

Abstract
Extracellular signal-regulated kinase 1/2 (ERK1/2) signalling is a key pathway in cardiomyocyte hypertrophy and survival in response to many different stress stimuli. We have previously characterized melusin as a muscle-specific chaperone protein capable of ERK1/2 signalling activation in the heart. Here, we show that in the heart, melusin forms a supramolecular complex with the proto-oncogene c-Raf, MEK1/2 (also known as MAPKK1/2) and ERK1/2 and that melusin-bound mitogen-activated protein kinases (MAPKs) are activated by pressure overload. Moreover, we demonstrate that both focal adhesion kinase (FAK) and IQ motif-containing GTPase activating protein 1 (IQGAP1), a scaffold protein for the ERK1/2 signalling cascade, are part of the melusin complex and are required for ERK1/2 activation in response to pressure overload. Finally, analysis of isolated neonatal cardiomyocytes indicates that both FAK and IQGAP1 regulate melusin-dependent cardiomyocyte hypertrophy and survival through ERK1/2 activation.
Key words: ERK1/2, FAK, IQGAP1, Melusin, Heart hypertrophy
Introduction
Melusin is a muscle-specific protein belonging to the cysteine- and histidine-rich domain (CHORD)-containing protein family (Shirasu et al., 1999). Melusin, in fact contains two CHORD sites in its N-terminal region, and a CS (CHORD-containing protein and Sgt1) domain in its C-terminal moiety, a domain frequently found in Hsp90 co-chaperones. We recently demonstrated that melusin is able to bind Hsp90 chaperone protein and possesses chaperone activity per se (Sbroggiò et al., 2008). Chaperones, as well as assisting protein folding and thus preventing aggregation and degradation, play an essential role in regulating signal transduction pathways assisting conformational changes required for protein activation (Pearl et al., 2008).
We have previously shown that melusin expression in the heart is upregulated by mechanical stress induced by surgical banding of the aorta (Sbroggiò et al., 2008), an intervention mimicking pathological condition of pressure overload such as aortic stenosis and hypertension. Forced melusin overexpression in the heart of transgenic mice leads to cardiomyocyte hypertrophy and protects from pressure overload-induced apoptosis, allowing the development of sustained compensatory cardiac hypertrophy and preventing the development of heart failure in conditions of long-standing pressure overload (De Acetis et al., 2005). Melusin overexpression in the mouse heart is able to strongly enhance ERK1/2 phosphorylation both in basal conditions and in response to pressure overload stimuli and this signalling pathway is required for melusin-induced cardiomyocyte hypertrophy (De Acetis et al., 2005).
Here, we demonstrate that melusin forms a complex with the mitogen-activated protein kinases (MAPKs), the proto-oncoprotein c-Raf, MEK1/2 (also known as MAPKK1/2) and extracellular signal-regulated kinase 1/2 (ERK1/2), the focal adhesion kinase (FAK) and IQ motif-containing GTPase activating protein 1 (IQGAP1; UniProtKB Q9JKF1), a MAPK scaffold protein. Moreover, using pharmacological kinase inhibitors and null mice, we show that FAK and IQGAP1 are required for melusin-dependent MAPK activation in response to aortic banding, and play an important role in melusin-induced cardiomyocyte hypertrophy and survival.
Results
Melusin enhances MAPK phosphorylation and interacts with Raf, MEK1/2 and ERK1/2
As shown in Fig. 1A, and as previously demonstrated, melusin overexpression in the heart of transgenic mice (melusin-TG) enhances ERK phosphorylation, both in basal conditions and in response to pressure overload induced by aortic banding (AB) (De Acetis et al., 2005). This result led us to investigate the possibility that melusin interacts with MAP kinases. Melusin was immunoprecipitated from hearts of melusin-TG mice that were subjected to AB or sham operation (basal conditions), and analysed by western blotting, which revealed the presence of c-Raf, MEK1/2 and ERK1/2 in the immunocomplex (Fig. 1B). In agreement with our previous results (Sbroggiò et al., 2008), we also detected the presence of Hsp90 (Fig. 1B). Interestingly, we found that the amounts of melusin-associated MEK1/2 and Hsp90 significantly increase in response to AB (Fig. 1B). The specificity of the co-precipitation is demonstrated by the fact that b-Raf, which is also expressed in the heart, was not detected in association with melusin (Fig. 1B).
Fig. 1.

Melusin-bound MAPKs are activated by aortic banding. (A) Western blot analysis of phosphorylated and total ERK1/2 in hearts from wild-type (WT) and melusin-overexpressing (MelTG) mice in basal condition (SH; sham operated) and after 10 minutes of aortic banding (AB). The graph shows densitometric quantification of western blot bands (n=6 mice/group). (B) Immunoprecipitation of melusin from MelTG hearts after AB for 10 minutes or sham operation. Melusin-null hearts were used as negative controls. Co-immunoprecipitated proteins were visualized by western blot analysis. Input: heart total protein extract loaded on the same western blot as reference for molecular masses. Quantification of the co-precipitated proteins is shown in the graph (n=10/group). (C) ERK1/2 kinase assays were performed using GST–ELK1 as a substrate on melusin immunocomplexes obtained from MelTG hearts after AB for 10 minutes or sham operation. ERK1/2 kinase activity was revealed by western blot analysis with anti-phosphorylated ELK1 (Ser338; n=6/group). (D) ERK1/2 kinase assay performed on melusin immunocomplex in absence or presence of MEK1/2 inhibitor PD89059 (1 μM; n=3/group). *P<0.05; ***P<0.001; IP, immunoprecipitation; KA, kinase assay.
Melusin-bound MAPKs are activated by AB
To investigate if melusin-bound MAPKs are involved in signal transduction triggered by AB, we subjected the immunoprecipitated samples to a kinase assay using recombinant ETS domain-containing protein ELK1 as a substrate for ERK1/2. Western blot analysis using anti-phosphorylated ELK1 antibodies demonstrated that melusin-bound ERK1/2 are activated following AB (Fig. 1C). Interestingly, the specific MEK1/2 inhibitors PD89059 (1 μM) and U0126 (0.1 μM) were able to abolish the AB-dependent ERK1/2 activation, indicating the involvement of MEK1/2 as an upstream kinase of ERK1/2 in this pathway (Fig. 1D and supplementary material Fig. S1).
