

Abstract
The Hippo tumor suppressor pathway restricts tissue growth by inactivating the transcriptional coactivator Yki. Although Sd has been implicated as a DNA-binding transcription factor partner for Yki and can genetically account for gain-of-function Yki phenotypes, how Yki regulates normal tissue growth remains a long-standing puzzle since Sd, unlike Yki, is dispensable for normal growth in most Drosophila tissues. Here we show that the yki mutant phenotypes in multiple developmental contexts are rescued by inactivation of Sd, suggesting that Sd functions as a default repressor and that Yki promotes normal tissue growth by relieving Sd-mediated default repression. We further identify Tgi as a cofactor involved in Sd's default repressor function and demonstrate that the mammalian orthologue of Tgi potently suppresses the YAP oncoprotein in transgenic mice. These findings fill a major gap in Hippo-mediated transcriptional regulation and open up new possibilities for modulating the YAP oncoprotein in cancer and regenerative medicine.
Introduction
The Hippo signaling pathway is a conserved regulator of organ size in Drosophila and mammals, and its dysregulation contributes to human cancers (Halder and Johnson, 2011; Harvey and Tapon, 2007; Pan, 2010; Zhao et al., 2010). This pathway functions through a kinase cascade involving Hippo (Hpo) and Warts (Wts) that ultimately phosphorylates and inactivates the transcriptional coactivator Yorkie (Yki; YAP/TAZ in mammals) (Huang et al., 2005). Understanding the mechanisms by which Yki regulates tissue growth and target gene expression has important implications for developmental and cancer biology.
As a transcriptional coactivator, Yki's ability to regulate target gene transcription depends on its interactions with DNA-binding transcription factors. The best characterized DNA-binding partner of Yki is Scalloped (Sd) (Goulev et al., 2008; Wu et al., 2008; Zhang et al., 2008; Zhao et al., 2008), a TEAD/TEF family transcription factor expressed in multiple imaginal discs (Campbell et al., 1992). Sd was identified as a Yki partner in multiple unbiased protein-protein interaction screens (Giot et al., 2003; Wu et al., 2008) and through its ability to bind a minimal Hippo Responsive Element (HRE) in the Hippo target gene diap1 (Wu et al., 2008). Furthermore, an unbiased genetic screen identified a point mutation in Yki that specifically disrupts Yki-Sd binding (Wu et al., 2008). Consistent with these molecular interactions, loss of sd fully rescues tissue overgrowth and elevated target gene transcription induced by excessive yki activity (Wu et al., 2008). A puzzling and unexpected finding from these studies is that while sd is required for all phenotypes induced by excessive yki activity, sd, but not yki, is dispensable for the normal growth and expression of Hippo target gene expression in most imaginal discs (Goulev et al., 2008; Huang et al., 2005; Wu et al., 2008; Zhang et al., 2008). In fact, despite that yki is required for the growth of all imaginal discs, the only place where sd is essential among all imaginal discs is the wing pouch, a region that gives rise to the adult wing blade (Campbell et al., 1992). In this context, Sd is known to function as a DNA-binding partner for Vestigial (Vg), a Tondu-domain containing transcriptional coactivator that is specifically expressed in the wing pouch (Halder et al., 1998; Simmonds et al., 1998). Here, the Sd-Vg complex functions as master switch for specifying the wing identity in the pouch region (Halder et al., 1998; Simmonds et al., 1998). Previous studies established that the Tondu domain of Vg mediates its binding to Sd (Simmonds et al., 1998; Vaudin et al., 1999). Conversely, the Vg- and Yki-binding region of Sd was both mapped to the C-terminal half of Sd (Simmonds et al., 1998; Wu et al., 2008), suggesting that Sd interacts with Vg and Yki in a mutually exclusive manner. Indeed, in the wing pouch, the Sd-Vg and the Sd-Yki complexes can regulate distinct target genes independent of each other, consistent with the notion that engagement of different coactivators influences the target specificity of a common DNA-binding transcription factor (Halder and Carroll, 2001; Wu et al., 2008).
The essentiality of yki and the apparent genetic dispensability of sd in most Drosophila tissues have been a long-standing puzzle in Hippo-mediated transcriptional regulation. The contrasting mutant phenotypes of sd and yki have led to the prevailing view that even though sd can fully account for gain-of-function yki phenotypes, Yki must engage other/additional DNA-binding partners in normal tissue growth. Indeed, Yki has been reported to interact with other DNA-binding transcription factors, such as the TALE-homeodomain protein Homothorax (Hth) (Peng et al., 2009) and the BMP/TGF-β signaling effector Smad (Oh and Irvine, 2011), although neither protein, like Sd, has been shown to genetically account for the yki mutant phenotype. An alternative model to account for the differing mutant phenotypes of sd and yki is to suppose that Sd functions by default as a transcriptional repressor in the absence of Yki and that Yki regulates normal tissue growth by antagonizing Sd's repressor function in a “relief-of-repression” manner. According to this model, the severe undergrowth of yki mutant clones is due to not simply loss of Yki-mediated transcriptional activation, but rather active repression of target gene transcription exerted by Sd the absence of Yki. In contrast, sd mutant clones may show a milder or even no growth defects compared to yki mutants since loss of Sd should result in de-repression of Hippo target genes. A critical genetic test to distinguish between these models is to analyze sd; yki double mutant clones. The default repression model predicts explicitly that the yki mutant phenotypes should be rescued, at least partially, by simultaneous inactivation of sd. In contrast, the model invoking multiple Yki partners predicts that sd; yki double mutant clones should show similar mutant phenotype as yki mutant clones. To date, such double mutant analysis has not been reported.
Here we show that, despite the lack of loss-of-function phenotypes for sd in most tissues, loss of sd rescues yki mutant phenotypes in multiple developmental contexts. This striking genetic epistasis uncovers a heretofore unrecognized function of Sd in transcriptional repression and demonstrates that the primary function of Yki in normal tissue growth is to relieve Sd-mediated transcriptional repression. This default repression model is further supported by the identification of Tgi, a Tondu-domain containing protein, as an Sd-binding cofactor that is required for Sd's default repression function. Importantly, the mammalian orthologue of Tgi potently suppresses the YAP oncoprotein in vivo, underscoring the generality of the mechanism uncovered in Drosophila.
Results
Sd functions as a default transcriptional repressor in the eye
To test whether loss of sd can rescue yki mutant phenotypes, we generated sd; yki double mutant clones in the developing eye imaginal disc, where sd, but not yki, is dispensable for growth. Since sd and yki are on different chromosomes, we used double FRT chromosomes carrying the respective mutations in trans to double FRT chromosomes carrying GFP and RFP markers together with an eye specific FLP source. Thus, sd; yki double mutant clones were generated in flies of the following genotype: eyFLP FRT19 GFP/FRT19 sd; FRT42 RFP/FRT42 ykiB5, where sd yki double mutant clones can be scored as negative for both GFP and RFP. We then quantified the total area of the double mutant tissue as a fraction of the whole eye disc as an indication of the mutant tissue's growth rate. As controls, we employed a similar double FRT strategy to generate and to quantify the area of sd or yki single mutant clones, using flies containing a mutation-carrying FRT chromosome and a wildtype FRT chromosome in trans to double FRT GFP RFP chromosomes -- eyFLP FRT19 GFP/FRT19; FRT42 RFP/FRT42 ykiB5 for yki mutant clones and eyFLP FRT19 GFP/FRT19 sd; FRT42 RFP/FRT42 for sd mutant clones. All quantification was done blindly without knowing the genotype of the discs being analyzed.
As shown previously, yki single (19; ykiB5) mutant clones grew poorly (Figure 1A), comprising only 0.5±0.1% of the eye discs (Figure 1F), and showed reduced Diap1 expression. As also shown previously, sd single (sd47M; 42) mutant clones (Figure 1B) grew normally, comprising 16.6±4.8% of the eye discs and showed no changes in Diap1 expression. Strikingly, sd47M; yki clones (Figures 1C) grew significantly more than yki mutant clones (p=1.8E-6), comprising 15.2 ± 2.7% of the eye disc, and showed normal Diap1 expression. In fact, there was no statistical difference between the fraction of sd47M; 42 and sd47M; yki mutant tissues relative to the whole eye discs (p=0.18; Figure 1F). To ensure that this rescue was not due to non-specific second hits on the sd47M chromosome, we repeated the analysis using sddel, a newly generated sd deletion allele (see Experimental Procedures). The sddel allele behaved similarly as sd47M in rescuing the growth defect and reduced Diap1 expression of yki mutant (Figures 1D–E and 1F).
Figure 1. Yki promotes normal growth by relieving Sd-mediated default repression in the eye.

