi: 10.1126/science.1151526. [DOI] [PubMed] [Google Scholar]
- Zwaan CM, Kaspers GJ, Pieters R, Hahlen K, Janka-Schaub GE, van Zantwijk CH, Huismans DR, de Vries E, Rots MG, Peters GJ, et al. Different drug sensitivity profiles of acute myeloid and lymphoblastic leukemia and normal peripheral blood mononuclear cells in children with and without Down syndrome. Blood. 2002;99:245–251. doi: 10.1182/blood.v99.1.245. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.