Identification of melusin binding partners
To investigate the mechanisms involved in the activation of melusin-bound ERK1/2, we further analyzed melusin co-immunoprecipitated material using both western blotting with phosphotyrosine antibodies and mass spectrometry of Coomassie-Blue-stained bands to identify possible additional signal transduction molecules. As shown in Fig. 2A, a prominent 125 kDa band was clearly stained when the melusin co-immunoprecipitated material was probed with anti-phosphotyrosine antibodies. Using a panel of antibodies to signalling molecules we identified this band as FAK, a well-known kinase acting downstream of integrins and involved in ERK1/2 pathway activation (Fig. 2B). The amount of FAK associated with melusin was not altered by AB (Fig. 2B).
Fig. 2.
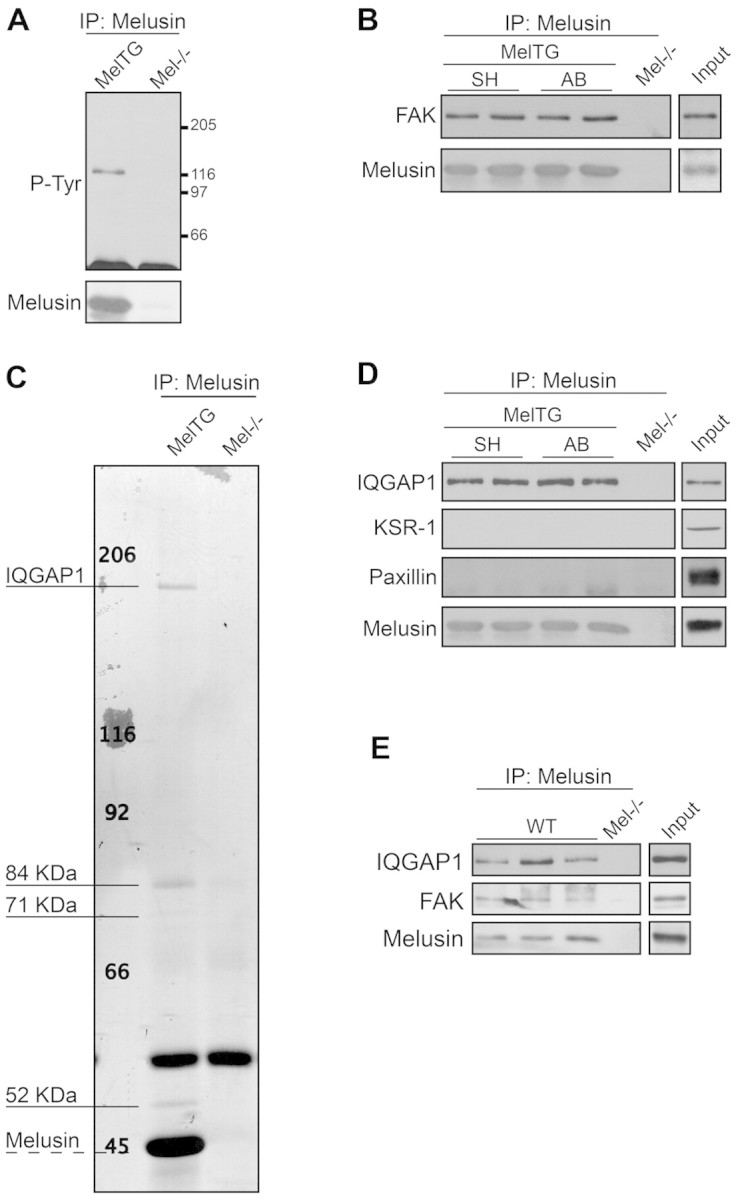
Melusin interacts with FAK and the MAPK scaffold IQGAP1. (A) Western blot analysis of tyrosine phosphorylated proteins in melusin immunocomplexes from MelTG hearts. (B) FAK co-immunoprecipitation revealed by western blots of melusin immunocomplexes isolated from MelTG hearts after AB for 10 minutes or sham operation (n=4 mice/group). Melusin-null hearts were used as negative controls. (C) Coomassie Blue staining of melusin immunoprecipitate from MelTG hearts. The indicated bands were cut out and subjected to mass spectrometry analysis. (D) Western blot of IQGAP1, KSR-1 and paxillin in melusin immunocomplexes isolated from MelTG hearts after AB for 10 minutes or sham operation (n=4 mice/group). (E) Western blot of IQGAP1 and FAK in melusin immunoprecipitate from wild-type (WT) hearts after AB for 10 minutes or sham operation (n=4/group). Melusin-null hearts were used as negative controls. Input: heart total protein extract.
When melusin co-immunoprecipitated proteins were analyzed by Coomassie Blue staining, four major bands were detected with molecular masses of 190 kDa, 84 kDa, 71 kDa and 52 kDa (Fig. 2C). Matrix-assisted laser desoption/ionisation-time of flight (MALDI-TOF) and liquid chromatography-nanospray-iontrap tandem (LC-nanospray-IT) analysis identified the co-precipitated band at 190 kDa as IQGAP1. IQGAP1 is a multidomain protein that acts as a scaffold protein for the MAPK pathway by binding to b-Raf (Ren et al., 2007), MEK1/2 and ERK1/2 (Roy et al., 2005), and thus represents a potentially interesting molecule in the complex. The other three additional bands are currently under investigation and are not related to ERK signalling. MAPKs and FAK were not detected in this assay, probably because of low sensitivity of the Coomassie Blue staining. The amount of IQGAP1 associated with melusin did not change after AB and the interaction with this molecule was specific because KSR1 and paxillin, two other proteins reported to act as scaffold for MAPKs, were not present in the co-precipitated material (Fig. 2D). IQGAP1 and FAK also co-immunoprecipitate with melusin from wild-type heart, further strengthening the physiological relevance of the interaction (Fig. 2E).
Because IQGAP1 has been originally described to bind several small GTPases (Bashour et al., 1997; Awasthi et al., 2010), we probed melusin immunoprecipitates with antibodies against Rac1, Cdc42, Rap1 and Ras. As shown in supplementary material Fig. S2, none of these proteins was detected in the complex.