(A–E) Eye discs containing mutant clones of the indicated genotypes were stained with α-Diap1. Note the small size and decreased Diap1 expression of 19; yki clones (A), but not sd; 42 (B and D) or sd; yki clones (C and E) (compare arrowheads).
(F) Quantification of the relative percentage of the mutant tissues in the eye (mean ± SEM). At least 12 eye discs were blindly scored for each genotype. Note the comparable areas occupied by sd; 42 and sd; yki mutant tissues.
The complete rescue of yki mutant clones by simultaneous loss of sd demonstrates that the loss-of-function phenotypes of yki, including poor growth and reduced Hippo target gene expression, result from active repression of target genes exerted by Sd in the absence of Yki. Thus, besides the well-established role for Sd in transcriptional activation driven by hyperactive Yki, the genetic epistasis between sd and yki uncovers a previously unrecognized role for Sd in transcriptional repression. Furthermore, the directionality of this epistasis demonstrates that repression, rather than activation, represents the default activity of Sd. Our default repression model makes explicit predictions about the existence of co-repressor(s) required for Sd's default repression function. The identification and characterization of an Sd-binding protein called Tondu-domain-containing Growth Inhibitor (Tgi), as detailed below, provide further molecular and genetic support for this overarching model.
Identification of Tgi as a component of the Hippo pathway
We identified Tgi, corresponding to CG10741, in two unbiased screens for potential components of the Hippo pathway (Figures S1, S2 and Table S1). First, Tgi was identified in a gain-of-function screen for genes whose overexpression results in decreased eye and wing size (Figure S1C–D). Quantification of wing hair density shows that Tgi-induced undergrowth results from a reduction in cell number, not cell size. In the second screen, Tgi was identified in a cell-based RNAi screen for genes whose RNAi knockdown leads to increased expression of a luciferase reporter driven by the minimal Sd-binding Hippo Responsive Element (HRE) derived from the Hippo target gene diap1 (Wu et al., 2008) (Figure S1E–F). RT-PCR and RNA in situ hybridization revealed widespread Tgi expression in whole adult flies, ovaries, wing discs, and eye discs and ubiquitous expression in both wing and eye discs (Figure S2A–B). Tgi contains two molecular features with potential significance in Hippo signaling (Figure S1B). First, it has two Tondu domains (T1 and T2), a known Sd-binding domain first described in Vg (Simmonds et al., 1998; Vaudin et al., 1999). Second, Tgi contains three PPxY motifs (P1, P2 and P3), a well-established ligand motif for WW domains, which are prevalently present in Hippo pathway components such as Yki, Sav and Kibra. The FlyBase annotates two possible transcripts for Tgi (RA and RB) (Figure S1A) that share these molecular features, and overexpression of either transcript produced similar tissue undergrowth (Figure S1D). We therefore used Tgi-RB for follow-up analysis.
To understand the molecular basis of Tgi-induced growth suppression, we analyzed Hippo target genes diap1 and ex. Flp-out clones (Basler and Struhl, 1994) with Tgi overexpression showed cell-autonomous decrease of diap1 and ex transcription (Figure 2A–B). Non-Hippo targets, such as Eya and Arm, and two well-characterized Vg targets, Vg and Distalless (Dll), were unaffected (Figures 2C–D and S2C–D). Unlike Tgi, overexpression of Vg, the only other Tondu domain-containing protein in Drosophila, did not result in reduced diap1 expression (Figure S2E–F). These results suggest that Tgi suppresses tissue growth by interfering with Hippo target transcription.
Figure 2. Tgi specifically represses Hippo target genes and functions epistatically to Wts.
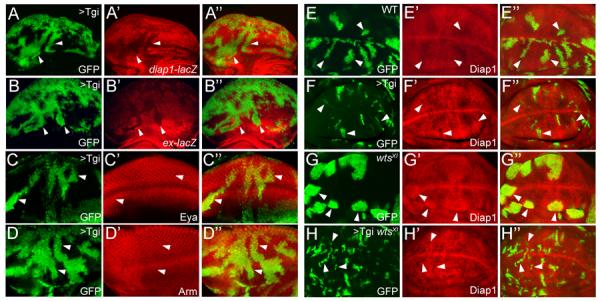
(A–D) Eye discs containing GFP-marked Tgi Flp-out clones (arrowheads), showing decreased diap1-lacZ (A) or ex-lacZ (B), and normal expression of Eya (C) or Arm (D).
(E–H) Wing discs containing GFP-marked MARCM clones of control (E), Tgi overexpression (F), wts mutant (G), and wts mutant with Tgi overexpression (H), and stained for Diap1 protein.
See Figure S1, S2 and Table S1 for data supplemental to Figure 2.
To position Tgi relative to the canonical Hippo kinase cascade, we used the MARCM technique (Lee and Luo, 1999) to generate wts mutant clones that simultaneously overexpressed Tgi. MARCM clones with Tgi overexpression were smaller than control clones and showed decreased Diap1 expression (Figure 2E–F). As expected, wts mutant clones were overgrown with elevated Diap1 levels (Figure 2G). Interestingly, wts mutant clones with Tgi overexpression phenocopied Tgi-overexpressing clones. Both were smaller than control clones and showed decreased Diap1 expression (Figure 2H), thus placing Tgi genetically downstream of Wts. A functional link between Tgi and Hippo signaling is also supported by extensive genetic interactions with Yki and Sd (Figure S2G–I).
Tgi-induced growth suppression requires the Tondu domains and is less dependent on the PPxY motifs
The primary sequence of Tgi suggests that it may physically interact with Sd (via the Tondu domains) and/or WW-domain-containing pathway components (via the PPxY motifs), among which Yki is a prime candidate given our genetic analysis placing Tgi downstream of Wts. Indeed, Tgi and Sd co-immunoprecipitated (co-IP) with each other in Drosophila S2R+ cells (Figure 3A). Tgi also interacted with Yki in co-IP assays (Figure 3B). Structure-function analysis revealed that the Tgi-Sd interaction requires the Tondu domains, as mutation of either Tondu domain (T1 or T2) attenuated and mutation of both Tondu domains (T12) abolished Tgi-Sd interaction (Figure 3A). Analysis of PPxY motif mutations identified P1 as the sole PPxY motif that mediates Tgi-Yki association (Figure 3B).
Figure 3. Tgi's growth suppressive function requires Sd.
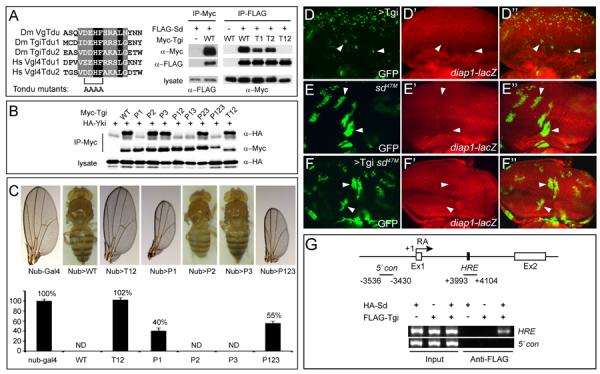
(A) Alignment of the Tondu domains from Drosophila Vg and Tgi and human Vgl4 (left), and co-IP between Tgi and Sd (right). Tondu domain mutation was made by changing the conserved D/ED/EHF sequence at the core of the domain to AAAA. For co-IP, the indicated epitope-tagged constructs were expressed in S2R+ cells and subjected to IP as indicated.
(B) Co-IP between Yki and wildtype Tgi or Tgi mutants with single, double or triple PPxY mutations. PPxY motif was mutated by changing Tyr to Ala. Note that mutation of P1, but not P2 or P3, abolished Tgi-Yki interactions, and all mutants containing P1 (P1, P12, P13 and P123) showed a similar abolishment of Tgi-Yki interactions. Also note that mutation of both Tondu domains (T12) did not affect Tgi-Yki binding.
(C) Wildtype or Tgi mutants were expressed by nub-Gal4. Representative adult wings (if wings were present) or whole flies (if no wings were present) are shown. The graph shows quantification of wing size relative to nub-Gal4/+ control (mean ± SEM). ND: not determined due to a no-wing phenotype.
(D–F) Eye discs containing GFP-marked MARCM clones of Tgi overexpression (D), sd mutant (E) or sd mutant with Tgi overexpression (F), and stained for diap1-lacZ.
(G) Tgi binds to the HRE of diap1 in an Sd-dependent manner. At the top is a schematic representation of the diap1 locus, showing exon 1 (Ex1) and exon 2 (Ex2) of the RA transcript, as well as the previously defined HRE (Wu et al., 2008). Chromatins were precipitated from lysates of S2R+ cells expressing the indicated combination of HA-Sd and FLAG-Tgi plasmids using anti-FLAG antibody. A ~100bp DNA (+3993–+4101) encompassing the HRE was amplified following ChIP. The region −3536 to −3430 (marked as 5'con) was used as a negative control region for PCR. The HRE amplicon was enriched only in the presence of both FLAG-Tgi and HA-Sd.
