

Abstract
The essential base excision repair protein, apurinic/apyrimidinic endonuclease 1 (APE1), plays an important role in redox regulation in cells and is currently targeted for development of cancer therapeutics. One compound that binds APE1 directly is (E)-3-(2-(5,6-dimethoxy-3-methyl-1,4-benzoquinonyl))-2-nonyl propenoic acid (E3330). Here, we revisit the mechanism by which this negatively charged compound interacts with APE1 and inhibits its redox activity. At high concentrations (mM), E3330 interacts with two regions in the endonuclease active site of APE1, as mapped by hydrogen/deuterium exchange mass spectrometry. However, this interaction lowers the melting temperature of APE1 consistent with a loss of structure in APE1, as measured by both differential scanning fluorimetry and circular dichroism. These results are consistent with other findings that concentrations of E3330 greater than 100 μM are required to inhibit APE1’s endonuclease activity. To determine the role of E3330’s negatively charged carboxylate in redox inhibition, we converted the carboxylate to an amide by synthesizing (E)-2-((4,5-dimethoxy-2-methyl-3,6-dioxocyclohexa-1,4-dien-1-yl)methylene)-N-methoxy-undecanamide (E3330-amide), a novel uncharged derivative. E3330-amide has no effect on the melting temperature of APE1, suggesting that it does not interact with the fully-folded protein. However, E3330-amide inhibits APE1’s redox activity in in vitro EMSA redox and cell-based transactivation assays producing lower IC50 values as compared to E3330, 8.5 μM vs. 20 μM and 7 μM vs. 55 μM, respectively. Thus, E3330’s negatively charged carboxylate is not required for redox inhibition. Collectively, our results provide additional support for a mechanism of redox inhibition involving interaction of E3330 or E3330-amide with partially unfolded APE1.
Apurinic/apyrimidinic endonuclease I (APE1) plays an important role in cellular response to oxidative stress by maintaining genomic integrity and regulating redox signaling (1, 2). Up-regulation of APE1 in cancer cells is associated with resistance to chemotherapeutic agents (3–5) while loss of APE1 expression results in cell-growth arrest, apoptosis (6), and impairment of mitochondrial function (7, 8). Thus, APE1 is emerging as a potential cancer therapeutic target (9, 10). Activities reported for APE1 include endonuclease activity essential for base excision repair (11), redox activity that regulates the DNA-binding activity of a number of important transcription factors (12), transcriptional repressor activity through indirect binding to Ca2+ responsive elements (13), and most recently cleavage of RNA containing abasic sites (14). Transcription factors that are regulated by APE1’s redox activity include AP-1, NF-κB, Erg-1, HIF-1α, p53, PAX, and others (12, 15–21). Currently, efforts to develop novel cancer therapeutics target either the endonuclease (repair) or the redox function of APE1 (10, 22).
APE1 was first reported as the redox factor responsible for reducing cellular Jun (c-Jun), thereby increasing its affinity for DNA (12). Subsequently, many other transcription factors were shown to be redox regulated by APE1 (12, 15–21). Three cysteine residues, 65, 93, and 99, in APE1 are necessary and sufficient for redox activity (23). Of these residues, 65 and 93 are buried, whereas 99 is solvent accessible. Further regulation of APE1’s activity under conditions of oxidative stress occurs through glutathionylation of C99, which inhibits both DNA-binding and endonuclease activity (24). Oxidation of APE1 also results in a specific disulfide bond formation cascade, implicating C65 as the nucleophilic Cys (23). This result is consistent with earlier results in which C65 was shown to play an important role in APE1’s redox activity (25). Through analysis of single cysteine-to-alanine substitutions in APE1 for each of the seven cysteines, C65A was identified as the only redox-inactive substitution (25). Redox activity associated with APE1 is found only in mammals; zebrafish APE contains five of the seven cysteine residues present in the human enzyme in structurally equivalent positions but still lacks redox activity. However, substitution of threonine 58, the zebrafish residue equivalent to C65, with cysteine confers redox activity in both in vitro and cell-based redox assays (26). More recently, APE1’s redox function and specifically C65 has been implicated in mediating localization of APE1 to the mitochondria and controlling cell proliferation (27).
