

Abstract
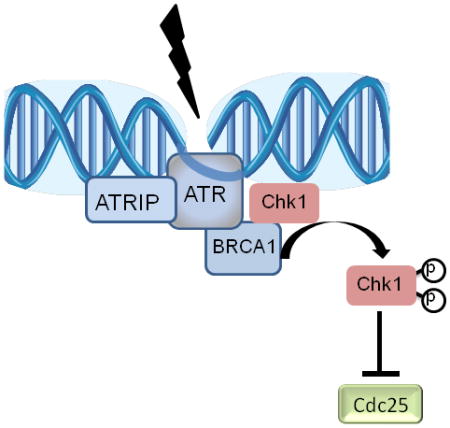
Chk1 phosphorylation by the PI3-like kinases ATR and ATM is critical for its activation and its role in prevention of premature mitotic entry in response to DNA damage or stalled replication. The breast and ovarian tumor suppressor, BRCA1, is among several checkpoint mediators that are required for Chk1 activation by ATM and ATR. Previously we showed that BRCA1 is necessary for Chk1 phosphorylation and activation following ionizing radiation. BRCA1 has been implicated in S-phase checkpoint control yet its mechanism of action is not well characterized. Here we report that BRCA1 is critical for Chk1 phosphorylation in response to inhibition of replication by either cisplatin or hydroxyurea. While Chk1 phosphorylation of S317 is fully dependent on BRCA1, additional proteins may mediate S345 phosphorylation at later time points. In addition, we show that a subset of phosphorylated Chk1 is released from the chromatin in a BRCA1-dependent manner which may lead to the phosphorylation of Chk1 substrate, Cdc25C, on S216 and to S-phase checkpoint activation. Inhibition of Chk1 kinase by UCN-01 or expression of Chk1 phosphorylation mutants in which the serine residues were substituted with alanine residues abrogates BRCA1-dependent cell cycle arrest in response replication inhibition. These data reveal that BRCA1 facilitates Chk1 phosphorylation and its partial chromatin dissociation following replication inhibition that is likely to be required for cycles-phase checkpoint signaling.
Keywords: Chk1, BRCA1, Cell Cycle Checkpoint, Hydroxyurea, Cisplatin, Replication Block
1. Introduction
Cells have to constantly ensure their genomic integrity because of continual environmental and metabolic challenges, including exposure to radiation, chemical agents, and oxidative stress. Cellular surveillance mechanisms are triggered when DNA is damaged or when the replication fork is disrupted to halt cell cycle progression until DNA is repaired(Jackson and Bartek, 2009). The cell cycle checkpoint signaling cascade includes DNA damage sensors, mediators that provide a platform for signal transduction, activated transducers and their effectors leading to cell cycle arrest, and repair and/or other metabolic actions needed. Failing to maintain genomic integrity results in accumulation of multiple deleterious mutations and may lead to cancer development (Jackson and Bartek, 2009, Abraham, 2001). Yet, DNA damaging agents are routinely used in cancer therapy to generate massive DNA damage based on the premise that tumor cells lacking functional surveillance mechanisms will undergo apoptotic cell death. Thus, understanding the DNA damage response is relevant for both cancer prevention and cancer treatment.
ATM and ATR are two protein kinases of the PI-3 protein kinase-like kinase (PIKK) family that activate signaling networks to stall cell cycle progression following DNA damage and replication stallment. Chk1 kinase is a downstream effector of ATR and ATM and is crucial for mediating G2/M arrest following double strand breaks (Ting and Lee, 2004, Yarden et al., 2002). In response to replication stress, Chk1 stabilizes stalled replication forks, delays S-phase replication, and delays mitotic entry (Sorensen and Syljuasen, 2012). More recently, Chk1 was also implicated in mitotic spindle checkpoint (Zachos et al., 2007).
Chk1 phosphorylation and activation following ionizing radiation or replication block was shown to be dependent on several mediators, including ATR-binding protein (ATRIP), TopBP1, claspin, and two protein complexes: the Rad17 and the 9-1–1(Kumagai et al., 2004, Liu et al., 2006, Delacroix et al., 2007, Gatei M, 2003). It has been hypothesized that these mediators provide a platform that facilitates Chk1 phosphorylation by ATM/ATR soon after DNA damage (Chen and Sanchez, 2004). BRCA1 is another mediator/adaptor that facilitates Chk1 activation in response to ionizing radiation (Yarden et al., 2002), but its role in mediating replication stress is less clear.
In this study, we explored the role of BRCA1 in Chk1 activation and signaling in response to S-phase checkpoint and replication arrest. We present herein evidence that BRCA1 is essential for S-phase checkpoint mediated via Chk1 phosphorylation following hydroxyurea (HU; a ribonucleotide reductase inhibitor) and cisplatin (a DNA cross-linking drug). Two serine residues at the carboxyl-terminus of Chk1: S317 and S345 are phosphorylated following impairment of replication progression (Zhao H and 2001). Phosphorylation of S317 is dependent on BRCA1 expression and this dependency is sustained for at least 24 hours following DNA damage. Phosphorylation of S345 is only transiently dependent on BRCA1 expression.
DNA damage –induced Chk1 phosphorylation has recently shown to regulate Chk1 cellular localization with some disagreement as to whether it enhances chromatin binding or causes its release from the chromatin (Smits et al., 2006, Jiang et al., 2003, Niida et al., 2007). In addition, recently it has been suggested that the uncoupling of essential and nonessential functions of Chk1 depends on differential phosphorylation (Wilsker et al., 2008). Here, we show that shortly following DNA damage and replication stress, a subpopulation of phosphorylated Chk1 dissociates from chromatin and expression of functional BRCA1 in the cells is required for proper release of Chk1 that is likely to promote Chk1’s downstream signaling to control cell cycle arrest.
2. Materials and Methods
2.1 Cell culture Drug Treatments, and Transfections
HCC1937, MCF7 cells were grown in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum (Gibco, Grand island, NY), 100 U/ml penicillin, 100 μg/ml streptomycin, and 2 mM L-glutamine in humidified incubator at 37°C and 5% CO2. To induce DNA double strand break damage, cells were treated with10 Gy IR by using a 137Cs source and were let to recover at 37°C for the indicated time points. To induce replication stress, hydroxyurea (HU) or cisplatin (Sigma, St. Louis, MO) were dissolved in ddH2O to a final concentration of 2 mM HU and 5 μg/ml cisplatin. To inhibit Chk1, cells were pretreated with 300 nM UCN-01 one hour prior to DNA damage. UCN-01 was a kind gift from Ed Sausville (NIH, Bethesda, MD). Transient transfections were carried out with Lipofectamine 2000 (Invitrogen, Grand Island, NY) according to manufacturer’s instructions. Different DNA damaging treatments were initiated 24 hours following transfection.