See Figure S3 for data supplemental to Figure 3.
To assess the functional relevance of Tondu-mediated Tgi-Sd interaction and PPxY-mediated Tgi-Yki interaction detected by co-IP assays, we created UAS transgenes expressing HA-tagged wildtype Tgi or mutant Tgi lacking Tondu or PPxY motifs using phiC31-mediated site-specific transformation (Bischof et al., 2007). Overexpression of wildtype Tgi using the wing specific driver nubbin-Gal4 resulted in a no-wing phenotype, whereas overexpression of TgiT12 (Sd-binding defective) resulted in a completely normal wing size (Figure 3C). In contrast, overexpression of TgiP1 (Yki-binding defective), or a triple mutant (TgiP123) lacking all three PPxY motifs, still resulted in significant growth inhibition (Figure 3C). Consistent with these findings, the T12 mutation completely eliminated Tgi's effect on Diap1 expression, while the P123 mutant still resulted in visible decrease in Diap1 expression (Figure S3A–D). Thus, while both interactions can be detected by co-IP and may potentially contribute to Tgi-induced growth suppression, the Tgi-Sd interaction clearly plays a more critical role than the Tgi-Yki interaction. As shown later in this paper, the lesser role for Tgi's PPxY motifs in Drosophila is also consistent with the absence of PPxY motifs in vertebrate homologues of Tgi.
Tgi is required for Sd-mediated default repression in the eye imaginal disc
Tgi-induced transcriptional repression, coupled with its nuclear localization (Figure S3E) and physical interactions with Sd, suggest that Tgi may function as a co-repressor that is required for Sd's default repressor function. This model makes several testable predictions. First, Tgi-overexpression phenotypes should depend on Sd function. We tested this prediction by examining sd mutant clones with Tgi-overexpression. Unlike Tgi overexpression clones, which showed decreased expression of Diap1 and greatly reduced clone size, sd mutant clones with Tgi overexpression showed normal Diap1 expression and clone size (Figure 3D–F). Consistent with these genetic findings, chromatin immunoprecipitation (ChIP) revealed that Tgi interacts with the diap1 HRE in an Sd-dependent manner (Figure 3G). Furthermore, Tgi inhibited Yki-mediated transcriptional activation of the Sd-binding diap1 HRE element as well as binding of Yki to the HRE element (Figure S3F).
Second, loss of Tgi, like loss of Sd, should rescue yki mutant eye phenotypes. We investigated this possibility by generating a null allele of tgi (tgiΔP) (Figure S1A). tgiΔP homozygotes die at 2nd to 3rd instar larval transition. tgiΔP mutant clones, like sd mutant clones, showed no detectable changes in clonal growth, expression of the Hippo targets or subcellular localization of Yki and Sd in the eye imaginal disc (Figure S4). Next, we investigated genetic epistasis between tgi and yki using a double FRT strategy similar to that used in the analysis of sd; yki double mutant, except that a different FRT combination (GFP marked FRT80 and RFP marked FRT42) was employed. If Tgi is involved in Sd-mediated default repression, one should also observe suppression of the yki mutant phenotype in yki; tgi double mutants. As expected, yki single (yki; 80) mutant clones (Figure 4A) grew poorly, comprising only 1.4 ± 0.4% of the eye discs, and showed reduced Diap1 expression. tgi single (42; tgi) mutant clones (Figure 4B) comprised 15 ± 3.6% of the eye disc and showed no changes in Diap1 expression. Importantly, yki; tgi double mutant clones (Figure 4C) grew significantly better than yki single (yki; 80) mutant clones (p=0.00011; Figure 4D), comprising 6.9± 1.4% of the eye discs. We note, however, the rescue of yki mutant clones by loss of tgi was incomplete, since yki; tgi double mutant tissues still grew less than tgi single (42; tgi) mutant tissues and still showed reduced Diap1 expression (Figure 4A–D). These data suggest that Tgi plays a partial role in Sd-mediated default repression in the eye disc and that additional Sd corepressors are likely required in this context. The fact that Tgi can be functional in the complete absence of Yki, as indicated by their genetic epistasis relationship, is consistent with our structure-function analysis showing a lesser role for the PPxY motifs in Tgi-induced growth suppression and target gene repression.
Figure 4. Tgi is required for default repression but not for Yki-mediated activation.
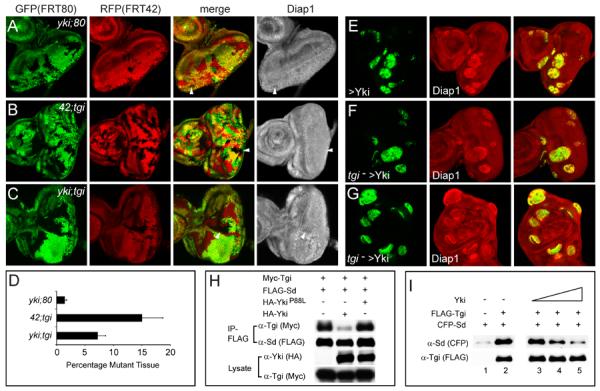
(A–D) Loss of tgi partially rescues yki loss-of-function phenotypes. Eye discs containing mutant clones of the indicated genotypes were stained with α-Diap1 (A–C). The areas occupied by the mutant tissues were quantified in (D) (mean ± SEM). 42D ykiB5; 80B tgiΔP mutant clones were significantly larger than 42D ykiB5; 80B+ mutant clones, but still statistically smaller than 42D+; 80B tgiΔP mutant clones (p= 0.00016), and still showed decreased Diap1 expression (arrowheads).
(E–G) Tgi is not required for Yki-mediated activation. Eye discs (E–F) and wing disc (G) containing MARCM clones (GFP-positive) of the indicated genotypes were stained for Diap1. Yki-overexpressing clones with or without tgi mutation showed similar round clone shape and elevated Diap1 levels.
(H) Yki inhibits Sd-Tgi binding in cultured cells. The indicated plasmids were transfected into S2R+ cells and subjected to immunoprecipitation by anti-FLAG. HA-Yki, but not HA-YkiP88L (Wu et al., 2008) inhibited Sd-Tgi interactions.
(I) Yki inhibits Sd-Tgi binding in vitro. Lanes 1–2: FLAG-Tgi expressed in S2R+ cells was immunoprecipitated with anti-FLAG conjugated beads. Beads with or without FLAG-Tgi were incubated with bacterially purified CFP-Sd protein, and the amount of CFP-Sd bound by the beads were probed with α-CFP antibody. CFP-Sd was recovered only by beads with FLAG-Tgi. Lanes 3–5: similar to lane 2, except that increasing concentration of bacterially purified Yki (0x, 0.5x and 1x relative to CFP-Sd, lanes 3–5) was added to the incubation mixture. Increasing concentration of Yki decreased the amount of CFP-Sd bound to FLAG-Tgi conjugated beads.
See Figure S4 for data supplemental to Figure 4.
Third, if Tgi functions specifically as a co-repressor for Sd, one should be able to genetically distinguish between Tgi and Sd, since while both proteins are involved in default repression, only Sd is required for transcriptional activation. We tested this prediction by examining the genetic requirement of Tgi in Yki-induced overgrowth, which absolutely requires Sd-mediated transcriptional activation. Unlike Sd (Wu et al., 2008), loss of Tgi does not affect tissue overgrowth or elevated Hippo target transcription in Yki-overexpression clones (Figure 4E–G). These results exclude the possibility that Tgi simply functions as a generic Sd-binding scaffold that promotes Sd function in non-specific ways such as stabilizing Sd, as such model would predict Tgi to be required for both loss- and gain-of-function Yki phenotypes.
Taken together, we conclude that Sd functions by default as a transcriptional repressor by engaging Tgi and that Yki regulates normal tissue growth by relieving Sd-mediated default repression. Implicit in this model is the prediction that Yki should compete with Tgi in binding to Sd. Indeed, Yki, but not a mutant form of Yki defective in Sd-binding, greatly diminished Sd-Tgi interactions in S2R+ cells (Figure 4H). Such competition was further confirmed in vitro using purified proteins (Figure 4I). Similar competitions were observed between mammalian counterparts of Yki, Sd and Tgi (see later).