FAK activity is required for ERK1/2 activation in melusin complexes after AB
Previous works have described a role of FAK in ERK1/2 activation in several cell types (Guo and Giancotti, 2004; Mitra et al., 2005; Vadali et al., 2007). We, therefore, investigated the possibility that FAK has a role in melusin-associated ERK1/2 family MAPK activation in response to AB. As shown in Fig. 3A and supplementary material Fig. S1, the FAK inhibitor PF573228 (0.1 μM) and inhibitor 14 (1 μM) abolished ELK1 phosphorylation in the ERK1/2 kinase assay, demonstrating that melusin-associated ERK1/2 activation in response to AB depends on FAK activity.
Fig. 3.

Melusin-bound ERK1/2 activation requires FAK and IQGAP1. (A) ERK1/2 kinase assays performed on melusin immunocomplexes obtained from MelTG hearts after AB for 10 minutes or sham operation, in the absence or presence of the FAK inhibitor PF573228 (0.1 μM). ERK1/2 kinase activity was revealed by western blot analysis with anti-phosphorylated ELK1 (Ser338; n=4/group). (B) FAK kinase assays performed on melusin immunocomplex obtained from MelTG hearts after AB for 10 minutes or sham operation. FAK kinase activity was assayed using GST–FAK378–406 as a substrate, and revealed by western blot analysis with anti-phosphorylated-FAK (Tyr397; n=3/group). Melusin-null hearts were used as negative controls. (C) FAK kinase activity measured on FAK immunocomplexes obtained from wild-type (WT) hearts after AB for 10 minutes or sham operation. FAK kinase activity was revealed by western blotting with anti-phosphorylated-FAK (Tyr397; n=4/group). (D) ERK1/2 kinase assay performed on melusin immunocomplex obtained from MelTG and MelTG/Iqgap1-null (MelTG/IQ−/−) hearts after AB for 10 minutes or sham operation. ERK1/2 kinase activity was revealed by western blot analysis with anti-phosphorylated ELK1 (Ser338; n=4/group). Melusin-null hearts were used as negative controls.
In agreement with these results, FAK kinase assays performed on melusin immunocomplexes demonstrated that melusin-bound FAK is activated by AB (Fig. 3B). In addition, direct immunoprecipitation of FAK from heart extracts showed an increased FAK kinase activity in response to AB (Fig. 3C), in agreement with previous reports (Franchini et al., 2000).
IQGAP1 contributes to ERK1/2 activation in melusin complexes after aortic banding.
Because IQGAP1 is a multidomain protein that acts as a scaffold for the MAPK pathway by binding to b-Raf (Ren et al., 2007), MEK1/2 and ERK1/2 (Roy et al., 2005), we investigated in greater detail the possible role of IQGAP1 in melusin induced-ERK1/2 activation. To this purpose we took advantage of Iqgap1-null mice (Li et al., 2000; Bahou et al., 2004) and of double transgenic mice overexpressing melusin in the heart in the absence of IQGAP1 (melusin-TG/Iqgap1-null). Interestingly, we noticed that absence of IQGAP1 abolished melusin-induced ERK1/2 over-phophorylation both in basal conditions and in response of aortic banding (supplementary material Fig. S3A).
This finding led us to investigate whether IQGAP1 is required for melusin–MAPK interaction. Co-immunoprecipitation of melusin from double transgenic melusin-TG/Iqgap1-null hearts, indicated that IQGAP1 is not required for melusin binding to c-Raf, MEK1/2 or ERK1/2 (supplementary material Fig. S3B).
However, melusin is not required for IQGAP1 binding to ERK1/2 family MAPKs, as shown by pull-down experiments with IQGAP1 fragments (supplementary material Fig. S3C) from wild-type and melusin-null hearts (supplementary material Fig. S3D). Moreover, analysis of melusin and IQGAP1 binding indicated a direct interaction through the CS domain of melusin and the IQ domain of IQGAP1 (supplementary material Fig. S3E–H). Thus, melusin and IQGAP1 can independently bind ERK1/2 family MAPKs and can directly bind to each other.
To investigate the role of IQGAP1 in melusin-bound ERK1/2 activation in response to AB, we immunoprecipitated melusin from melusin-TG and melusin-TG/Iqgap1-null mice hearts in basal conditions and upon AB. We then subjected the immunoprecipitated samples to an ERK1/2 kinase assay using recombinant ELK1 as a substrate. As shown in Fig. 3D, the absence of IQGAP1 clearly reduced ERK1/2 activity, demonstrating that ERK1/2 activation in response to AB depends on the presence of IQGAP1.
Melusin overexpression enhances ERK1/2 phosphorylation in cultured neonatal cardiomyocytes through FAK and IQGAP1
Analysis of neonatal cardiomyocytes from melusin-TG mice demonstrated that melusin overexpression induces significant upregulation of ERK1/2 phosphorylation (Fig. 4A). We therefore investigated the possibility that FAK activity is required for the melusin-dependent MAPK phosphorylation in cardiomyocytes. Wild-type and melusin-TG primary mouse cardiomyocytes were treated with the FAK inhibitor PF573228 (3 μM) for 30 minutes, and total protein extracts were then subjected to western blot analysis with anti-phosphorylated ERK1/2. As shown in Fig. 4B, both in wild-type and melusin-TG cardiomyocytes, FAK inhibition reduced ERK1/2 phosphorylation to the same degree. At the concentration used, PF573228 did not inhibit MEK1/2 activity as tested on MDA-MB-231 cells in which the ERK1/2 pathway is activated by constitutively active Ras (not shown). This result indicates that increased ERK1/2 phosphorylation due to melusin overexpression is dependent on FAK activity. The efficacy of the inhibition was further confirmed by analyzing the phosphorylation of tyrosine (Y) 397, a well-known autophosphorylated residue on FAK (Fig. 4B).
Fig. 4.

Melusin enhances ERK1/2 phosphorylation through FAK and IQGAP1. (A) Western blot analysis of phosphorylated and total ERK1/2 in neonatal cardiomyocytes obtained from wild-type and MelTG mice. The graph shows the densitometric quantification of western blot bands (n=5/group). (B) Phosphorylation and total protein levels of MEK1/2, ERK1/2 and FAK measured by western blotting on wild-type and MelTG cardiomyocytes untreated or treated with 3 μM PF573228. Anti-phosphorylated FAK (Tyr397), anti-phosphorylated MEK1/2 (Ser217/Ser221) and anti-phosphorylated ERK1/2 (Thr202/Tyr204) were used. Densitometric quantification of western blot bands is shown in the graph (n=4/group). (C) Western blot analysis of phosphorylated and total ERK1/2 on neonatal cardiomyocytes obtained from wild-type, MelTG and MelTG/Iqgap1-null (MelTg/IQ−/−) mice. The graph shows the densitometric quantification of western blot bands (n=4/group). *P<0.05; **P<0.01; ***P<0.001.