2.2 Plasmids
Full-length wild type Chk1 cDNA was obtained by reverse transciption–PCR-amplification from HeLa-total RNA with primer pairs with BamHI site at the 5′ end and XbaI at the 3′ end: forward 5′-ATAGATGGATCCATGGCAGTGCCCTTTGTG-3′ and reverse: 5′-TAGTAATCTAGATCATGTGGCAGGAAGCCA-3′. Following TA cloning (Invitrogen) the cDNA digested with the above mentioned enzymes and subcloned in frame into pEGFP-C1 (Clontech, Mountain View, CA) that was digested with BglII and XbaI. Phosphorylation mutants were generated by a site-directed mutageneis kit (Agillent, Santa Clara, CA) according to manufacturer instructions. For S317A mutation, the following primer set was used: forward 5′-GTGAAGTACTCCAGTGCTCAGCCAGAACCCCGC-3′, reverse: 5′-GCGGGGTTCTGGCTGAGCACTGGAGTACTTCAG-3′. For S345A mutation, the following primer set was used: forward: 5′-CAAGGGATCAGCTTTGCTCAGCCCACA TGTCC -3′ and reverse: 5′-GGACATGTGGGCTGAGCAAAGCTGATCCCTTG-3′. Ad-BRCA1 and the control virus were described earlier and infections were carried out as described (Yarden et al., 2002). BRCA1 and scrambled-control shRNA plasmids construction in the pSUPER.retro.puro vector were described (Berns et al., 2004). Primer sequences for BRCA1 silencing were: 5′-GTTCAGAGGCAACGAAACT-3′ and for scrambled control: 5′GCTATTGCTGACAGTGTAG-3′. All plasmids were sequence verified.
2.3 Cell Extracts, Western Blots, and Antibodies
Whole cell extracts were prepared as previously described (Yarden and Brody, 1999). Briefly, cells were harvested and washed twice with ice-cold PBS and then lysed in NTEN buffer (150 mM NaCl, 20 mM Tris-Cl pH=7.5, 1 mM EDTA, 1 mm Sodium Fluoride (NaF) [Sigma, St. Louis, MO], 1mm Sodium Vanedate (Na3VO4) [Sigma, St. Louis, MO], and protease inhibitors [EMD, Billerica, MA]). Extracts were clarified by centrifugation at 16,000 xg for 20 min at 4°C, and soluble fractions were collected. Protein concentrations were determined by BCA assay (ThermoScientific, Rockford, IL). 50 μg of total cell lysate was separated on SDS-PAGE gel and transferred onto nitrocellulose membranes (Invitrogen, Grand Island, NY). Membranes were blocked with 5% BSA and then incubated with the indicated antibodies at room temperature for 1 h. Signals were detected using an ECL Western Blotting Kit (ThermoScientific, Rockford, IL). The primary antibodies used in this study are anti Chk1, anti-Chk2, anti- GAPDH (Santa-Cruz Biotechnology, Santa Cruz, CA), anti–Chk1-S317, anti-Chk1-S345, anti-Cdc25C Ser-216 (Cell Signaling Technology, Danvers, MA), anti-BRCA1 (Ab-1, EMD, Billerica, MA), anti-Orc2, and anti-Tubulin (ThermoScientific, Rockford, IL). To verify protein phosphorylation, HU-treated cell extracts were incubated with increasing amounts of the enzyme λ-phosphatase (New England Biolabs, Ipswich, MA) and in the presence or absence of 2 mM Na3VO4 (Sigma, St. Louis, MO) prior to separation via SDS-PAGE.
2.4 Chromatin Fractionation
Chromatin fractionation was performed essentially as described (Zou et al., 2002, Mendez and Stillman, 2000). The preparation was carried from duplicate samples to enable presentation of all fractions. Approximately 1 × 107 cells were washed in PBS and resuspended in 200 μl of solution A (10 mM Hepes [pH 7.9], 10 mM KCl, 1.5 mM MgCl2, 0.34 M sucrose, 10% glycerol, 1 mM DTT, and protease and phosphatase inhibitors). Triton X-100 was added to a final concentration of 0.1%, cells were incubated on ice for 5 min, and the cytoplasmic (S1) and nuclear fractions (P1) were harvested by centrifugation at 1,300 × g for 4 min. Isolated nuclei were then washed in solution A, lysed in 150 μl solution B (3 mM EDTA, 0.2 mM EGTA, 1 mM DTT, and protease and phosphatase inhibitors), and incubated on ice for 10 min. The soluble nuclear (S2) and chromatin fractions (p2) were harvested by centrifugation at 1,700 × g for 4 min. Isolated chromatin was washed once with solution B and spun down at high speed (10,000 × g for 1 min). Finally, chromatin was resuspended in 150 μl of SDS sample buffer and sheared by sonication. Protein concentrations were determined by BCA assay (ThermoScientific, Rockford, IL). Equal amounts of protein from whole cell extracts or from the different cellular fractions were mixed with 5 × laemmli buffer (Bio-Rad, Hercules, CA) and samples were loaded onto SDS-PAGE and transferred to nitrocellulose membranes. Membranes were blocked, incubated with the indicated antibodies, and developed with enhanced chemiluminescence (ThermoScientific, Rockford, IL). Equal protein loading and transfer efficiency were determined by Ponceau Red staining (Sigma, St. Louis, MO) after electro-transfer and α-Tubulin/GAPDH detection.
2.5 Cell Cycle Analysis
MCF7 cells were transfected with empty pEGFP vector (clontech, Mountain View, CA) or with pEGFP-Chk1 vectors encoding for wild type or mutant Chk1recombinant proteins. On the following day, cells were exposed to 10 Gy of IR and were let to recover for additional 24 hours. HCC1937 cells were infected with either Ad-BRCA1 or Ad-control vectors and 24 hours later were treated with 2 mM HU. Cells were harvested 1 hour or 24 hours following DNA damage treatments. One group of cells was pretreated with UCN-01 as described above 1 hour prior to HU treatment. Cell cycle analysis of IR- or HU- treated and untreated cells was carried out following fixation in 70% EtOH and staining with 40 μg/ml propidium iodide by fluorescence-activated cell sorting (FACS) using a FACSCalibur instrument (Beckon Dickinson, Mountain View, CA,) and BD CellQuest™ Pro (Yarden et al., 2002).
2.6 Immunofluorescence
Cells were grown on glass coverslips and treated with 2 mM HU for 2 hrs. Cells were washed once with PBS and subsequently fixed with 4% paraformaldehyde for 15 minutes and permeablized with 0.2% Triton –X-100 for 15 minutes. Samples were blocked in PBS with 10% goat serum and then stained as described (Yarden and Brody, 1999). Polyclonal anti-Cdc25C antibody was diluted 1:350 and detected by anti-rabbit IgG conjugated with Dylight 594 diluted 1:1000 (ThermoScientific, Rockford, IL). Images were viewed with × 63 objective lens with an Olympus Ax70 microscope and captured by a CCD digital camera.