Sd functions as a default transcriptional repressor in ovarian follicle cells
The genetic epistasis presented above provides compelling evidence that Sd functions by default as a transcriptional repressor. To investigate whether default repression is unique to the eye, we turned to oogenesis, where Hippo signaling is required in the posterior follicle cells (PFCs) for a mitosis-to-endoreplication switch between stage 6 and 7 (Meignin et al., 2007; Polesello and Tapon, 2007; Yu et al., 2008). Prior to this switch, Hippo signaling is presumably off/low in the follicle cells, leaving Yki in an active state to activate a mitotic transcriptional program. After this switch, Hippo signaling becomes activated in PFCs, rendering Yki inactive and therefore turning off the mitotic transcriptional program (Figure S5A). Thus, oogenesis offers a unique tissue context in which one can readily test our model in both Yki-on (stages 1–6, mitotic) and Yki-off (stage 7–9, endoreplicative) follicle cells.
We first analyzed stage 7–9 follicle cells, corresponding to the Hippo-on/Yki-off state. A hallmark of Hippo pathway tumor suppressor mutants is the persistent expression of cut in PFCs (Meignin et al., 2007; Polesello and Tapon, 2007; Yu et al., 2008). Unlike sd or tgi clones, yki mutant clones were hard to recover in stage 7 egg chambers and could not be recovered beyond stage 7. The rare yki mutant clones recovered at stage 7 showed no cut expression like wildtype PFCs, consistent with the Yki-off state of the endoreplicative follicle cells (Figure 5A). Strikingly, in stage 7 egg chambers, we observed persistent expression of cut in PFCs mutant clones of sd (100%, n=28) or tgi (97%, n=39) (Figure 5B–C). Furthermore, unlike yki clones, sd; yki or yki; tgi double mutant clones could be readily recovered beyond stage 7, and persistent cut expression could be detected in these double mutant clones even at stage 9 (Figure 5D–E). The fact that sd or tgi mutant PFCs show similar cut expression as inactivation of upstream Hippo tumor suppressors demonstrates that in the endoreplicative Yki-off follicle cells, Sd and Tgi are required to repress cut expression. Consistent with these defects, some egg chambers containing sd or tgi mutant PFC clones showed mislocalization of Gurken (Grk) protein in the oocyte (Figure S5B–F).
Figure 5. Default repression mediated by Sd and Tgi in ovarian follicle cells.

(A–E) Loss of sd or tgi results in persistent Cut expression in stage 7–9 PFCs. Mutant clones of the indicated genotypes were marked as GFP-negative and stained for Cut. PFC clones are highlighted by white dots. (A–C) stage 7 egg chambers. Note the persistent expression of Cut in PFC clones of sd or tgi, but not yki. (D–E) stage 9 egg chambers, showing persistent Cut expression in sd; yki or yki; tgi double mutant PFC clones.
(F–J) Loss of sd or tgi rescues yki mutant phenotype in early stage follicle cells. Mosaic stage 5 egg chambers were stained for Cut. Mutant clones of the respective genotypes were marked as GFP-negative and highlighted by white dots. Note the reduced Cut expression in yki clones, but not sd, tgi, sd; yki, or yki; tgi clones.
See Figure S5 for data supplemental to Figure 5.
Next, we examined stages 1–6 follicle cells, where Yki is presumably active as in third-instar eye discs. Consistent with the Yki-on state of early follicle cells, yki mutant clones showed reduced cut expression (77%, n=39) (Figure 5F). In contrast, we observed no changes of cut expression in mutant clones of sd (93%, n=14) (Figure 5G) or tgi (100%, n=20) (Figure 5H). Thus, the requirement of yki and the dispensability of sd or tgi in early follicle cells resemble the requirement/dispensability of the respective genes in the eye. Importantly, as in the eye, inactivation of sd or tgi significantly rescued the yki mutant phonotype in early follicle cells, as 60% of sd; yki double mutant clones (n=25) (Figure 5I) and 44% of yki; tgi double mutant clones (n=18) (Figure 5J) showed normal cut expression. Taken together, we conclude that in both mitotic and endoreplicative follicle cells, Sd functions as a default repressor by engaging Tgi.
Sd and Tgi are required for ectopic apoptosis induced by hyperactive Hippo signaling in the eye and the wing
We reasoned that if loss of sd or tgi can reverse mutant phenotypes resulting from a null mutation of yki, it should also reverse phenotypes caused by hyperactive Hippo signaling, a condition that is associated with Yki phosphorylation and inactivation. We first tested this hypothesis in the eye by taking advantage of a genetic background of hyperactive Hippo signaling in which overexpression of Ex by the GMR-Gal4 driver induces a stripe of dying cells posterior to the morphogenetic furrow in third instar eye discs. Indeed, RNAi knockdown of Sd or Tgi, but not a control gene (GFP), significantly suppressed Ex-induced ectopic cell death (Figure 6A–G) without affecting Ex transgene expression (Figure S6). Thus, although neither sd nor tgi is essential in normal eye development, both genes are required for ectopic cell death induced by hyperactive Hippo signaling. These data are entirely consistent with our analysis of sd; yki and tgi; yki double mutant clones in the eye, and provide further support for the default repressor function of the Sd-Tgi complex.
Figure 6. Sd and Tgi are required for ectopic apoptosis induced by hyperactive Hippo signaling.
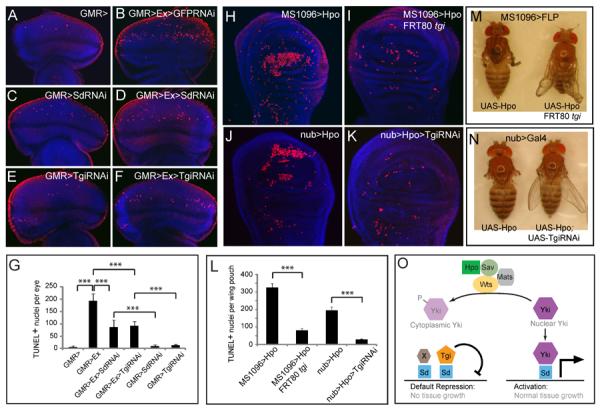
(A–G) Eye discs were stained for TUNEL (red) and DAPI (blue) (A–F), and TUNEL positive nuclei were quantified (G) (mean ± SEM, n=8). TUNEL positive nuclei in the whole eye disc were counted by projecting a Z-stack of serial confocal optical sections into one focal plane. *** denotes a p-value < 1.5E-5. The complete genotypes are: (A) UAS-Dicer2; GMR-Gal4; (B) UAS-Dicer2; GMR-Gal4 UAS-Ex; UAS-GFPRNAi; (C) UAS-Dicer2; GMR-Gal4; UAS-Sd RNAi; (D) UAS-Dicer2; GMR-Gal4 UAS-Ex; UAS-SdRNAi; (E) UAS-Dicer2; GMR-Gal4/UAS- TgiRNAi; (F) UAS-Dicer2; GMR-Gal4 UAS-Ex/UAS-TgiRNAi.
(H–L) Wing discs were stained for TUNEL and DAPI (H–K) and TUNEL positive nuclei in the wing pouch region was quantified by Z-stack projections of confocal sections (L). The complete genotypes are: (H) MS1096-Gal4 UAS-Flp; UAS-Hpo; (I) MS1096-Gal4 UAS-Flp; UAS-Hpo; FRT80BtgiΔP/FRT80 RpS17; (J) UAS-Dicer2; nub-Gal4/UAS-Hpo; (K) UAS-Dicer2; nub-Gal4/UAS-Hpo; UAS-TgiRNAi. (L) shows quantification of TUNEL-positive nuclei in the wing pouch (mean ± SEM, n=8). *** denotes a p-value < 2.5E-5.
(M) and (N): Adult flies corresponding to animals analyzed in (H–I) and (J–K), respectively. Note the extremely reduced wing size in flies expressing UAS-Hpo by MS1096-Gal4 or nub-Gal4 (animal to the left; showing tiny wing remnants), and the appreciable wing size of flies in which Hpo overexpression was combined with loss of Tgi (animal to the right).
(O) A model for how Hippo signaling switches Sd between Tgi-mediated repression and Yki-mediated activation. Additional Sd co-repressors (protein X in the diagram) likely exist since Tgi plays a partial role in Sd-mediated default repression in the eye disc. See text for details.
See Figure S6 for data supplemental to Figure 6.
Next, we tested whether the Sd-Tgi complex is required for ectopic cell death induced by hyperactive Hippo signaling in the wing disc. Since Sd is essential in the wing due to its function in the Sd-Vg complex, we cannot specifically interrogate the requirement of Sd with respect to Hippo signaling. We therefore asked whether Tgi, an Sd co-repressor specific to Hippo signaling, is required for ectopic apoptosis induced by hyperactive Hippo signaling in the wing. We used two different strategies to reduce Tgi function in the wing, one by RNAi knockdown and the other by wing-specific FLP/FRT-mediated recombination of tgiΔP coupled with a Minute mutation. In both cases, we found that Tgi is required for Hpo-induced ectopic cell death and reduction in wing size (Figure 6H–N). Thus, although Tgi is dispensable in the wing as in the eye disc, it is nonetheless required under conditions of hyperactive Hippo signaling.