Considering that MEK1/2 is the only known direct activator of ERK1/2, our findings suggested that FAK is upstream of MEK1/2. To investigate this we studied the effect of the FAK inhibitor on MEK1/2 phosphorylation. As shown in Fig. 4B, melusin overexpression increased MEK1/2 phosphorylation to a comparable extent as that observed with ERK1/2. Notably, this increased phosphorylation was abolished when cardiomyocytes were treated with the FAK inhibitor PF573228 (Fig. 4B), pointing to a causal relationship between FAK and MEK1/2.
To investigate the role of IQGAP1 in melusin-dependent ERK phosphorylation we analyzed primary cardiomyocytes from wild-type, melusin-TG and melusin-TG/Iqgap1-null mice. As shown in Fig. 4C the enhancement in ERK1/2 phosphorylation due to melusin overexpression is dependent on IQGAP1.
FAK and IQGAP1 are required for melusin-induced cardiomyocyte hypertrophy and survival
We previously demonstrated that melusin overexpression in cardiomyocytes in vivo induces hypertrophy and protects against apoptosis after long-standing AB (De Acetis et al., 2005). Moreover, because the ERK1/2 pathway has been shown to be involved in melusin-dependent cardiomyocyte hypertrophy (De Acetis et al., 2005), we investigated the role of FAK and IQGAP1 in these phenomena using FAK inhibition and taking advantage of melusin-TG/Iqgap1-null mice. In accordance with our previous results, melusin-TG neonatal cardiomyocytes were significantly larger than wild-type cells (Fig. 5A). We also demonstrated that treatment of melusin-TG primary cardiomyocytes with the FAK inhibitor PF573228 totally abolished the melusin-dependent hypertrophy (Fig. 5A). Similar to previous results (De Acetis et al., 2005), the MEK1/2 inhibitor PD89059 also reversed the effect of melusin overexpression on cell area (Fig. 5A). To exclude an unspecific effect of the FAK inhibitor, we knocked down FAK in cardiomyocytes using a lentivirus carrying a short hairpin RNA (shRNA) against FAK. As shown in Fig. 5B, FAK expression was downregulated to 20% of the level of control-infected cells, as quantified by densitometry of the western blot bands. FAK knock-down in melusin-TG cardiomyocytes reduced cell areas to the level of wild-type cardiomyocytes (Fig. 5C), confirming the results obtained with the pharmacological inhibition of FAK. Moreover, primary neonatal cardiomyocytes obtained from melusin-TG/Iqgap1-null mice had a significantly reduced cell area with respect to melusin-TG cardiomyocytes and comparable with wild-type cells (Fig. 5D). Notably, neonatal cardiomyocytes isolated from Iqgap1-null hearts had a cell area comparable with that of wild-type cells (supplementary material Fig. S4A).
Fig. 5.

FAK and IQGAP1 are required for melusin-induced cardiomyocyte hypertrophy and survival. (A) The effect of the FAK inhibitor PF573228 and the MEK1/2 inhibitor PD89059 on cardiomyocyte surface areas, measured on at least 100 α-actinin-positive cells for each group (five cultures/group). (B) Representative western blots of FAK protein levels in cardiomyocytes infected with lentivirus encoding Fak shRNA or control viruses. Vinculin was used as a control for loading and RNA interference specificity. (C) Cardiomyocyte surface areas of cells treated as in B, measured on at least 100 α-actinin-positive, GFP-positive cells for each group (four cultures/group). (D) Wild-type, MelTG and MelTG/Iqgap1-null cardiomyocyte surface areas measured on at least 100 α-actinin-positive cells for each group (five cultures/group). (E) Percentage of apoptotic cardiomyocytes treated as in A, as indicated by TUNEL nuclear staining, measured on at least 100 α-actinin-positive cells for each group (five cultures/group). (F) Percentage of apoptotic cardiomyocytes treated as in B and C, as indicated by TUNEL nuclear staining and measured on at least 100 α-actinin-positive, GFP-positive cells for each group (4 cultures/group). (G) Percentage of apoptotic cardiomyocytes after treatment with H2O2, as indicated by TUNEL nuclear staining, measured on at least 100 α-actinin-positive cells for each group (five cultures/group). ***P<0.001; °°°P<0.001 versus untreated cells (NT).
These results demonstrate that both FAK and IQGAP1 are required for melusin-dependent hypertrophic effects on cardiomyocytes. We thus decided to investigate the role of FAK and IQGAP1 on melusin-dependent protection from apoptosis (De Acetis et al., 2005). To this end wild-type and melusin-TG neonatal cardiomyocytes were treated with H2O2 and apoptotic cells were detected with TUNEL assays. As shown in Fig. 5E, melusin-TG cardiomyocytes showed a significantly reduced number of apoptotic cells when treated with H2O2 compared with wild-type cardiomyocytes. Interestingly, both the FAK inhibitor PF57322 and the MEK1/2 inhibitor PD89059 were equally able to abolish the protective effect (Fig. 5E). Furthermore, FAK knock-down abolished the melusin-mediated protection from apoptosis, closely replicating the effects of FAK pharmacological inhibition (Fig. 5F). Moreover, melusin-TG/Iqgap1-null neonatal mouse cardiomyocytes had a significantly higher mortality rate than melusin-TG cardiomyocytes, when treated with H2O2 (Fig. 5G). The absence of IQGAP1 in wild-type cardiomyocytes also resulted in increased apoptotic death in response to oxidative stress (supplementary material Fig. S4B) further demonstrating a role of IQGAP1 in cardiomyocyte survival (Sbroggiò et al., 2011). These results demonstrated that FAK and IQGAP1 act on the same melusin-activated signal transduction pathway in promoting cell survival.
In conclusion, we have demonstrated that both FAK and IQGAP1 are needed for the melusin-activated MAPK pathway leading to hypertrophy and survival.