3. Results
3.1 BRCA1-Dependent Chk1 Phosphorylation following Replication Stress
Given that Chk1 is required for S-phase checkpoint activation in response to DNA replication inhibitors, we considered whether the mediator, BRCA1, is required for Chk1 function in a similar fashion to its role following ionizing radiation (IR)(Yarden et al., 2002). HCC1937, breast cancer cells null of functional BRCA1, were infected with wild type full-length cDNA of BRCA1 or a control-empty adenovirus expression vector. BRCA1-deficient and proficient cells were treated with 2 mM HU and Chk1 expression was analyzed by immunoblot analysis 1 hour and 24 hours after treatment. As a control for specificity of Chk1 response, cells were pretreated with 300 nM Chk1-selective inhibitor, UCN-01 (a gift from Dr. Ed Sausville, NCI) for 1 hour prior to HU (Sausville, 2003). Immunoblot analysis with antibody against total Chk1 protein detects also phosphorylated Chk1 as the slower migrating band of Chk1. Baseline phosphorylation of Chk1 is detected in control untreated cells but is further induced in a BRCA1-dependent manner in HCC1937-BRCA1 cells and not in parental HCC1937 or HCC1937 vector-only cells as early as one hour after treatment of cells with HU. Chk1 induced phosphorylation is sustained for at least 24 hours following exposure to HU. No phosphorylation of Chk1 detected when BRCA1-expressing cells were pretreated with Chk1 inhibitor, UCN-01 (Fig 1A).
Fig. 1. BRCA1 expression is required for Chk1 phosphorylation on S317 and S345.
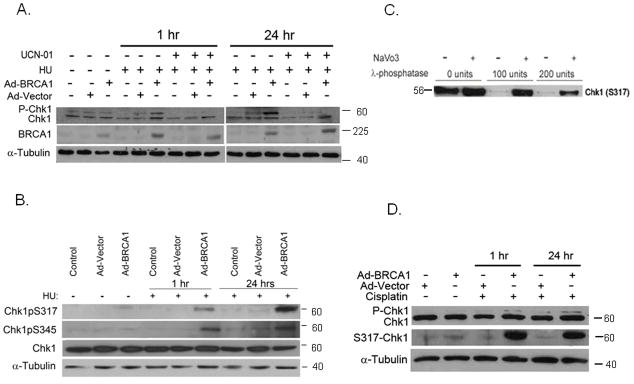
A. HCC1937, BRCA1-null breast cancer cells, are mock infected or infected with either control adenovirus vector (Ad-vector) or with full-length, wild type BRCA1-containing adenovirus (Ad-BRCA1). After 24 hours, cells were pretreated with 300 nM UCN-01 and then with 2 mM HU and harvested at 1 hr or 24 hr following treatment. Chk1 and α-Tubulin expression was determined by western blot analysis.
B. HCC1937 cells were transfected as before. Expression of S345 and S317 Chk1 (phosphorylated Chk1) and total Chk1was determined by western blot analysis relative to α-Tubulin expression.
C. HCC1937-BRCA1 cells were treated with 2mM of HU and then harvested after 1 hour. Total cell extracts were treated in vitro with increasing amounts of the enzyme λ-phosphatase in the presence of the phosphatase inhibitor NA-Vanadate (2 mM) for 1 hr prior to western blot analysis with an antibody against phosphorylated Chk1.
D. HCC1937 cells were infected with Ad-vector or Ad-BRCA1 as described and 24 hr later treated with Cisplatin (5 μg/ml) for either 1 or 24 hours. Chk1, S317-Chk1 and α-Tubulin expression was determined by western blot analysis.
Two conserved SQ phosphorylation sites at the regulatory C-terminus Chk1 appear to be particularly important for DNA damage response: S317/Q318 and S345/Q346. Using Chk1 phosphorylation-specific antibodies directed against S317 or S345 epitopes, we show that these S317 and S345 sites are phosphorylated shortly after exposure to HU in a BRCA1-dependent manner. While BRCA1-dependent phosphorylation on S317 is sustained for at least 24 hours post-HU treatment, S345 phosphorylation may not depend solely on BRCA1 expression and additional proteins may be important for its continued phosphorylation state at later time points (Fig 1B).
To confirm the specificity of Chk1 phosphorylation signal, total cell extract harvested from HU-treated BRCA1-expressing cells (as described above) was in vitro treated with increasing amounts of the phosphorylation inhibitor, λ-phosphatase, in the presence or absence of the phosphatase inhibitor NaVO3. Chk1-pS317 phosphorylation was blocked in the presence of λ-phosphatase and restored in the presence of NaVO3 indicating the specificity of the signal detected by the antibody directed against S317 (Fig 1C).
Next, we analyzed whether BRCA1 plays a role in mediating Chk1 phosphorylation in response to an additional inhibitor of the replication, cisplatin, that induces bulky adducts in the DNA. We show that Chk1 is phosphorylated only in BRCA1-expressing cells that were treated with 5μg/ml Cisplatin for 1 hour but not in BRCA1-deficient cells (Fig 1D). Following 24 hr treatment, more prominent phosphorylation of Chk1 is detected in BRCA1-expressing cells while less significant phosphorylation of Chk1 is independent of BRCA1 (Fig 1D).
To confirm the generality of BRCA1 role in Chk1 and checkpoint activation, we silenced BRCA1 expression in MCF7 cells with specific BRCA1-shRNA or with scrambled-BRCA1 shRNA and exposed the cells to 2 mM HU. Chk1 expression remains constant regardless of BRCA1-specific shRNA transfection or exposure to replication block (Fig 2A). However, phosphorylation of both serine residues S317 and S345 following exposure to 2 mM HU is dependent on BRCA1 expression as no induction was detected in BRCA1-silenced MCF7 cells (Fig 2A). However, at 24 hours following HU exposure, phosphorylation of S345 was detected also in BRCA1-deficient cells, suggesting that additional mediators are involved in regulation of Chk1 phosphorylation at later time points. The inhibitory phosphorylation of Cdc25C, the downstream target of Chk1, on Serine residue 216 was more prominently detected in BRCA1-expressing cells than in BRCA1-silenced cells, despite somewhat lower levels of total Cdc25C protein, suggesting that BRCA1 expression is required for Chk1 activation. Chk2 phosphorylation following 2 mM HU was independent of BRCA1 expression (Fig 2B). Similarly, we investigated the role of BRCA1 on checkpoint signaling following treatment of cells with 5 μg/ml cisplatin. Chk1 expression did not change in response to BRCA1 silencing or cisplatin. However, Chk1 phosphorylation on S317 and S345 was specifically induced only in BRCA1-expressing cells and not in BRCA1-silenced cells (Fig 2B). Here again, the inhibitory phosphorylation of Cdc25C on S216 was more noticeable in BRCA1-expressing cells than in BRCA1-silenced cells (Fig 2B). Taken together, we show that early phosphorylation of Chk1following replication stress induced by HU or cisplatin is dependent on BRCA1 expression.