Functional conservation of Vgl-4, the mammalian orthologue of Tgi
The mammalian genome encodes four Tondu domain-containing proteins, Vestigial like 1 (Vgl1) through 4. Among them, Vgl4 is the only protein with two Tondu domains and phylogenetic analysis unambiguously identifies Vgl4 as the mammalian orthologue of Tgi (Figure S7A). Unlike Tgi, Vgl4 does not contain PPxY motifs (Figure S1B). Consistent with these molecular features, Vgl4 co-immunoprecipitated with TEAD2 but not YAP in HEK293 cells (Figures 7A and S7B). Like their Drosophila counterparts, YAP potently inhibited TEAD-Vgl4 interaction, as shown by co-IP in HEK293 cells (Figure 7B) and in vitro binding using purified proteins (Figure 7C). To assess a possible role for Vgl4 in Hippo signaling, we ectopically expressed human Vgl4 in Drosophila. Like Tgi, overexpression of Vgl4 by nub-Gal4 resulted in a decrease in wing size in a Tondu domain-dependent manner (Figure 7D). Furthermore, overexpression of Vgl4 by GMR-Gal4, although having no visible effect on eye size by itself, significantly suppressed the eye overgrowth resulting from YAPS127A, an activated form of the human YAP (Figure 7E).
Figure 7. Conserved function of Vgl-4 in Hippo signaling and organ size control.

(A) Vgl4 binds to TEAD2 but not YAP. The indicated plasmids were transfected into HEK293 cells and analyzed by immunoprecipitation.
(B) YAP inhibits TEAD2-Vgl4 binding in cultured cells. The indicated plasmids were transfected into HEK293 cells and subjected to immunoprecipitation by anti-Myc. HA-YAP, but not the TEAD binding-defective HA-YAPS94A (Zhao et al., 2008), inhibited TEAD2-Vgl4 interactions.
(C) YAP inhibits in vitro binding between TEAD2 and Vgl4. Lanes 1–2: HA-Vgl4 expressed in HEK293 cells was immunoprecipitated on Protein G beads. Protein G beads with or without HA-Vgl4 were incubated with purified His-TEAD2, and the amount of His-TEAD2 bound by the beads were probed with α-His antibody. His-TEAD2 was recovered only by protein G beads with HA-Vgl4 (compare lanes 1 and 2). Lanes 3–5: similar to lane 2, except that increasing concentration of purified YAP (0×, 0.5× and 1× relative to TEAD2, lanes 3–5) was added to the incubation mixture. Increasing concentration of YAP decreased the amount of TEAD2 bound to HA-Vgl4 conjugated protein G beads.
(D) nub-Gal4 was used to express attB-UAS-Vgl4 or attB-UAS-Vgl4T12 inserted into the same chromosomal locus. The graph shows quantification of wing size relative to nub-Gal4/+ control (mean ± SEM, n=8). Vgl4-expressing wings were smaller than control (69 ± 1% vs.100 ± 1%; p=3.4E-7). Vgl4T12-expressing wings were slighted smaller than control (95 ± 1% vs.100 ± 1%; p-value: 0.004).
(E) Vgl4 suppressed eye overgrowth induced by YAP127A in Drosophila.
(F) livers from wildtype (WT), ApoE-rtTA/TRE-Vgl4 (ApoE>Vgl4), ApoE-rtTA/TRE-YAP (ApoE>YAP), ApoE-rtTA/TRE-YAP/TRE-Vgl4 (ApoE>YAP+Vgl4) mice, treated with 0.2g/L Dox for 4 weeks starting at 3 weeks of age (4W0.2, top row), or 1g/L Dox for 8 weeks starting at birth (8WB1.0, bottom row). Note the enlarged size of ApoE>YAP livers in both conditions and the widespread development of HCC (arrowheads) in 8WB1.0 condition. Both were suppressed in ApoE>YAP+Vgl4 livers.
(G) Quantification of liver-to-body weight ratio for animals analyzed in (F). Besides 4W0.2 and 8WB1.0, an additional condition in which animals were treated with 0.2g/L Dox for 2 weeks starting at 3 weeks of age (abbreviated as 2W0.2) is also included. Values are mean ± SEM, n>=3 for each data point.
(H) Following 2 weeks of 0.2 g/L Dox treatment starting at 3 weeks of age, liver lysates from the indicated genotypes were probed with an antibody specific to human YAP (top gel), an antibody reactive to both mouse and human YAP (middle gel) and an antibody against actin (bottom gel). Three mice were analyzed for each genotype.
(I) Vgl4 suppressed Nf2-deficient phenotype in the liver. Littermates of the following genotypes were treated with Dox starting from E11.5 and analyzed at birth with CK staining (arrowheads). WT: ROSA26-loxP-STOP-loxP-rtTA/+; NF2flox2/+ >Vgl4: Alb-Cre; Nf2flox2/flox2; ROSA26-loxP-STOP-loxP-rtTA; TRE-Vgl4 NF2CKO: Alb-Cre; Nf2flox2/flox2; ROSA26-loxP-STOP-loxP-rtTA/ ROSA26-loxP-STOP-loxP-rtTA NF2CKO >Vgl4: Alb-Cre; Nf2flox2/flox2; ROSA26-loxP-STOP-loxP-rtTA; TRE-Vgl4
See Figure S7 and Table S2 for data supplemental to Figure 7.
Consistent with Vgl4's ability to suppress YAP-driven overgrowth in Drosophila, in HEK293 cells, Vgl4 potently suppressed YAP-mediated transactivation of Gal4-TEAD (Figure S7C) as well as a luciferase reporter driven by the promoter of Ctgf, a direct transcription target of YAP-TEAD (Zhao et al., 2008) (Figure S7D). In both cell-based transcription assays, mutation of Vgl4's Tondu domains abolished its repressive activity (Figure S7C–D), in agreement with an absolute requirement for Tondu domains in Vgl4-TEAD interactions (Figures S7E). Taken together, these findings suggest that Vgl4 may play a conserved role in mammalian Hippo signaling.
Vgl4 potently suppresses the YAP oncoprotein in mammals
The widespread overexpression/activation of YAP in human cancers has stimulated much interest in developing therapeutic interventions against this oncoprotein (Liu-Chittenden et al., 2012). Based on our findings that Vgl4 suppresses tissue overgrowth driven by activated YAP oncoprotein in Drosophila, we reasoned that Vgl4 may be exploited as an inhibitor of YAP in mammals. To test this hypothesis, we generated TRE-Vgl4 transgenic mice that express human Vgl4 under the control of a tetracycline-responsive element (TRE). We then examined Vgl4's ability to suppress YAP oncogenic activity using two mouse models of YAP gain-of-function, achieved by transgenic YAP overexpression or inactivation of a tumor suppressor upstream of YAP.
We have previously reported a transgenic mouse model, ApoE-rtTA/TRE-YAP, which allows overexpression of human YAP in mouse livers in a Doxycycline (Dox)-dependent manner (Dong et al., 2007). Induction of YAP overexpression starting at 3 weeks of age leads to a rapid expansion of liver size, whereas induction of YAP overexpression starting at birth leads to hepatomegaly accompanied by widespread development of HCC (Dong et al., 2007). To test the efficacy of Vgl4 in blocking YAP-induced overgrowth and tumorigenesis, we generated mice containing both TRE-Vgl4 and ApoE-rtTA/TRE-YAP. In mice subjected to Dox treatment starting at 3 weeks of age, Vgl4 greatly suppressed YAP-induced liver overgrowth (Figure 7F–G), whereas in mice subjected to Dox treatment starting at birth, Vgl4 greatly suppressed YAP-induced hepatomegaly and completely abolished YAP-induced HCC formation (Figure 7F–G). These results are particularly striking given that overexpression of Vgl4 alone by ApoE-rtTA in an otherwise wildtype background did not reduce liver size (Figure 7F–G). Consistent with the phenotypic suppression, Vgl4 greatly attenuated the transcription of many genes that are known to be induced in YAP transgenic livers, such as Afp, survivin, Ctgf, Epcam and Osteopontin (Opn) (Dong et al., 2007) (Figure S7F). Importantly, Vgl4 overexpression did not reduce YAP protein levels in the double transgenic livers (Figure 7H), excluding the possibility that Vgl4 reduced YAP transgene expression or YAP protein stability.