Other approaches to provide mechanistic details concerning APE1’s redox activity used a redox inhibitor (E)-3-(2-(5,6-dimethoxy-3-methyl-1,4-benzoquinonyl))-2-nonyl propenoic acid (E3330). E3330 was first reported to bind APE1 with a Kd of 1.6 × 10−9 M (28), which later studies find to be far too small(29, 30). As the redox activity of APE1 represents a unique target, E3330 has been evaluated for its potential as a chemotherapeutic agent, making the nature of E3330’s interaction with APE1 of considerable interest and the subject of two recent biophysical studies.
In one of those studies examining the binding of APE1 and E3330, we reported that E3330 interacts with a partially unfolded form of APE1, as monitored by NEM footprinting and mass spectrometry (29). Incubating APE1 in the absence of E3330, we found NEM modification of the two solvent-accessible Cys residues, C99 and C138. Over 24 h at room temperature, barely detectable labeling of buried Cys residues was observed. However, in the presence of E3330, 60% of the enzyme had all seven Cys residues labeled with NEM in the same time frame. This result suggests that E3330 interacts with a partially unfolded state of APE1 long enough for the reaction of Cys and NEM to occur. Other evidence of APE1 unfolding that is essential for function includes the finding that localization of APE1 to mitochondria involves exposure of the C-terminal region 289–318, which serves as the mitochondrial targeting sequence (31). This exposure would necessarily involve unfolding of the protein structure as it forms an integral part of the protein structure.
In another recent study, NMR was used to define interactions of E3330 with APE1. In this study, several residues in proximity to the repair active site of the enzyme showed backbone perturbations consistent with an interaction of E3330 and APE1, specifically at G231, M270, M271, N272, A273, V278, W280, and D308. However, the Kd reported for this interaction, 390 μM at room temperature, indicates the binding affinity is very weak. A mechanism for redox inhibition was then proposed in which E3330 binds specifically to the repair active site of APE1, preventing a conformational change of the protein required for its redox activity (30). This is a surprising conclusion given that high concentrations of E3330 are required to inhibit the enzyme’s endonuclease activity, even as determined by this group who found that significant inhibition was observed at 100 μM but did not report an IC50 (30) whereas much lower concentrations are required to inhibit APE1’s redox activity. Thus, this study (30) concluded that E3330 acts as a redox inhibitor by binding to the repair active site, thereby stabilizing APE1’s structure and preventing it from unfolding. In this scenario, there is no proposed role for E3330’s quinone group, known to be critical for redox inhibition. We concluded that E3330 shifts the equilibrium between folded and unfolded states of APE1 toward unfolded states and inhibits the redox activity through direct interaction with the critical redox residue Cys 65 exposed in a partially unfolded state of the protein leading to its inactivation by disulfide bond formation (29).
In an effort to resolve the disagreement, we pursued additional experimental approaches to determine the nature of the E3330’s interaction with APE1 and report here new insights on the inhibition mechanism. Key questions that we sought to answer were the following. Does E3330 bind specifically to a single site within the repair active site of APE1? Does the interaction of E3330 with APE1 stabilize its structure and thereby prevent a conformational change required for its redox activity? And finally, is the negatively charged carboxylate group of E3330 required for redox inhibition?
Experimental Procedures
Preparation of APE1 proteins
Full-length human APE1 (APE1) was expressed and purified as previously described (29). In brief, the protein was expressed as an N-terminal hexa-His SUMO fusion protein. Following lysis using a French press, the crude extract was bound to a Ni-NTA column, and APE1 was released by cleavage with the SUMO-specific Ulp1 protease. The protein was then subjected to SP-Sepharose ion exchange and gel filtration (Superdex 75) chromatographic purification steps.