Discussion
Our previous findings demonstrated a key role of melusin in triggering a compensatory hypertrophic program and in preventing left ventricle dilation and heart failure in response to long standing pressure overload. Melusin is a chaperone molecule regulated by mechanical stress (Sbroggiò et al., 2008), capable of activating AKT and ERK signalling, two pathways required for melusin-induced cardiomyocyte hypertrophy and survival (De Acetis et al., 2005). Here we show that melusin is able to interact with the MAP kinases and it is part of a molecular complex that includes the FAK, the scaffold protein IQGAP1 and the chaperone protein Hsp90. FAK and IQGAP1 were identified as key molecules in the complex responsible for ERK1/2 activation in response to pressure overload. To our knowledge this is the first report describing a supramolecular complex involved in the activation of the ERK1/2 cascade in hearts challenged by pressure overload.
The MAPK pathway involves a cascade of three kinases (Raf, MEK1/2 and ERK1/2) that sequentially activate each other by phosphorylation. A key role in regulating such cascades is played by scaffold proteins, which enhance signal efficiency by assembling the molecules involved in signal transduction in close proximity. IQGAP1 is a widely expressed multidomain protein regulating different aspects of cell physiology and capable of binding to distinct signalling molecules (Brown and Sacks, 2006). Emerging evidence indicates that IQGAP1 acts as a scaffold for the MAPK cascade by binding b-Raf, MEK1/2 and ERK1/2, and regulating their activation in response to EGF in fibroblasts and epithelial cells (Brown and Sacks, 2009). We recently showed that IQGAP1 also acts as a scaffold of the MAPK pathway in the heart by binding to c-Raf, MEK1/2 and ERK1/2 (Sbroggiò et al., 2011). The role of IQGAP1 is highlighted by the fact that in its absence, ERK1/2 activation is impaired (see Fig. 3D), consistent with the property of scaffold molecules to spatially organize the components of the MAPK cascade.
Activation of ERK1/2 in response to AB is also dependent on MEK1/2 and FAK kinase activities. In fact, FAK is also part of the melusin complex, and specific inhibition of this kinase abolished ERK1/2 activation in response to AB. FAK is a ubiquitously expressed non-receptor tyrosine kinase acting as a primary integrin effector at focal adhesion sites (Giancotti and Ruoslahti, 1999; Parsons, 2003). Using kinase assays, we demonstrated that melusin-bound FAK is activated in response to AB, in agreement with previous findings (Franchini et al., 2000). Several reports in the literature have indicated that FAK is a crucial transducer for ERK1/2 activation downstream of integrins and growth factor receptors (Guo and Giancotti, 2004; Mitra et al., 2005; Vadali et al., 2007). The activation of ERK1/2 by FAK involves the recruitment of the adaptor protein Grb-2 to the FAK C-terminal region, leading to the activation of the small GTPase Ras through the guanosine exchange factor Sos. Ras, however, was not detected in the melusin supramolecular complex. Moreover, other GTPases known to be involved in ERK1/2 signalling and to interact with IQGAP1, such as Rac1, Cdc42 (Bashour et al., 1997) and Rap1 (Awasthi et al., 2010) were not detectable in the complex, indicating that these GTPases are not involved in FAK- and melusin-dependent ERK1/2 activation. Interestingly, however, our results demonstrated that FAK activity is required for melusin-dependent MEK1/2 phosphorylation, indicating that FAK activates ERK1/2 through MEK1/2. FAK-mediated and Ras-independent ERK1/2 activation has been reported in a number of experimental systems (Chen et al., 1996; Hood et al., 2003; Slack-Davis et al., 2003), but further studies are required to clarify the ability of FAK to activate the MAPK pathway in a Ras-independent way.
Comparison of the supramolecular complexes isolated from basal level and pressure-overloaded hearts indicated that the amounts of FAK, IQGAP1, c-Raf and ERK1/2 were unaltered following pressure overload. However, recruitment of MEK1/2 and Hsp90 to the complex was significantly enhanced by pressure overload. Because melusin binds Hsp90 and possesses chaperone activity per se, we recently proposed a role for melusin as a co-chaperone in the Hsp90 machinery (Sbroggiò et al., 2008). Hsp90 is able to bind and protect a wide range of substrates or ‘client proteins’ from degradation, and has a key role in signal transduction because of its ability to maintain proteins in a three-dimensional conformation that favours their activation (Pearl et al., 2008). Therefore, a plausible hypothesis is that melusin, by interacting with FAK, IQGAP1, c-Raf, MEK1/2 and ERK1/2 regulates the MAPK pathway through its own chaperone activity and Hsp90 recruitment. In line with this hypothesis is the fact that FAK, c-Raf and MEK1/2 are known Hsp90 client proteins (Schulte et al., 1997; Sbroggiò et al., 2008). We hypothesize that the scaffold protein IQGAP1 and the chaperone proteins melusin and Hsp90, are both needed to fully activate the ERK1/2 pathway in response to pressure overload in the heart. In fact, ERK1/2 activation requires chaperones for stimulus-dependent conformational changes and scaffold-mediated assembly of the three MAP kinases signalling complex (Casar et al., 2009). An hypothetical model of the melusin supramolecular complex is illustrated in Fig. 6.
Fig. 6.

Hypothetical model of the melusin supramolecular complex involved in ERK1/2 activation in cardiomyocytes. The protein complex indicated above schematically illustrates the molecular interactions described in the results section. The model does not take into account the stoichiometry of the interactions and it is not comprehensive of all possible interactions among the different protein in the complex. Chaperones are in blue, kinases are in orange and the scaffold protein is in green. The stars indicate the activated state of the kinases.
Analysis of isolated cardiomyocytes from melusin-TG mice, indicated that melusin-dependent ERK1/2 activation leads to cardiomyocyte hypertrophy and protection from apoptosis in response to stress stimuli, confirming and further extending our previous data obtained from rat primary cardiomyocytes (De Acetis et al., 2005). Moreover, FAK and IQGAP1, in addition to being required for melusin-dependent ERK1/2 activation, are crucial in melusin-induced cardiomyocyte hypertrophy and survival.