Fig. 2. BRCA1 expression is required for Chk1 signaling.
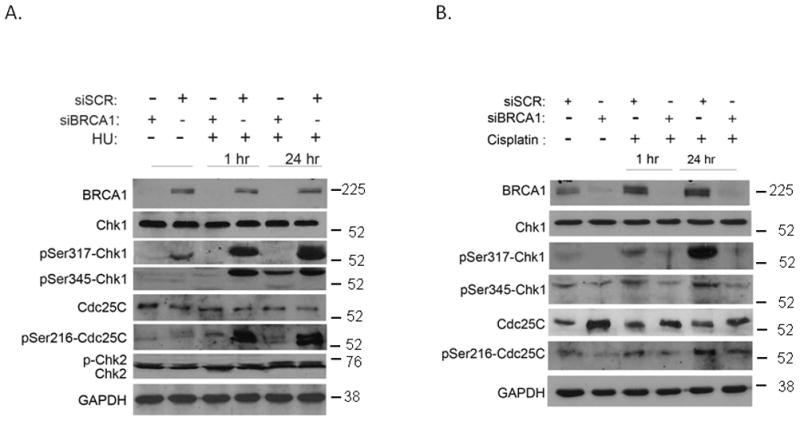
A. MCF7 cells were infected with scrambled or BRCA1-specific shRNA retroviral vectors and 24 hours later were treated with 2 mM HU for 1 hour or 24 hours. The expression of BRCA1, Chk1, Chk2, Cdc25C, and GAPDH was analyzed by western blot as indicated.
B. Control and BRCA1-silenced MCF7 cells were treated with 5 μg/ml Cisplatin for either 1 hour or 24 hours. Expression and phosphorylation levels of Chk1, Cdc25C, and GAPDH were determined by western blot analysis as indicated. Experiments were repeated three times and a typical experiment was presented.
3.2 BRCA1-Dependent Chk1 Activation Is Required for S-Phase Arrest
To determine whether BRCA1-dependent phosphorylation of Chk1 is important for S-phase checkpoint, we analyzed the cell cycle distribution of BRCA1-deficient and proficient HCC1937 cells following exposure to 2mM HU. BRCA1-deficient cells displayed an impaired cell cycle checkpoints control and were unable to arrest in response to perturbations in DNA replication. Expression of full-length wild type BRCA1 restored checkpoint function and cells were arrested at S-phase following exposure to HU (Fig 3). Pretreatment of BRCA1-proficient cells with 300 nM UCN-01 for 1 hour before the exposure of cells to HU compromised the S-phase arrest, suggesting that, at least in part, Chk1 is important for BRCA1-mediated cell cycle arrest during S phase (Fig 3).
Fig. 3. BRCA1 is required for HU-induced S-phase arrest.
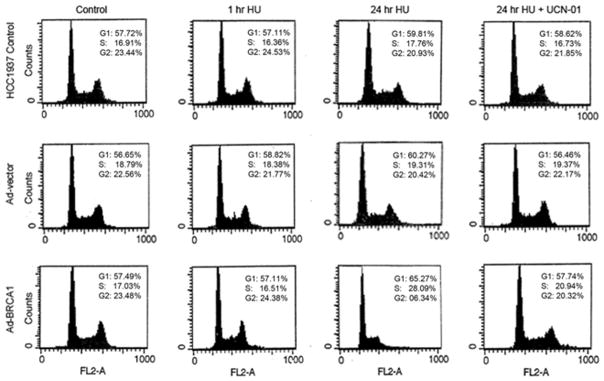
C. BRCA1-deficient and proficient HCC1937 cells were pretreated with 300 nM UCN-01for 1 hour prior to treatment with 2 mM HU for 24 hours. Cells were harvested and analyzed for cell cycle progression. Experiment was repeated three times and a typical experiment is presented.
3.3 Differential Requirements of Individual SQ Residues Phosphorylation within Chk1
To further understand the relationship between the two SQ phosphorylation sites, S317 and S345, we analyzed the phosphorylation kinetics of recombinant GFP-Chk1 proteins in which serine residues were replaced with alanine residues at positions S317A and S345A. Transfection efficiency was equal for wild type and mutant Chk1 constructs (Fig 4A) and confirmed to be more than 85% (data not shown). Transfected cells expressing either wild type or mutant GFP-Chk1 proteins were treated with 2 mM HU and harvested at different time points after treatment beginning as soon as 10 minutes and up to 2 hours. Immunoblot analyses with antibodies directed against the total Chk1 protein or against the S317 phosphorylation epitope show an increase in Chk1 S317 phosphorylation in the endogenous and GFP-Chk1 following HU (Fig 4B). S317 phosphorylation was stimulated in the endogenous Chk1 protein and in the recombinant GFP-Chk1 protein as soon as 10 minutes following exposure to HU (Fig 4B). As expected, no stimulation of S317 phosphorylation was detected when cells were transfected with S317A mutant GFP-Chk1. To our surprise, phosphorylation of the endogenous Chk1 protein was not stimulated following HU in these cells (Fig 4B). Interestingly, levels of S317 phosphorylation did not increase in cells expressing the GFP-Chk1S345A mutant protein following HU (Fig 4B), suggesting that Chk1 phosphorylation is a cooperative process and phosphorylation of S345 may precede phosphorylation of S317 and both are required for Chk1-dependent DNA damage responses (Katsuragi and Sagata, 2004).
Fig. 4. Analysis of Serine phosphorylation sites in Chk1 in response to HU.