We have previously shown that inactivation of NF2/Merlin, an upstream tumor suppressor in the Hippo pathway, results in bile duct overproliferation due to activation of endogenous YAP (Zhang et al., 2010). Therefore, the NF2/Merlin-deficient livers provide a distinct genetic context in which oncogenic YAP activity is caused by activation of endogenous YAP rather than transgene overexpression. To test whether Vgl4 can suppress NF2/Merlin-deficient liver phenotypes, we generated Alb-Cre; Nf2flox2/flox2; ROSA26-loxP-STOP-loxP-rtTA; TRE-Vgl4 mice, in which Vgl4 can be expressed in a Dox-dependent manner in Nf2-mutant livers. An important feature of this genetic strategy is that it combines Cre-mediated Nf2 inactivation with rtTA-mediated Vgl4 expression: the Alb-Cre inactivates Nf2 in the liver and meanwhile activates rtTA expression by excising a loxP-flanked transcriptional STOP signal (Belteki et al., 2005); the resulting liver-specific expression of rtTA from the ROSA26 locus, in turn, activates Vgl4 expression in a Dox-dependent manner. Since Alb-Cre drives liver-specific Cre expression starting at E13.5 (Geisler et al., 2008), we subjected timed pregnant mothers bearing such animals to Dox treatment starting at E11.5, and biliary epithelial cells (BECs) were visualized by cytokeratin (CK) staining at birth. As shown previously (Zhang et al., 2010), Nf2 mutant livers showed a massive increase in the abundance of CK-positive BECs (Figure 7I). Strikingly, this phenotype was completely suppressed by Vgl4 expression (Figure 7I). Taken together, we conclude that Vgl4 can be exploited as a potent inhibitor the YAP oncoprotein in mammals.
Discussion
Yki regulates normal tissue growth by relieving Sd-mediated default repression
The transcriptional coactivator Yki occupies a pivotal position in the Hippo signaling pathway. Loss of upstream tumor suppressors, or simply overexpression of Yki, leads to gain-of-function of Yki that drives massive tissue overgrowth in an Sd-dependent manner. Although the overgrowth phenotypes driven by hyperactive Yki underscore the normal function of the tumor suppressors acting upstream of Yki, they do not inform us about how physiological levels of Yki regulates normal tissue growth. Indeed, a long-standing conundrum in answering the latter question centers on the essentiality of yki and the apparent genetic dispensability of sd in most Drosophila tissues. Thus, how physiological levels of Yki controls normal tissue growth has remained an unresolved issue.
The differential mutant phenotype of sd and yki led to the view that additional DNA-binding transcription factors, either by themselves or in addition to Sd, may function together with Yki in normal tissue growth. In retrospect, such models were proposed with the assumption that Sd can only mediate transcriptional activation. While the supposition of additional Yki partners provides a plausible solution to the different genetic requirement of sd and yki in normal growth, it has obvious shortcomings. First, since Sd can fully account for all phenotypic consequences resulting from Yki gain-of-function (Wu et al., 2008), such models must invoke complicated mechanisms for Yki to switch between binding partners at different Yki activity levels. Second, in other transcription-based developmental pathway such as Notch and Wnt signaling, the signal-responsive transcriptional coactivator (i.e., Notch intracellular domain for Notch and Arm/β-catenin for Wnt signaling, respectively) usually engages a dedicated DNA-binding transcription factor (Su(H)/RBPjκ for Notch and Pangolin/TCF for Wnt signaling, respectively) (Barolo and Posakony, 2002). The supposition of multiple Yki partners would make the Hippo pathway an exception to this general rule.
The realization that Sd functions as a default repressor offers a simple explanation for the differential mutant phenotype of sd and yki. The complete rescue of yki growth defects in the eye by sd mutations demonstrates that the yki mutant phenotypes are largely due to Sd-mediated repression of target genes, instead of loss of Yki-mediated activation. Thus, physiological levels of Yki promote normal tissue growth not by “activating” transcription, but rather by relieving the repressor activity of Sd (Figure 6O). Conversely, the lack of mutant phenotypes for sd in the eye discs can be explained by de-repression of Hippo target genes in the absence of Sd's repressor function. This model also provides a simple solution for the seemingly bewildering observation that loss of Sd is epistatic to both loss- and gain-of-function of Yki -- in the absence of Sd, the transcriptional state of target genes should be the same (de-repressed) irrespective of Yki activity. Whether a similar default repression mechanism operates in mammals remains to be tested, although this may be technically challenging given the presence of multiple mammalian Sd and Yki homologues.
Default repression as a general mechanism in developmental signaling
Default repression has been noted as a common theme in transcription-based developmental pathways such as Hedgehog, Notch and Wnt (Affolter et al., 2008; Barolo and Posakony, 2002). Sd-mediated default repression is similar to these precedents, but in an antithetic manner, since Hippo signaling induces inactivation rather than activation of the signal-responsive coactivator Yki. Irrespective of whether a signal-responsive coactivator is activated or inactivated by signaling, these pathways all involve a transcriptional switch, whereby a pathway-specific coactivator converts the DNA-binding transcription factor from a state of transcriptional repression to activation. Another commonality is that inactivation of a pathway-specific DNA-binding transcription factor (e.g. Sd for Hippo, Ci for Hedgehog, Su(H) for Notch, TCF for Wnt) results in a quantitatively milder phenotype than genetic loss of signaling or coactivator. Interestingly, in every case, such genetic discrepancies have historically led to proposals pathway bifurcation at the level of transcription factors. However, through double mutant analyses akin to those employed in the current study, it was shown that the milder phenotypes caused by the loss of the DNA-binding transcription factor can be best accounted for by its default repressor activity (Cavallo et al., 1998; Methot and Basler, 2001; Morel and Schweisguth, 2000). Why is default repression such a pervasive mechanism in developmental signaling? It was suggested that default repression increases signaling robustness and fidelity by ensuring signal-regulated transcription while at the same time preventing spurious target gene expression in the absence of the signal-regulated coactivator (Affolter et al., 2008; Barolo and Posakony, 2002). Such precision and robustness are of utmost importance for Hippo signaling, as a cell's decision to proliferate, survive or die must be tightly controlled.
Loss of Sd or Tgi leads to gain-of-Yki phenotypes in the late ovarian follicle cells but no visible phenotypes in the eye discs. Such context dependency is not unique to the Hippo pathway. For example, the default repressor function of Ci is critical for Hedgehog signaling in imaginal discs but dispensable in embryogenesis (Methot and Basler, 1999). In Notch signaling, the corepressor Hairless is required for neurogenesis in imaginal discs but dispensable in the embryos (Schweisguth and Lecourtois, 1998). We suggest that whether loss of Sd/Tgi results in a visible gain-of-Yki phenotype depends on the status of Yki activity. In endoreplicative follicle cells, Yki is inactive due to high levels of Hippo signaling. Under this context, Sd's default repression function prevails, and accordingly, loss of Sd or Tgi leads to de-repression. In contrast, in 3rd instar eye discs or mitotic follicle cells, Yki is active and is indeed genetically indispensable. In these contexts, the Sd-Tgi repressor activity is kept largely inactive by the active Yki, which explains why loss of Sd or Tgi does not lead to phenotypic consequences. Consistent with this reasoning, an essential role for Sd and Tgi can be genetically uncovered when their mutations are combined with yki mutation, or when Yki activity is artificially lowered by hyperactive Hippo signaling.
Recent studies demonstrated that the Hippo signaling pathway was already present in unicellular ancestors of Metazoa, as illustrated by the ability of the Sd-Yki complex from unicellular organisms such as Capsaspora to induce dramatic eye overgrowth in Drosophila (Sebe-Pedros et al., 2012). Interestingly, the Sd co-repressor Tgi/Vgl4 is conserved in Capsaspora, and comparative genomic analysis reveals a striking co-existence between Tgi and Yki throughout evolution (Table S2). Thus, default repression may represent a fundamental aspect of Hippo signaling with a deep evolutionary origin.