These findings on isolated cardiomyocytes are in agreement with in vivo data obtained from transgenic mice. In fact, Fak-null (DiMichele et al., 2006), melusin-null (unpublished data) and Iqgap1-null mice (Sbroggiò et al., 2011) all show an impaired wave of ERK1/2 phosphorylation in response to AB. Moreover, absence of either FAK, melusin or IQGAP1 strongly impairs the AB-induced cardiomyocyte hypertrophy, exacerbating cardiac dysfunction in response to long-term AB (Brancaccio et al., 2003; DiMichele et al., 2006; Sbroggiò et al., 2011).
Overall, these data underlie an important role of ERK1/2 in adaptive cardiac remodelling (Kehat and Molkentin, 2010; Kehat et al., 2011) and indicate a key role for melusin, FAK and IQGAP1 in this pathway.
Materials and Methods
Mice
The use of animals was in compliance with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health and was approved by the Animal Care and Use Committee of Turin University.
Mice were anesthetized with sodium pentothal (50 mg/kg, intraperitoneally) and, after 20 minutes pressure overload was imposed to the left ventricle through the surgical ligation of the abdominal aorta (aortic banding). After 10 minutes of aortic banding, mice were sacrificed and hearts were collected. Sham-operated animals underwent the same surgical procedures without aortic stenosis.
Cell culture
Cardiomyocytes were isolated from 1-day-old neonatal mice of the following types: melusin-TG in the FVB genetic background, Iqgap1-null in the 129SV genetic background and double transgenic melusin-TG/Iqgap1-null in the FVB/129SV mixed background. Neonatal hearts were pre-digested overnight at 4°C in Hank's balanced salt solution (HBSS) with 0.5 mg/ml trypsin (Sigma), followed by four sequential dissociation cycles with 240 IU/ml collagenase type II (Worthington) in HBSS. Fibroblasts were removed by two rounds of pre-plating for 2 hours on plastic tissue culture dishes. Cardiomyocytes were plated on gelatin- and fibronectin-coated dishes or glass coverslips and maintained in DMEM–M199 medium, 10% horse serum (Gibco) and 5% FBS (Gibco). Final cultures contained >90% cardiomyocytes as determined by immunofluorescence staining for sarcomeric α-actinin (antibody from Sigma). When indicated, cells were treated with the inhibitor PD890959 (50 μM; Calbiochem) or PF537228 (3 μM; Tocris) after 3 days in culture. For western blot analyses of signalling pathways, cells were lysed for 30 minutes after treatment with inhibitors. For measurements of cell area, cells were fixed after 2 days of treatment with the inhibitors. For analysis of apoptotic death cells were treated with 1 μM H2O2 for 10 minutes and then cells were maintained in normal culture medium in the absence or presence of inhibitors.
FAK knock-down was performed by infecting cardiomyocytes with pGIPZ lentiviral particles expressing FAK shRNA together with green fluorescent protein (GFP) reporter (Open Biosystems; clone ID V3LMM_440799) 24 hours after plating. Empty pGIPZ lentiviral vector, expressing GFP reporter, was used as control. For measurements of cell area and apoptosis, cells were fixed 4 days after infection.
Cardiomyocyte surface area and TUNEL assay
Neonatal cardiomyocytes were cultured on coverslips and maintained as described above for 5 days after plating, and they were then fixed with 3% paraformaldheyde (PFA) in PBS. To measure the surface area of cells, cells were treated with immunofluorescent antibodies against α-actinin (sarcomeric) and phalloidin. Cell areas were measured on images captured at 400× magnification using Axio Vision (Zeiss) software.
Cardiomyocyte apoptosis was assayed using the In Situ Cell Death Detection Kit TMR red (Roche), following the manufacturer's instructions. To ensure exclusive counting of cardiomyocytes nuclei, cells were stained with anti-α-actinin (sarcomeric) antibodies and nuclei were counterstained with DAPI. Cell area measurements and apoptotic cell counts on FAK-knockdown cardiomyocytes were only performed on α-scarcomeric-actinin- and GFP-positive cells, indicative of lentivirus-infected cardiomyocytes.
Immunoprecipitations and western blotting
For immunoprecipitation experiments, hearts were homogenized in lysis buffer (50 mM HEPES, 100 mM NaCl, 0.1% sodium deoxycholate, 0.1% Triton X-100, 1 mM EGTA, 2 mM EDTA), containing Roche complete protease inhibitor cocktail, 10 mM NaF, 1 mM PMSF and 1 mM Na3VO4. Protein extracts were clarified with three sequential centrifugations for 20 minutes at 20,000 g, at 4°C. Immunoprecipitations were performed for 2 hours at 4°C using 5 mg of total heart proteins and 15 μg of anti-melusin (clone 5E1) (Sbroggiò et al., 2008) antibody, covalently conjugated to CNBr–Sepharose beads (GE Healthcare). After immunoprecipitation, beads were washed 10 times in lysis buffer and resuspended in Laemmli buffer.
For western blotting, hearts were lysed in Tris-buffered saline with 1% Triton X-100, plus phosphatase and protease inhibitors as indicated above. Neonatal cardiomyocytes were lysed in RIPA buffer. Western blot band quantifications were performed with Quantity One software (Bio-Rad).
For western blot analysis antibodies against the following proteins were used: ERK1/2, MEK1/2, phospho-T202/Y204 ERK1/2, phospho-S217/S221 MEK1/2, phospho-Y397 FAK, phospho-S338 ELK1, Ras, Cdc42 (Cell Signaling), IQGAP1, Raf, b-Raf, p-Tyr (PY99), Rap1 (Santa Cruz), myc clone 9E10, vinculin (Sigma), GST (Invitrogen), Hsp90 (Stressgene), KSR-1, paxillin, Rac1 (BD transduction), FAK clone 2A7 (Millipore), and the mouse monoclonal anti-melusin 5E1 (Sbroggiò et al., 2008). A second monoclonal antibody against melusin (clone C3) was produced in our laboratory by immunizing melusin-null mice with recombinant GST–mouse melusin and its reactivity was characterized by western blotting and immunoprecipitation. The epitope was mapped in the N-terminal CHORD I–II region.