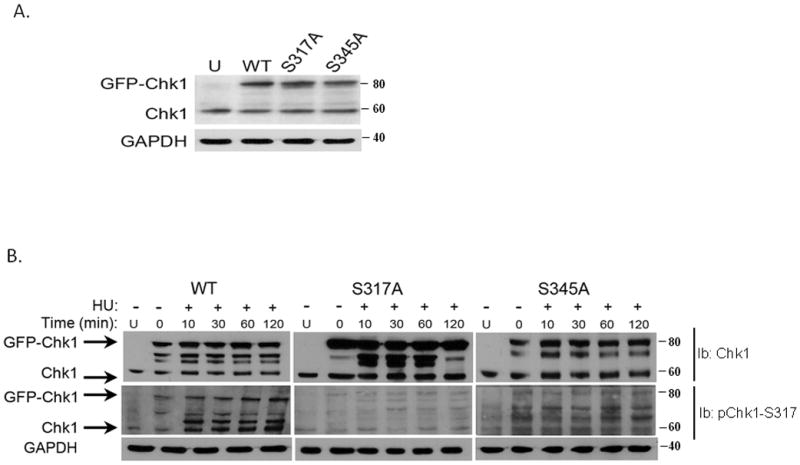
A. Recombinant wild type pEGFP-Chk1 and the phosphorylation mutants: pEGFP-Chk1-S317A and pEGFP-Chk1-S345A plasmids were transiently transfected into MCF7 cells and expression of endogenous and recombinant proteins was detected by western blot analysis.
B. MCF7 cells were transiently transfected with wild type or phosphorylation mutants of Chk1 and 24 hours after transfection, cells were treated with 2 mM HU for the indicated time points. The expression and phosphorylation levels of Chk1 were examined by western blot analysis with phospho-Chk1-specific antibodies and anti-Chk1 antibody.
To determine whether expression of Chk1 phosphorylation mutants S317A and S345A will affect cellular response to DNA damage, we analyzed the cell cycle distribution of MCF7 cells expressing empty vector (pEGFP), wild type GFP-Chk1, or GFP-Chk1 mutants S317A and S345A 24 hours following IR. As expected, cells expressing wild type Chk1 or an empty vector control arrest at the G2/M checkpoint and do not progress through the cell cycle (Fig 5A). However, cells expressing the GFP-S317A mutant are unable to arrest and the distribution of these cells at the different cell cycle phases resembles untreated cells. Cells that express the GFP-S345A mutant exhibit an intermediate response and there is some increased occupancy of cells at the G2/M phase that coincide with decreased occupancy of cells in the G1 phase of the cell cycle (Fig 5A). The intermediate phenotype may result from this mutant ability to suppress the phosphorylation of the recombinant protein but not the endogenous Chk1.
Fig. 5. Phosphorylation mutants of Chk1 abrogate the cell cycle checkpoint control in response to IR or HU.
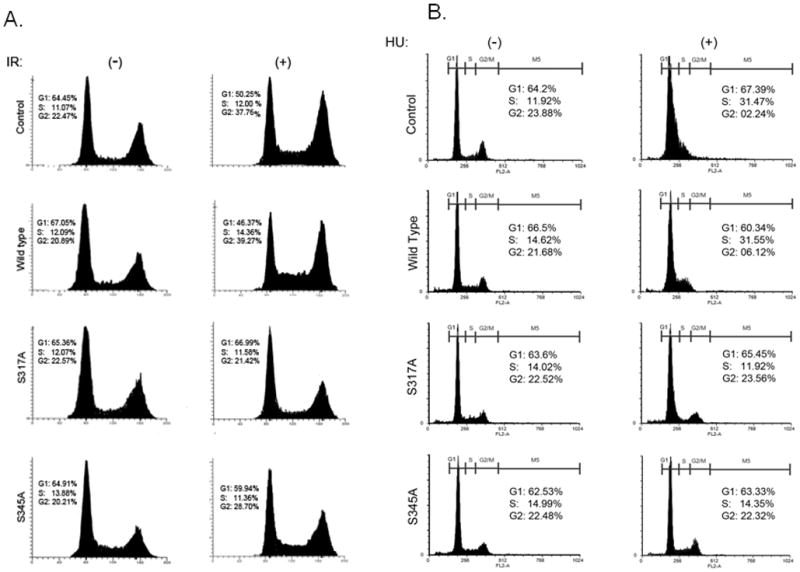
A. MCF7 cells were transiently transfected with wild type and phosphorylation mutants of Chk1 and 24 hours after transfection were exposed to 10 Gy of IR and followed by analysis of cell cycle distribution.
B. MCF7 cells were transiently transfected with wild type and phosphorylation mutants of Chk1 and 24 hours after transfection were exposed to 2 mM of HU and followed by analysis of cell cycle distribution.
Similarly, following expression of wild type and mutant GFP-Chk1, mutant S317A and S345A- expressing cells were unable to arrest at S-phase in response to HU, and thus exhibited an impaired cellular checkpoint (Fig 5B).
3.4 Chk1 Phosphorylation is Required for Its Dissociation from Chromatin following DNA Damage
Previously, we showed that BRCA1 colocalizes with Chk1 in nuclear foci. Upon DNA damage induced by IR, we noticed dispersement of Chk1 from the distinct foci to smaller foci (Yarden et al., 2002). In light of a recent debate on the cellular localization of Chk1 following DNA damage in which Jiang et al. (Jiang et al., 2003) reported that Chk1 binds to DNA breaks, while Smits et al. and Shimada et al. (Smits et al., 2006, Shimada et al., 2008) reported that Chk1 dissociates from the chromatin upon DNA damage, we were interested to find out whether BRCA1 plays a role in Chk1 subnuclear localization. We fractionated extracts of MCF7 cells by the centrifugation-based method that was developed by Mendez and Stillman (Mendez and Stillman, 2000). We analyzed the cytoplasmic (S1), nuclear (P1) soluble nuclear (S2) and chromatin-enriched (P2) fractions for the presence of Chk1 protein before exposure to DNA damage and at different time points following exposure to IR or HU (Fig 6A and 6B). Chk1 was detected by immunoblot analysis mostly in P1 and P2 fractions and to a lesser extent in the fractions S1 and S2 of naive untreated cells (Fig 6A). We did not observe significant changes in total Chk1 localization following exposure of cells to 10 Gy IR or 2 mM HU, and the majority of the protein was still localized in the insoluble chromatin fraction of the nucleus (P1+P2). However, following DNA damage (HU and IR), there was a time-dependent increase of the phosphorylated subpopulation of Chk1 in the soluble fraction of the nucleus (S2), as determined by immunoblot analysis with antibody directed to the Ser317 epitope (Fig 6A and 6B), suggesting that phosphorylated Chk1 dissociates from the chromatin following DSBs and replication stress. To determine whether BRCA1 affects the affinity of Chk1 to chromatin, we expressed the wild type recombinant protein GFP-Chk1 in BRCA1-deficient and proficient MCF7 cells (Fig 6B and 6C) and analyzed the localization of Chk1 in the different cellular fractions following HU by immunoblotting Chk1 with GFP antibody. As expected, GFP-Chk1 like endogenous Chk1 can be found both in the soluble and insoluble fractions of the nucleus in BRCA1-proficient cells (Fig 6B). Yet, no immobilization of Chk1 from the insoluble to the soluble fraction of the nucleus was observed in BRCA1-silenced cells following exposure of cells to 2 mM HU (Fig 6C). No phosphorylation on Ser317 was detected (data not shown).