Hydrogen/Deuterium Exchange Mass Spectrometry
Differential, solution phase HDX experiments were performed to detect conformational changes of full length APE1 induced by E3330 binding as previously described (32). A solution of 25 μM full-length APE1 was incubated with 16 mM E3330 for at least 1 h on ice before HDX analysis. Each exchange reaction was initiated by incubating 2 μL of 25 μM protein complex (with or without E3330) with 18 μL of D2O protein buffer for a predetermined time (10, 30, 60, 120, 900, 3600, and 14400 s) at 4 °C. The final protein concentration was 2.5 μM, and the E3330 concentration was 1.6 mM after D2O dilution. These conditions resulted in 80% of the protein forming a complex, accepting that the Kd is 390 μM, as measured by NMR (30). The exchange reaction was quenched by mixing with 30 μL of 3 M urea, 1% TFA at 1 °C. The mixture was passed across a custom-packed pepsin column (2 mm × 2 cm) at 200 μL/min, and the digested peptides were captured on a 2 mm × 10 mm C8 trap column (Agilent) and desalted (total time for digestion and desalting was 3 min). Peptides were then separated across a 1.0 mm × 50 mm C18 column (1.9 μ Hypersil Gold, Thermo Scientific) with a linear gradient of 4%–40% CH3CN, 0.1% formic acid, over 5 min. Protein digestion and peptide separation were conducted on columns immersed in an ice-water bath to reduce D/H back exchange. Mass spectrometric analyses were carried out at a capillary temperature at 225 °C, and data were acquired at a measured mass resolving power of 100,000 at m/z 400. Duplicates were performed for each on-exchange time point.
Peptide Identification and HDX Data Processing
MS/MS experiments were performed with a LTQ Orbitrap mass spectrometer (ThermoFisher). Production spectra were acquired in a data-dependent mode, and the six most abundant ions were selected for the product ion analysis. The MS/MS *.raw data files were converted to *.mgf files and then submitted to Mascot (Matrix Science, London, UK) for peptide identification. Peptides included in the peptide set used for HDX had a MASCOT score of 20 or greater. The MS/MS MASCOT search was also performed against a decoy (reverse) sequence, and ambiguous identifications were ruled out. The production spectra of all of the peptide ions from the MASCOT search were further manually inspected, and only those verifiable were used in the coverage.
Differential scanning fluorimetry (DSF)
DSF assays were performed using a Roche Light Cycler 480 with an excitation wavelength of 483 nm and emission of 568 nm. Reactions were carried out in 100 mM HEPES pH 7.0, 150 mM NaCl, and 4x SYPRO orange (Invitrogen Cat # S6651)/0.08% DMSO. Each reaction contained a final protein concentration of 2 μM. E3330 was dissolved in DMSO or absolute ethanol to give final solvent concentrations of 2%, not including DMSO from the SYPRO dye. Titrations of APE1 with E3330 (10 μM-1 mM) or E3330-amide (16 μM-4 mM) were performed in the presence and absence of 1 mM MgCl2. Titrations of APE1 with Mg2+ alone were performed by using a concentration range of 10 μM – 10 mM MgCl2. SYPRO orange was added after a 15 min incubation with all other reagents. Reactions were subsequently covered prior to taking measurements.
Circular Dichroism
Far-UV CD spectra were recorded on a Jasco J-810 spectropolarimeter (JASCO Corp) in a 225–260 nm interval. Experiments were performed employing a protein concentration of 5.0 μM in 10 mM phosphate buffer at pH 7.0 in the absence and in the presence of E3330 at 500 μM (1% DMSO), using a 0.1 cm path-length cuvette. Thermal denaturation profiles were obtained by measuring the temperature dependence of the signal at 230 nm in the 20–80 °C range with a resolution of 0.5 °C and 1.0 nm bandwidth. Peltier temperature controller was used to set up the temperature of the sample; the heating rate was 1°C/min. Data were collected at 0.2 nm resolution, 20 nm/minute scan speed, 4 sec response and were reported as unfolded fraction vs. temperature.
Synthesis of E3330 and E3330-amide
Both E3330 and E3330-amide were synthesized by the University of Michigan Vahlteich Medicinal Chemistry Core Facility (Dr. Hollis Showalter). E3330 was synthesized as previously described (33). The synthesis of E3330-amide is described below (see Scheme 1).
Scheme 1.
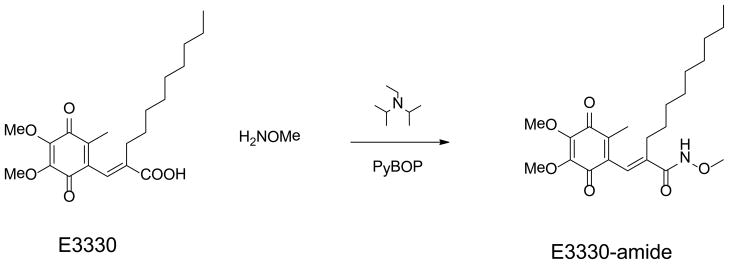
Synthetic scheme for conversion of E3330 to E3330-amide, (E)-2-((4,5-dimethoxy-2-methyl-3,6-dioxocyclohexa-1,4-dien-1-yl)methylene)-N-methoxy-undecanamide.