Kinase assays
The kinase activity of the melusin-bound ERK1/2 was detected by performing a cold kinase assay, using recombinant GST–ELK1 (Cell Signaling) as the ERK1/2 substrate, on the melusin immunoprecipitation as described above. Briefly, immunoprecipitated samples were washed, the washing buffer was completely removed and resuspended in 18 μl of kinase buffer (25 mM Tris-HCl pH 7.5, 5 mM β-glycerophosphate, 2 mM dithiothreitol, 1 mM Na3VO4, 10 mM MgCl2, 200 μM ATP) and 2 μg recombinant GST–ELK1. For FAK kinase assays, a GST fusion recombinant protein containing a Y397 FAK autophorylation site (pGEX-FAK amino acids 378–406) was used as a substrate (Chen and Chen, 2006). Where indicated, samples were pre-incubated in kinase buffer plus 1 μM PD890959 (Calbiochem), 0.1 μM PF537228, 0.1 μM U0126 or 1 μM FAK inhibitor 14 (Tocris) for 10 minutes at room temperature, before the addition of the substrate. Reactions were carried out at 30°C for 30 minutes and stopped by adding an equal volume of 2× Laemmli buffer, followed by 5 minutes incubation at 95°C.
Mass spectrometry
Coomassie-Blue-stained pieces of gel were trypsin-digested and analyzed by MALDI-TOF and LC-nanospray-IT mass spectrometry. MALDI mass spectra were acquired on an Ultraflex II TOF-TOF instrument (Bruker Daltonics, Bremen, Germany) equipped with a 200 Hz all-solid-state laser. LC-nanospray-IT experiments were performed on a HP 1100 nanoLC system coupled to a XCT-Plus nanospray-ion trap mass spectrometer (Agilent Italia). A database search using the data from MALDI-TOF experiments was performed against the UniProtKB/SwissProt-TrEMBL database using the Aldente search algorithm (http://www.expasy.org/tools/aldente/); for nanoLC-nanospray-ion trap experiments we used the NCBInr database and Mascot search algorithm (http://www.matrixscience.com/search_form_select.html).
Recombinant proteins
Expression vectors encoding glutathione S-transferase (GST) and maltose binding protein (MBP) fused to melusin were prepared as previously described (Sbroggiò et al., 2008). To produce recombinant MBP-fused IQGAP1 fragments, the nucleotide sequences encoding the protein fragments indicated in supplementary material Fig S3C were cloned in pMAL C2 vector.
MBP and GST fusion proteins were produced in Escherichia coli BL21 bacterial strain and purified as described below.
Pull-down assays
Frozen heart samples were homogenized with an Ultra-Turrax (VWR, West Chester, PA) in Tris-buffered saline (TBS) plus 1% Triton X-100 with protease and phosphatase inhibitors, centrifuged three times for 20 minutes at 20,000 g at 4°C, and ~3 mg total proteins were used for pull-down assays. MBP-fused IQGAP1 and melusin fragments or MBP alone as control, were purified from bacterial protein extracts using an amylose resin (GE Healthcare) for 1 hour at 4°C. Resin was washed five times with TBS plus 1% Triton X-100, and 5 μg recombinant protein was incubated (with agitation) with 3 mg of heart total proteins for 2 hours. Resin was washed 10 times with TBS plus 1% Triton X-100 and resuspended in Laemmli buffer.
Pull-down experiments, to evaluate the direct interaction between melusin and IQGAP1, were performed using MBP–IQGAP1 and GST–melusin fusion proteins, together with MBP and GST alone as controls. Resin-bound proteins were washed five times with interaction buffer (50 mM Tris-HCl pH 7.4, 150 mM NaCl, 1% Triton X-100) and only the GST fusion proteins were eluted from the resin with 10 mM glutathione. The purified proteins were then quantified using SDS-PAGE and Coomassie Blue staining. Resin-bound MBP–IQGAP1 fragments and eluted GST–melusin fragments (4 μmol of each) were incubated for 2 hours at 4°C in interaction buffer. Following incubation, resins were washed 10 times with interaction buffer and resuspended in Laemmli buffer.
Statistical analyses
The data are presented as means ± s.e.m. Differences between experimental groups were evaluated for statistical significance using one or two-way ANOVA with Bonferroni's correction. For all analyses, a minimum value of P<0.05 was considered significant. All statistical analyses were performed using GraphPad Prism 4 (GraphPad Software version 4.0).
Acknowledgements
We wish to thank Tiziana Cravero and Cristina Rubinetto for technical assistance in antibody preparation, and Roberta Ferretti and R. Srinivasan for comments on the manuscript. We also thank Flavio Cristofani and Maddalena Iannicella for assistance and support with the animal experiments. We thank Prof. Chen-Hong Chen for kindly providing the pGEX–FAK (amino acids 378-406).
Footnotes
Funding
This work was supported by grants from EUGeneHeart [grant number LSHM-CT-2005-018833 to G.T.]; Regione Piemonte POR F.E.S.R. 2007/2013 “DRUIDI: Piattaforme Innovative per le Scienze della Vita” [grant number 129-9/6/2009 to G.T. and M.B]; Regione Piemonte CIPE 2006 [grant number RU/01/02 to M.B.]; and Fondazione Veronesi [grant number 2010 to G.T.]