Fig. 6. Chk1 dissociation from chromatin requires its phosphorylation.
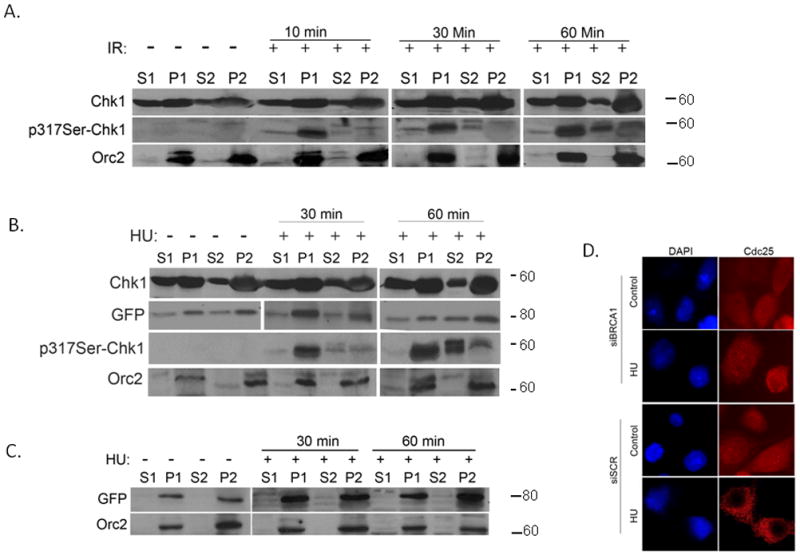
A. MCF7 cells were exposed to 10 Gy of IR and then were harvested at the indicated recovery times. Cells were fractionated as described by Zou et al. and Mendez et al. (Zou et al., 2002, Mendez and Stillman, 2000) to obtain the cytoplasmic and nuclear fractions and were further fractionated to obtain the nucleoplasm and the insoluble chromatin fraction. The occupancy of Chk1 in the different fractions was examined by western blot analysis with anti-Chk1 and phospho-specific Chk1 antibodies. Orc2 confirmed the quality of the nuclear fractionations.
B. MCF7 cells were transfected with pEGFP-Chk1 and 24 hours later were exposed to 2 mM HU. Cells were harvested at the indicated recovery times. The occupancy of endogenous Chk1 and GFP-Chk1 in the chromatin was examined by western blot analysis with anti-Chk1 and anti-GFP antibodies. The occupancy of phosphorylated Chk1 was examined with phospho-specific Chk1 antibody against S317 as described above. Orc2 confirmed the quality of the nuclear fractionations.
C. BRCA1-silenced MCF7 cells were transfected with pEGFP-Chk1 and 24 hours after transfection, the cells were treated with 2 mM HU. Cells were harvested at the indicated time points and the occupancy of GFP-Chk1 in the different fractions was examined with an anti-GFP antibody.
D. MCF7 cells were transfected with shBRCA1 or shSCR silencing vectors. Twenty four hours after transfection cells were left untreated or were treated with 2mM HU. After 2 hours cells were fixed and immunostained with Cdc25C antibody.
Taken together, our results suggest that Chk1 is associated with chromatin prior to DNA damage or stress. Exposure to DNA damage or replication stress triggers the rapid dissociation of a subpopulation of phosphorylated Chk1 from the chromatin. In the absence of BRCA1 and proper phosphorylation, Chk1 dissociation from the chromatin is impaired. Our data establish that BRCA1 is critical for Chk1 phosphorylation and migration from chromatin to the soluble fraction of the nucleus (Fig 6C). To determine whether the downstream target of Chk1, Cdc25C, is immobilized in response to replication stress we carried out immunoflouresence assays. Unlike Chk1, Cdc25C does not associate with chromatin and it shuttles between the soluble fraction of the nucleus and the cytosol upon the cell cycle. Here we show that Cdc25C is evenly distributed between the nucleus and the cytosol in MCF7 cells silenced for BRCA1 and treated with HU. No down regulation of Cdc25C expression levels was detected (Fig 6D). Conversely, in MCF7 cells that were transfected with the non-silencing, scrambled, shRNA and express wild type BRCA1, Cdc25C expression levels were down regulated and the protein is sequested to the cytosol (Fig. 6D). This data suggest that Chk1 phosphorylation and activation is likely to be required for Cdc25C immobilization following replication stress. However, other mechanisms such BRCA1 E3 ligase mediated ubiquitination and degradation via and the proteasome cannot be excluded (unpublished data).
4. Discussion
Chk1 activation is a critical step in the cellular response to DNA double strand breaks and replication stress. Cell cycle checkpoint control, mediated by Chk1 activation, allows for DNA repair, maintenance of replication fork stability, and, most importantly, suppression of unscheduled mitotic entry. Yet, Chk1 is not only activated in response to exogenous DNA damaging agents, but exhibits basal level of activity during normal S phase progression (Sorensen and Syljuasen, 2012). In this study we investigated the role of the breast and ovarian tumor suppressor, BRCA1, in regulation of Chk1 phosphorylation and activation as well as its impact on cell cycle checkpoint regulation. Upon DNA damage/replication block, Chk1 is phosphorylated by DNA damage sensors, members of the PIKK family, and it relays the signal to multiple downstream substrates that promote a variety of cellular functions. In the context of replication stress, it is believed that ATR is the prominent initiating signal. The mechanism of how ATR contacts and phosphorylates Chk1 during the DNA damage response is unclear. Data from a variety of model systems have demonstrated that different mediators including Claspin, TopBP1, Hus1, rad17, and, most recently, Timeless and Tipin are required for ATR-mediated Chk1 phosphorylation in response to replication block (Liu et al., 2006, Wang et al., 2006, Kemp et al.). Deficiency in any of these mediator proteins abrogates Chk1 phosphorylation and checkpoint activation leading to the hypothesis that the mediator proteins provide a platform for the interaction between ATR and Chk1. Previously, we demonstrated that the tumor suppressor protein, BRCA1, associates with Chk1 and is required for Chk1 and G2/M checkpoint activation following IR (Yarden et al., 2002). Here we show that BRCA1 is important for activation of Chk1 and intra-S phase checkpoint following replication stress.
It has been suggested that ATR-mediated phosphorylation at the Carboxyl terminus SQ/TQ sites of Chk1 cause unfolding of the protein so the auto-inhibitory domain cannot inhibit the kinase domain. It is possible that a multi-mediator complex either loosens the inhibitory intramolecular interaction or stabilizes the open conformation of Chk1 to sustain its activation. Alternatively, the specificity of the different mediators could reflect tissue type and model systems.