Discovery of Tgi/Vgl4 opens new avenues for modulating the YAP oncoprotein
It is interesting to compare Tgi with Vg, the only other Tondu domain-containing protein in Drosophila. Vg contains a single Tondu domain, is expressed in a tissue-specific manner, and acts as a transcriptional coactivator for Sd. In contrast, Tgi contains two Tondu domains, is widely expressed in multiple tissues, and functions as a transcriptional corepressor for Sd. Likewise, in vertebrates, the single-Tondu-domain proteins Vgl1, Vgl2 and Vgl3 are each expressed in restricted tissues and act as transcriptional coactivators for TEAD factors (Faucheux et al., 2010; Gunther et al., 2004; Maeda et al., 2002). In contrast, Vgl4, which contains two Tondu domains, is expressed ubiquitously in all tissues (Faucheux et al., 2010) and inhibits TEAD/YAP-mediated transcription (this study). We suggest that the ubiquitous expression of Tgi reflects the widespread role of Hippo signaling in animal development, while the restricted expression of Vg follows its role as a selector gene in specific tissue context. This hypothesis is supported by the co-existence of Yki and Tgi homologues in diverse phylogenetic lineages down to single cell organisms (Table S2). In contrast, Vg homologues appeared much later in evolution, subsequent to the emergence of Ecdysozoa (Table S2). Another interesting characteristic of Tgi is that its repressor activity is specific to Hippo/Yki targets since Tgi overexpression does not affect the expression of Vg targets (Figure S2C–D), despite that Vg and Yki engage the same DNA-binding transcription factor, Sd. Conversely, Vg overexpression does not suppress Hippo targets (Figure S2E–F). What determines the target selectivity of Tgi versus Vg? What molecular features dictate the repressor function of Tgi versus the activator function of Vg? How did single-Tondu-domain and double-Tondu domain proteins evolve to regulate distinct cellular processes? These are interesting questions that warrant further investigation.
Using transgenic mice, we provide direct evidence that Vgl4 can be exploited as a potent inhibitor of the YAP oncoprotein in vivo. Since Vgl4 directly targets the transcriptional machinery at the end of the Hippo pathway, this strategy can potentially be applied in a wide spectrum of human cancers, irrespective of whether YAP is activated through gene amplification/overexpression or genetic/epigenetic silencing of upstream tumor suppressors, as illustrated in our a proof-of-concept experiment involving YAP overexpression or inactivation of Merlin/NF2 in mice. Therefore, finding ways to enhance Vgl4 function in vivo, possibly through chemicals that enhance Vgl4's repressor activity or Vgl4-based protein and gene therapies, may be beneficial in the treatment of a wide array of human cancers. Conversely, there may be situation in which attenuating Vgl4 function (and therefore enhancing YAP function) may be desirable, such as promoting tissue regeneration or stem cell expansion. Thus, our identification of Tgi/Vgl4 provides a new entry point for modulation of Hippo signaling activity in cancer and regenerative medicine.
Experimental Procedures
Over-expression screen for growth regulators
The Gene Search (GS) system (Toba et al., 1999) was used to conduct an overexpression screen for new growth regulators. Flies containing a GS P-element on the CyO balancer were crossed to flies containing an active Δ2–3 transposase. The resultant dysgenic male progeny were then crossed to w1118 females. The resulting individual white+ male progeny were then crossed to the eye-specific GMR-Gal4 flies and screened for changes in eye size. Those same white+ males were later crossed to the wing-specific Vg-Gal4, and we selected insertions that resulted in consistent changes in both eye and wing size. P-element integration sites were determined by inverse PCR (BDGP: http://www.fruitfly.org/about/methods/inverse.pcr.html). Besides Tgi, we recovered insertion lines in known regulators of organ growth such as bantam, yki, dIAP1 and imp-L2.
RNAi screen using HRE-luciferase reporter
S2R+ cells were propagated in Drosophila Schneider's Medium (GIBCO) supplemented with 10% FBS and antibiotics according to standard protocols (Wu et al., 2003). Cells were transfected with Yki and Sd expression plasmids, the HRE-luciferase reporter (containing 24 tandem copies of the 26-bp minimal HRE from the diap1 gene) and the Pol III-Renilla reporter as an internal control. 24 hours later, transfected S2R+ cells were seeded into whole genome dsRNA-containing 96-well plates (Open Biosystems). Cells were then grown for 4 days to allow for target protein depletion. After depletion, HRE-luciferase reporter gene activity was measured using the Dual-Luciferase Assay System (Promega). After data normalization to the Renilla internal control, the change in HRE-reporter activity within each well was determined and assigned a Z-score. Wells with Z-scores of either less than −3.0 or greater than +3.0 were categorized as “hits”.
Drosophila genetics
To generate a null mutation in CG10741, the P-element P[gy2]EY11498 (Bloomington Drosophila Stock Center) was mobilized using Δ2–3 transposase and progeny of that cross were screened for the loss of y+ expression. The resultant y− lines were screened for imprecise excision by PCR using one primer upstream of the fourth and fifth exons (5'-GTCGTTCTTTATGGCTTC) of cg10741 and a second primer annealing to both the intergenic region between cg10741 and cg17689 and the 5' most end of cg17689 (5'-GATACGCCGAGGATTGCTC). In wild-type flies, these primers generate a PCR product of 3929 bp. In tgiΔP flies, these primers amplify a band of approximately 1.75 kb corresponding to removal of the entirety of the fourth and fifth exons, which account for greater than 90% of the protein coding sequence. tgiΔP was then recombined with FRT80B for follow-up analysis. sddel was generated using the FRT/FLP-mediated genomic deletion strategy (Parks et al., 2004), taking advantage of two FRT-containing Exelexis lines (PBac{RB}sde00587 and PBac{RB}sde02321) flanking the sd locus. sddel deletes all the coding exons of Sd without removing any nearby genes (coordinates X:15705513 to X:15719444).
Supplementary Material
Highlights
Yki controls normal tissue growth by relieving Sd-mediated default repression
Discovery of Tgi as a cofactor involved in Sd-mediated transcriptional repression
Vgl4, the human ortholog of Tgi, is a potent inhibitor of the YAP oncoprotein in vivo
Acknowledgments
This study was supported in part by grants from the National Institutes of Health to D.P. (EY015708). D.P. is an investigator of the Howard Hughes Medical Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Affolter M, Pyrowolakis G, Weiss A, Basler K. Signal-induced repression: the exception or the rule in developmental signaling? Dev. Cell. 2008;15:11–22. doi: 10.1016/j.devcel.2008.06.006. [DOI] [PubMed] [Google Scholar]
- Barolo S, Posakony JW. Three habits of highly effective signaling pathways: principles of transcriptional control by developmental cell signaling. Genes Dev. 2002;16:1167–1181. doi: 10.1101/gad.976502. [DOI] [PubMed] [Google Scholar]
- Basler K, Struhl G. Compartment boundaries and the control of Drosophila limb pattern by hedgehog protein. Nature. 1994;368:208–214. doi: 10.1038/368208a0. [DOI] [PubMed] [Google Scholar]
- Belteki G, Haigh J, Kabacs N, Haigh K, Sison K, Costantini F, Whitsett J, Quaggin SE, Nagy A. Conditional and inducible transgene expression in mice through the combinatorial use of Cre-mediated recombination and tetracycline induction. Nucleic Acids Res. 2005;33:e51. doi: 10.1093/nar/gni051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bischof J, Maeda RK, Hediger M, Karch F, Basler K. An optimized transgenesis system for Drosophila using germ-line-specific phiC31 integrases. Proc. Natl. Acad. Sci. U. S. A. 2007;104:3312–3317. doi: 10.1073/pnas.0611511104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell S, Inamdar M, Rodrigues V, Raghavan V, Palazzolo M, Chovnick A. The scalloped gene encodes a novel, evolutionarily conserved transcription factor required for sensory organ differentiation in Drosophila. Genes Dev. 1992;6:367–379. doi: 10.1101/gad.6.3.367. [DOI] [PubMed] [Google Scholar]
- Cavallo RA, Cox RT, Moline MM, Roose J, Polevoy GA, Clevers H, Peifer M, Bejsovec A. Drosophila Tcf and Groucho interact to repress Wingless signalling activity. Nature. 1998;395:604–608. doi: 10.1038/26982. [DOI] [PubMed] [Google Scholar]