Supplementary material available online at http://jcs.biologists.org/lookup/suppl/doi:10.1242/jcs.091140/-/DC1
References
- Awasthi A., Samarakoon A., Chu H., Kamalakannan R., Quilliam L. A., Chrzanowska-Wodnicka M., White G. C., 2nd, Malarkannan S. (2010). Rap1b facilitates NK cell functions via IQGAP1-mediated signalosomes. J. Exp. Med. 207, 1923-1938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahou W. F., Scudder L., Rubenstein D., Jesty J. (2004). A shear-restricted pathway of platelet procoagulant activity is regulated by IQGAP1. J. Biol. Chem. 279, 22571-22577 [DOI] [PubMed] [Google Scholar]
- Bashour A. M., Fullerton A. T., Hart M. J., Bloom G. S. (1997). IQGAP1, a Rac- and Cdc42-binding protein, directly binds and cross-links microfilaments. J. Cell Biol. 137, 1555-1566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brancaccio M., Fratta L., Notte A., Hirsch E., Poulet R., Guazzone S., De Acetis M., Vecchione C., Marino G., Altruda F., et al. (2003). Melusin, a muscle-specific integrin beta1-interacting protein, is required to prevent cardiac failure in response to chronic pressure overload. Nat. Med. 9, 68-75 [DOI] [PubMed] [Google Scholar]
- Brown M. D., Sacks D. B. (2006). IQGAP1 in cellular signaling: bridging the GAP. Trends Cell Biol. 16, 242-249 [DOI] [PubMed] [Google Scholar]
- Brown M. D., Sacks D. B. (2009). Protein scaffolds in MAP kinase signalling. Cell Signal. 21, 462-469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casar B., Arozarena I., Sanz-Moreno V., Pinto A., Agudo-Ibanez L., Marais R., Lewis R. E., Berciano M. T., Crespo P. (2009). Ras subcellular localization defines extracellular signal-regulated kinase 1 and 2 substrate specificity through distinct utilization of scaffold proteins. Mol. Cell Biol. 29, 1338-1353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q., Lin T. H., Der C. J., Juliano R. L. (1996). Integrin-mediated activation of MEK and mitogen-activated protein kinase is independent of Ras [corrected]. J. Biol. Chem. 271, 18122-18127 [DOI] [PubMed] [Google Scholar]
- Chen S. Y., Chen H. C. (2006). Direct interaction of focal adhesion kinase (FAK) with Met is required for FAK to promote hepatocyte growth factor-induced cell invasion. Mol. Cell Biol. 26, 5155-5167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Acetis M., Notte A., Accornero F., Selvetella G., Brancaccio M., Vecchione C., Sbroggio M., Collino F., Pacchioni B., Lanfranchi G., et al. (2005). Cardiac overexpression of melusin protects from dilated cardiomyopathy due to long-standing pressure overload. Circ. Res. 96, 1087-1094 [DOI] [PubMed] [Google Scholar]
- DiMichele L. A., Doherty J. T., Rojas M., Beggs H. E., Reichardt L. F., Mack C. P., Taylor J. M. (2006). Myocyte-restricted focal adhesion kinase deletion attenuates pressure overload-induced hypertrophy. Circ. Res. 99, 636-645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franchini K. G., Torsoni A. S., Soares P. H., Saad M. J. (2000). Early activation of the multicomponent signaling complex associated with focal adhesion kinase induced by pressure overload in the rat heart. Circ. Res. 87, 558-565 [DOI] [PubMed] [Google Scholar]
- Giancotti F. G., Ruoslahti E. (1999). Integrin signaling. Science 285, 1028-1032 [DOI] [PubMed] [Google Scholar]
- Guo W., Giancotti F. G. (2004). Integrin signalling during tumour progression. Nat. Rev. Mol. Cell Biol. 5, 816-826 [DOI] [PubMed] [Google Scholar]
- Hood J. D., Frausto R., Kiosses W. B., Schwartz M. A., Cheresh D. A. (2003). Differential alphav integrin-mediated Ras-ERK signaling during two pathways of angiogenesis. J. Cell Biol. 162, 933-943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kehat I., Molkentin J. D. (2010). Extracellular signal-regulated kinase 1/2 (ERK1/2) signaling in cardiac hypertrophy. Ann. NY Acad. Sci. 1188, 96-102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kehat I., Davis J., Tiburcy M., Accornero F., Saba-El-Leil M. K., Maillet M., York A. J., Lorenz J. N., Zimmermann W. H., Meloche S., et al. (2011). Extracellular signal-regulated kinases 1 and 2 regulate the balance between eccentric and concentric cardiac growth. Circ. Res. 108, 176-183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S., Wang Q., Chakladar A., Bronson R. T., Bernards A. (2000). Gastric hyperplasia in mice lacking the putative Cdc42 effector IQGAP1. Mol. Cell Biol. 20, 697-701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra S. K., Hanson D. A., Schlaepfer D. D. (2005). Focal adhesion kinase: in command and control of cell motility. Nat. Rev. Mol. Cell Biol. 6, 56-68 [DOI] [PubMed] [Google Scholar]
- Parsons J. T. (2003). Focal adhesion kinase: the first ten years. J. Cell Sci. 116, 1409-1416 [DOI] [PubMed] [Google Scholar]
- Pearl L. H., Prodromou C., Workman P. (2008). The Hsp90 molecular chaperone: an open and shut case for treatment. Biochem. J. 410, 439-453 [DOI] [PubMed] [Google Scholar]
- Ren J. G., Li Z., Sacks D. B. (2007). IQGAP1 modulates activation of B-Raf. Proc. Natl. Acad. Sci. USA 104, 10465-10469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy M., Li Z., Sacks D. B. (2005). IQGAP1 is a scaffold for mitogen-activated protein kinase signaling. Mol. Cell Biol. 25, 7940-7952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sbroggiò M., Ferretti R., Percivalle E., Gutkowska M., Zylicz A., Michowski W., Kuznicki J., Accornero F., Pacchioni B., Lanfranchi G., et al. (2008). The mammalian CHORD-containing protein melusin is a stress response protein interacting with Hsp90 and Sgt1. FEBS Lett. 582, 1788-1794 [DOI] [PubMed] [Google Scholar]
- Sbroggiò M., Carnevale D., Bertero A., Cifelli G., De Blasio E., Mascio G., Hirsch E., Bahou W. F., Turco E., Silengo L., et al. (2011). IQGAP1 regulates ERK1/2 and AKT signaling in the heart and sustains functional remodeling upon pressure overload. Cardiovasc Res. 91, 456-464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulte T. W., An W. G., Neckers L. M. (1997). Geldanamycin-induced destabilization of Raf-1 involves the proteasome. Biochem Biophys. Res. Commun. 239, 655-659 [DOI] [PubMed] [Google Scholar]
- Shirasu K., Lahaye T., Tan M. W., Zhou F., Azevedo C., Schulze-Lefert P. (1999). A novel class of eukaryotic zinc-binding proteins is required for disease resistance signaling in barley and development in C. elegans. Cell 99, 355-366 [DOI] [PubMed] [Google Scholar]
- Slack-Davis J. K., Eblen S. T., Zecevic M., Boerner S. A., Tarcsafalvi A., Diaz H. B., Marshall M. S., Weber M. J., Parsons J. T., Catling A. D. (2003). PAK1 phosphorylation of MEK1 regulates fibronectin-stimulated MAPK activation. J. Cell Biol. 162, 281-291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vadali K., Cai X., Schaller M. D. (2007). Focal adhesion kinase: an essential kinase in the regulation of cardiovascular functions. IUBMB Life 59, 709-716 [DOI] [PubMed] [Google Scholar]