Analysis of Chk1 phosphorylation kinetics revealed a complex regulation. We found that two of Chk1 regulatory phosphorylation sites S317 and S345 are rapidly phosphorylated in response to replication block in a BRCA1-dependent manner. Yet, we detected residual phosphorylation in naive cells as well as in BRCA1-deficient cells. This phosphorylation may account for the basal level of activity during normal replication (Sorensen and Syljuasen, 2012). While phosphorylation of S317 is sustained for 24 hours and is dependent on BRCA1 expression, S345 eventually became phosphorylated even in the absence of BRCA1 expression. Moreover, substitution of SQ site S345 with non-phosphorylatable alanine residue, S345A, interfered with phosphorylation of S317 site in response to replication block. Taken together, our results suggest that basal level of S345 phosphorylation is key for enabling phosphorylation on adjacent SQ site and for promoting induction of Chk1 following inhibition of replication. Our results are in agreement with recent findings reported by Walker et al. (Walker et al., 2009) that S345A substitution rendered Chk1 incapable of restoring checkpoint response in DT40 cells. However, unlike Walker’s suggestion that substitution of SQ site S317 has a more subtle effect on cell cycle checkpoint, we found that S317A substitution prevents not only its own phosphorylation, but also interferes with the phosphorylation of the endogenous Chk1 molecules. Furthermore, it completely abrogated IR-induced G2/M checkpoint activation. These results could suggest intermolecular interactions of Chk1 as a homodimer and a dominant effect of the mutant protein. Shann and Hsu (Shann and Hsu, 2001) showed that carboxyl-terminal half of rat Chk1 can physically interact and inhibit the kinase domain activity of full length Chk1. Yet Katsuragi and Sagata (Katsuragi and Sagata, 2004) reported that transfection of the carboxyl domain of Xenopus Chk1 (adjacent to the SQ sites) to Xenopus oocytes can inhibit the ectopically expressed kinase domain but not the full length of Chk1. Further studies might be required because it was reported recently that strong denaturing conditions of RIPA buffer also induced Chk1 kinase activity of wild type and S345A mutant protein relative to regular lysis buffer (Katsuragi and Sagata, 2004).
Despite carrying similar functions in checkpoint control and in the DNA damage response as Chk1 (Ahn JY, 2000), we did not observe any dependency of Chk2 phosphorylation on BRCA1 expression. These results should not be a surprise because it was shown that Chk2 acts upstream of BRCA1 in the signaling cascade and in fact phosphorylates BRCA1 in an ATM manner (Lee et al., 2000).
Our data is in agreement with data reported by Smits et al and Shimida et al and clearly show that Chk1 associates with chromatin, even in naive, unperturbed cells. Expression of BRCA1 does not modulate the chromatin binding of total Chk1 in naive cells, but rather regulates the release of phosphorylated Chk1 from the chromatin shortly after exposure to DNA damage and replication inhibitors. In the absence of BRCA1 expression, Chk1 phosphorylation is attenuated and thus no release of Chk1 from chromatin to the nucleoplasm is observed, which may explain the faulty checkpoint activation. Our results are mostly in agreement with Smits et al. (Smits et al., 2006) and Shimada et al. (Shimada et al., 2008) that reported that in U2OS cells Chk1 is released from the chromatin in response to DNA damage. However, inconsistent with these results, we report here that in MCF7 cells only a subpopulation of Chk1 that is phosphorylated is released from the chromatin while unphosphorylated Chk1 remains bound to chromatin. It will be interesting to identify additional functions of Chk1 in the context of chromatin. Taken together, we report here that the kinetics of Chk1 phosphorylation shortly after DNA damage and replication arrest is dependent on expression of functional BRCA1. The kintetics of Chk1 phosphorylation and activation is complex and a cooperative mechanism between the two main phosphorylation sites S345 and S317 may be required for displacement from the chromatin and full cell cycle arrest. The dissociation of phosphorylated Chk1 from the chromatin provides a mechanism for relaying the damage/stress recognition to downstream targets such as Cdc25 phosphatases that shuttle between the cytoplasm and the nucleus to activate cell cycle checkpoint control (Graves et al., 2001). Once released from the chromatin, phosphorylated Chk1 may be primed for recycling to ensure checkpoint recovery.
Acknowledgments
We thank Drs. Tiffany A. Frey and Sarah W. Kehoe for assistance in generating the Chk1 constructs. We thank Dr. Lawrence C. Brody and members of our lab for helpful discussion during the course of this work. This work was supported by a career development award from the Israel Cancer Research Fund to RIY and by grants from The Israeli Health Ministry and Israel Cancer Association to RIY and MZP.
Abbreviations
- ATM
Ataxia Telangiectasia Mutated
- ATR
Ataxia Telangiectasia Mutated and RAD3-related kinase
- EGFP
Enhanced Green Fluorescence Protein
- HU
Hydroxyurea
- IR
Ionizing radiation
- PI
Propidium Idodide
- PI3K
Phosphoinosiditide 3-Kinases
References
- Abraham Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev. 2001;15:2177–2196. doi: 10.1101/gad.914401. [DOI] [PubMed] [Google Scholar]
- Ahn JYSJ, Piwnica-Worms H, Canman CE. Threonine 68 phosphorylation by ataxia telangiectasia mutated is required for efficient activation of Chk2 in response to ionizing radiation. Cancer Res. 2000;60(21):5934–5936. Related Articles, Links. [PubMed] [Google Scholar]
- Berns K, Hijmans EM, Mullenders J, Brummelkamp TR, Velds A, Heimerikx M, Kerkhoven RM, Madiredjo M, Nijkamp W, Weigelt B, Agami R, Ge W, Cavet G, Linsley PS, Beijersbergen RL, Bernards R. A large-scale RNAi screen in human cells identifies new components of the p53 pathway. Nature. 2004;428:431–437. doi: 10.1038/nature02371. [DOI] [PubMed] [Google Scholar]
- Chen Y, Sanchez Y. Chk1 in the DNA damage response: conserved roles from yeasts to mammals. DNA Repair (Amst) 2004;3:1025–1032. doi: 10.1016/j.dnarep.2004.03.003. [DOI] [PubMed] [Google Scholar]