- Dong J, Feldmann G, Huang J, Wu S, Zhang N, Comerford SA, Gayyed MF, Anders RA, Maitra A, Pan D. Elucidation of a universal size-control mechanism in Drosophila and mammals. Cell. 2007;130:1120–1133. doi: 10.1016/j.cell.2007.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faucheux C, Naye F, Treguer K, Fedou S, Thiebaud P, Theze N. Vestigial like gene family expression in Xenopus: common and divergent features with other vertebrates. Int. J. Dev. Biol. 2010;54:1375–1382. doi: 10.1387/ijdb.103080cf. [DOI] [PubMed] [Google Scholar]
- Geisler F, Nagl F, Mazur PK, Lee M, Zimber-Strobl U, Strobl LJ, Radtke F, Schmid RM, Siveke JT. Liver-specific inactivation of Notch2, but not Notch1, compromises intrahepatic bile duct development in mice. Hepatology. 2008;48:607–616. doi: 10.1002/hep.22381. [DOI] [PubMed] [Google Scholar]
- Giot L, Bader JS, Brouwer C, Chaudhuri A, Kuang B, Li Y, Hao YL, Ooi CE, Godwin B, Vitols E, et al. A protein interaction map of Drosophila melanogaster. Science. 2003;302:1727–1736. doi: 10.1126/science.1090289. [DOI] [PubMed] [Google Scholar]
- Goulev Y, Fauny JD, Gonzalez-Marti B, Flagiello D, Silber J, Zider A. SCALLOPED interacts with YORKIE, the nuclear effector of the hippo tumor-suppressor pathway in Drosophila. Curr. Biol. 2008;18:435–441. doi: 10.1016/j.cub.2008.02.034. [DOI] [PubMed] [Google Scholar]
- Gunther S, Mielcarek M, Kruger M, Braun T. VITO-1 is an essential cofactor of TEF1-dependent muscle-specific gene regulation. Nucleic Acids Res. 2004;32:791–802. doi: 10.1093/nar/gkh248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halder G, Carroll SB. Binding of the Vestigial co-factor switches the DNA-target selectivity of the Scalloped selector protein. Development. 2001;128:3295–3305. doi: 10.1242/dev.128.17.3295. [DOI] [PubMed] [Google Scholar]
- Halder G, Johnson RL. Hippo signaling: growth control and beyond. Development. 2011;138:9–22. doi: 10.1242/dev.045500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halder G, Polaczyk P, Kraus ME, Hudson A, Kim J, Laughon A, Carroll S. The Vestigial and Scalloped proteins act together to directly regulate wing-specific gene expression in Drosophila. Genes Dev. 1998;12:3900–3909. doi: 10.1101/gad.12.24.3900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey K, Tapon N. The Salvador-Warts-Hippo pathway - an emerging tumour-suppressor network. Nat. Rev. Cancer. 2007;7:182–191. doi: 10.1038/nrc2070. [DOI] [PubMed] [Google Scholar]
- Huang J, Wu S, Barrera J, Matthews K, Pan D. The Hippo signaling pathway coordinately regulates cell proliferation and apoptosis by inactivating Yorkie, the Drosophila Homolog of YAP. Cell. 2005;122:421–434. doi: 10.1016/j.cell.2005.06.007. [DOI] [PubMed] [Google Scholar]
- Lee T, Luo L. Mosaic analysis with a repressible cell marker for studies of gene function in neuronal morphogenesis. Neuron. 1999;22:451–461. doi: 10.1016/s0896-6273(00)80701-1. [DOI] [PubMed] [Google Scholar]
- Liu-Chittenden Y, Huang B, Shim JS, Chen Q, Lee SJ, Anders RA, Liu JO, Pan D. Genetic and pharmacological disruption of the TEAD-YAP complex suppresses the oncogenic activity of YAP. Genes Dev. 2012;26:1300–1305. doi: 10.1101/gad.192856.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda T, Chapman DL, Stewart AF. Mammalian vestigial-like 2, a cofactor of TEF-1 and MEF2 transcription factors that promotes skeletal muscle differentiation. J. Biol. Chem. 2002;277:48889–48898. doi: 10.1074/jbc.M206858200. [DOI] [PubMed] [Google Scholar]
- Meignin C, varez-Garcia I, Davis I, Palacios IM. The salvador-warts-hippo pathway is required for epithelial proliferation and axis specification in Drosophila. Curr. Biol. 2007;17:1871–1878. doi: 10.1016/j.cub.2007.09.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Methot N, Basler K. Hedgehog controls limb development by regulating the activities of distinct transcriptional activator and repressor forms of Cubitus interruptus. Cell. 1999;96:819–831. doi: 10.1016/s0092-8674(00)80592-9. [DOI] [PubMed] [Google Scholar]
- Methot N, Basler K. An absolute requirement for Cubitus interruptus in Hedgehog signaling. Development. 2001;128:733–742. doi: 10.1242/dev.128.5.733. [DOI] [PubMed] [Google Scholar]
- Morel V, Schweisguth F. Repression by suppressor of hairless and activation by Notch are required to define a single row of single-minded expressing cells in the Drosophila embryo. Genes Dev. 2000;14:377–388. [PMC free article] [PubMed] [Google Scholar]
- Oh H, Irvine KD. Cooperative regulation of growth by Yorkie and Mad through bantam. Dev. Cell. 2011;20:109–122. doi: 10.1016/j.devcel.2010.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan D. The hippo signaling pathway in development and cancer. Dev. Cell. 2010;19:491–505. doi: 10.1016/j.devcel.2010.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parks AL, Cook KR, Belvin M, Dompe NA, Fawcett R, Huppert K, Tan LR, Winter CG, Bogart KP, Deal JE, et al. Systematic generation of high-resolution deletion coverage of the Drosophila melanogaster genome. Nat. Genet. 2004;36:288–292. doi: 10.1038/ng1312. [DOI] [PubMed] [Google Scholar]
- Peng HW, Slattery M, Mann RS. Transcription factor choice in the Hippo signaling pathway: homothorax and yorkie regulation of the microRNA bantam in the progenitor domain of the Drosophila eye imaginal disc. Genes Dev. 2009;23:2307–2319. doi: 10.1101/gad.1820009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polesello C, Tapon N. Salvador-warts-hippo signaling promotes Drosophila posterior follicle cell maturation downstream of notch. Curr. Biol. 2007;17:1864–1870. doi: 10.1016/j.cub.2007.09.049. [DOI] [PubMed] [Google Scholar]
- Schweisguth F, Lecourtois M. The activity of Drosophila Hairless is required in pupae but not in embryos to inhibit Notch signal transduction. Dev. Genes Evol. 1998;208:19–27. doi: 10.1007/s004270050149. [DOI] [PubMed] [Google Scholar]
- Sebe-Pedros A, Zheng Y, Ruiz-Trillo I, Pan D. Premetazoan origin of the hippo signaling pathway. Cell Rep. 2012;1:13–20. doi: 10.1016/j.celrep.2011.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmonds AJ, Liu X, Soanes KH, Krause HM, Irvine KD, Bell JB. Molecular interactions between Vestigial and Scalloped promote wing formation in Drosophila. Genes Dev. 1998;12:3815–3820. doi: 10.1101/gad.12.24.3815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toba G, Ohsako T, Miyata N, Ohtsuka T, Seong KH, Aigaki T. The gene search system. A method for efficient detection and rapid molecular identification of genes in Drosophila melanogaster. Genetics. 1999;151:725–737. doi: 10.1093/genetics/151.2.725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaudin P, Delanoue R, Davidson I, Silber J, Zider A. TONDU (TDU), a novel human protein related to the product of vestigial (vg) gene of Drosophila melanogaster interacts with vertebrate TEF factors and substitutes for Vg function in wing formation. Development. 1999;126:4807–4816. doi: 10.1242/dev.126.21.4807. [DOI] [PubMed] [Google Scholar]
- Wu S, Huang J, Dong J, Pan D. hippo encodes a Ste-20 family protein kinase that restricts cell proliferation and promotes apoptosis in conjunction with salvador and warts. Cell. 2003;114:445–456. doi: 10.1016/s0092-8674(03)00549-x. [DOI] [PubMed] [Google Scholar]
- Wu S, Liu Y, Zheng Y, Dong J, Pan D. The TEAD/TEF family protein Scalloped mediates transcriptional output of the Hippo growth-regulatory pathway. Dev. Cell. 2008;14:388–398. doi: 10.1016/j.devcel.2008.01.007. [DOI] [PubMed] [Google Scholar]
- Yu J, Poulton J, Huang YC, Deng WM. The hippo pathway promotes Notch signaling in regulation of cell differentiation, proliferation, and oocyte polarity. PLoS. ONE. 2008;3:e1761. doi: 10.1371/journal.pone.0001761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Ren F, Zhang Q, Chen Y, Wang B, Jiang J. The TEAD/TEF family of transcription factor Scalloped mediates Hippo signaling in organ size control. Dev. Cell. 2008;14:377–387. doi: 10.1016/j.devcel.2008.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang N, Bai H, David KK, Dong J, Zheng Y, Cai J, Giovannini M, Liu P, Anders RA, Pan D. The Merlin/NF2 Tumor Suppressor Functions through the YAP Oncoprotein to Regulate Tissue Homeostasis in Mammals. Dev. Cell. 2010;19:27–38. doi: 10.1016/j.devcel.2010.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B, Li L, Lei Q, Guan KL. The Hippo-YAP pathway in organ size control and tumorigenesis: an updated version. Genes Dev. 2010;24:862–874. doi: 10.1101/gad.1909210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B, Ye X, Yu J, Li L, Li W, Li S, Yu J, Lin JD, Wang CY, Chinnaiyan AM, Lai ZC, Guan KL. TEAD mediates YAP-dependent gene induction and growth control. Genes Dev. 2008;22:1962–1971. doi: 10.1101/gad.1664408. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.