- Delacroix S, Wagner JM, Kobayashi M, Yamamoto K, Karnitz LM. The Rad9-Hus1-Rad1 (9-1-1) clamp activates checkpoint signaling via TopBP1. Genes Dev. 2007;21:1472–1477. doi: 10.1101/gad.1547007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatei MSK, Sorensen C, Syljuasen R, Falck J, Hobson K, Savage K, Lukas J, Zhou BB, Bartek J, Khanna KK. Ataxia-telangiectasia-mutated (ATM) and NBS1-dependent phosphorylation of Chk1 on Ser-317 in response to ionizing radiation. J Biol Chem. 2003;278:14806–14811. doi: 10.1074/jbc.M210862200. [DOI] [PubMed] [Google Scholar]
- Graves PR, Lovly CM, Uy GL, Piwnica-Worms H. Localization of human Cdc25C is regulated both by nuclear export and 14-3–3 protein binding. Oncogene. 2001;20:1839–1851. doi: 10.1038/sj.onc.1204259. [DOI] [PubMed] [Google Scholar]
- Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461:1071–1078. doi: 10.1038/nature08467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang K, Pereira E, Maxfield M, Russell B, Goudelock DM, Sanchez Y. Regulation of Chk1 includes chromatin association and 14-3–3 binding following phosphorylation on Ser-345. J Biol Chem. 2003;278:25207–25217. doi: 10.1074/jbc.M300070200. [DOI] [PubMed] [Google Scholar]
- Katsuragi Y, Sagata N. Regulation of Chk1 kinase by autoinhibition and ATR-mediated phosphorylation. Mol Biol Cell. 2004;15:1680–1689. doi: 10.1091/mbc.E03-12-0874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemp MG, Akan Z, Yilmaz S, Grillo M, Smith-Roe SL, Kang TH, Cordeiro-Stone M, Kaufmann WK, Abraham RT, Sancar A, Unsal-Kacmaz K. Tipin-replication protein A interaction mediates Chk1 phosphorylation by ATR in response to genotoxic stress. J Biol Chem. 285:16562–16571. doi: 10.1074/jbc.M110.110304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumagai A, Kim SM, Dunphy WG. Claspin and the activated form of ATR-ATRIP collaborate in the activation of Chk1. J Biol Chem. 2004;279:49599–49608. doi: 10.1074/jbc.M408353200. [DOI] [PubMed] [Google Scholar]
- Lee JS, Collins KM, Brown AL, Lee CH, Chung JH. hCds1-mediated phosphorylation of BRCA1 regulates the DNA damage response. Nature. 2000;404:201–204. doi: 10.1038/35004614. [DOI] [PubMed] [Google Scholar]
- Liu S, Bekker-Jensen S, Mailand N, Lukas C, Bartek J, Lukas J. Claspin operates downstream of TopBP1 to direct ATR signaling towards Chk1 activation. Mol Cell Biol. 2006;26:6056–6064. doi: 10.1128/MCB.00492-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendez J, Stillman B. Chromatin association of human origin recognition complex, cdc6, and minichromosome maintenance proteins during the cell cycle: assembly of prereplication complexes in late mitosis. Mol Cell Biol. 2000;20:8602–8612. doi: 10.1128/mcb.20.22.8602-8612.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niida H, Katsuno Y, Banerjee B, Hande MP, Nakanishi M. Specific role of Chk1 phosphorylations in cell survival and checkpoint activation. Mol Cell Biol. 2007;27:2572–2581. doi: 10.1128/MCB.01611-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sausville EA. Cyclin-dependent kinase modulators studied at the NCI: pre-clinical and clinical studies. Curr Med Chem Anticancer Agents. 2003;3:47–56. doi: 10.2174/1568011033353560. [DOI] [PubMed] [Google Scholar]
- Shann YJ, Hsu MT. Cloning and characterization of liver-specific isoform of Chk1 gene from rat. J Biol Chem. 2001;276:48863–48870. doi: 10.1074/jbc.M108253200. [DOI] [PubMed] [Google Scholar]
- Shimada M, Niida H, Zineldeen DH, Tagami H, Tanaka M, Saito H, Nakanishi M. Chk1 is a histone H3 threonine 11 kinase that regulates DNA damage-induced transcriptional repression. Cell. 2008;132:221–232. doi: 10.1016/j.cell.2007.12.013. [DOI] [PubMed] [Google Scholar]
- Smits VA, Reaper PM, Jackson SP. Rapid PIKK-dependent release of Chk1 from chromatin promotes the DNA-damage checkpoint response. Curr Biol. 2006;16:150–159. doi: 10.1016/j.cub.2005.11.066. [DOI] [PubMed] [Google Scholar]
- Sorensen CS, Syljuasen RG. Safeguarding genome integrity: the checkpoint kinases ATR, CHK1 and WEE1 restrain CDK activity during normal DNA replication. Nucleic Acids Res. 2012;40:477–486. doi: 10.1093/nar/gkr697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ting NS, Lee WH. The DNA double-strand break response pathway: becoming more BRCAish than ever. DNA Repair (Amst) 2004;3:935–944. doi: 10.1016/j.dnarep.2004.03.026. [DOI] [PubMed] [Google Scholar]
- Walker M, Black EJ, Oehler V, Gillespie DA, Scott MT. Chk1 C-terminal regulatory phosphorylation mediates checkpoint activation by de-repression of Chk1 catalytic activity. Oncogene. 2009;28:2314–2323. doi: 10.1038/onc.2009.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Zou L, Lu T, Bao S, Hurov KE, Hittelman WN, Elledge SJ, Li L. Rad17 phosphorylation is required for claspin recruitment and Chk1 activation in response to replication stress. Mol Cell. 2006;23:331–341. doi: 10.1016/j.molcel.2006.06.022. [DOI] [PubMed] [Google Scholar]
- Wilsker D, Petermann E, Helleday T, Bunz F. Essential function of Chk1 can be uncoupled from DNA damage checkpoint and replication control. Proc Natl Acad Sci U S A. 2008;105:20752–20757. doi: 10.1073/pnas.0806917106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarden RI, Brody LC. BRCA1 interacts with components of the histone deacetylase complex. Proc Natl Acad Sci U S A. 1999;96:4983–4988. doi: 10.1073/pnas.96.9.4983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarden RI, Pardo-Reoyo S, Sgagias M, Cowan KH, Brody LC. BRCA1 regulates the G2/M checkpoint by activating Chk1 kinase upon DNA damage. Nat Genet. 2002;30:285–289. doi: 10.1038/ng837. [DOI] [PubMed] [Google Scholar]
- Zachos G, Black EJ, Walker M, Scott MT, Vagnarelli P, Earnshaw WC, Gillespie DA. Chk1 is required for spindle checkpoint function. Dev Cell. 2007;12:247–260. doi: 10.1016/j.devcel.2007.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao HP-WHRA, Links ATR-mediated checkpoint pathways regulate phosphorylation and activation of human Chk1. Mol Cell Biol. 2001;21(13):4129–4139. doi: 10.1128/MCB.21.13.4129-4139.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou L, Cortez D, Elledge SJ. Regulation of ATR substrate selection by Rad17-dependent loading of Rad9 complexes onto chromatin. Genes Dev. 2002;16:198–208. doi: 10.1101/gad.950302. [DOI] [PMC free article] [PubMed] [Google Scholar]