General Chemical Methods
1H NMR spectra were recorded on a Bruker 500 MHz spectrometer. Chemical shifts are reported in δ (parts per million, ppm), by reference to the hydrogen residues of deuterated solvent as the internal standard (CDCl3: δ = 7.28 ppm; CD3OD: δ = 3.31). Signals were described as s, d, t, and m, for singlet, doublet, triplet, and multiplet, respectively. Coupling constants (J) are given in Hertz (Hz). Mass spectra for compound characterization were recorded on a Micromass LCT time-of-flight instrument utilizing the electrospray ionization mode. Reactions were monitored by thin-layer chromatography (TLC) using pre-coated silica gel 60 F254 plates. Chromatography was performed using bulk silica gel F60, 43–60 μm. Reagents and monomers were purchased from common vendors and were used without purification. Glassware was oven-dried before use for reactions run under anhydrous conditions.
(E)-2-((4,5-dimethoxy-2-methyl-3,6-dioxocyclohexa-1,4-dien-1-yl)methylene)-N-methoxy-undecanamide (E3330-amide)
Benzotriazol-1-yl-oxytripyrrolidinophosphonium hexafluorophosphate (2.48 g, 4.76 mmol) was added to a stirred solution of (E)-2-((4,5-dimethoxy-2-methyl-3,6-dioxocyclohexa-1,4-dien-1-yl)methylene)undecanoic acid (1.2 g, 3.17 mmol), O-methylhydroxylamine hydrochloride (0.34g, 4.12 mmol) and diisopropylethylamine (1.02 g, 7.93 mmol) in dichloromethane (10.5mL) under N2 at room temperature. A mild exotherm was noted, and a clear red solution resulted. After 22 h, TLC [hexane/ethyl acetate/acetic acid 60:50:1] indicated the reaction to be complete. The mixture was diluted with more dichloromethane and washed with water, then saturated brine, then dried over magnesium sulfate. The solvent was removed in vacuo, leaving ared syrup. Chromatography on a column of silica gel under pressure, eluting with hexane/ethyl acetate/acetic acid 60:50:1, afforded the pure product as a thick, clear, blood-red syrup (1.0 g, 77%). ESI MS: m/z 408, [M + H]+ and 430, [M+Na]+. 1H NMR (500 MHz, CDCl3): δ 6.49 (s, 1H), 4.06 (s, 3H), 4.02 (s, 3H) 3.87 (s, 3H), 2.13 (t, J = 10.5, 2H), 1.97 (s, 3H), 1.38 (m, 2H), 1.32-1.15 (m, 14H), 0.88 (t, J = 7.1, 3H).
Electrophoretic mobility shift assays (EMSA)
E3330 or E3330-amide was pre-incubated with 2 μL purified APE1 (reduced with 1.0 mM DTT for 10 min and then diluted to a concentration of 0.06 mM with 0.2 mM DTT in PBS) in an EMSA reaction buffer 10 mM Tris [pH 7.5], 50 mM NaCl, 1 mM MgCl2, 1 mM EDTA, 5% [vol/vol] glycerol) with a total volume of 16 μL for 30 min. The EMSA assay was performed as previously described (23, 33).
Transactivation Assays
Panc1 cells (3) with pGreenFire- NFκB gene (luciferase gene with the NFκB–responsive promoter) stably inserted were treated with increasing amounts of E3330 or E3330-amide and activity assayed at 40 h. The cells were lysed, and the Firefly luciferase activities were assayed as previously described (33). The luciferase activity was normalized to the total cell number measured by MTT assay. All of the experiments were performed in triplicate and repeated at least three times in independent experiments.
Cell Survival/Growth Assays
3-(4, 5-dimethylthiazol-2-yl)-5-(3-carboxymeyhoxyphenyl)-2-(4-sulfophenyl)-2Htetrazolium (MTT) dye assay for cell growth was performed as previously published (34, 35).
Oligonucleotide gel-based APE1 endonuclease activity assays
Oligonucleotide gel-based APE1 endonuclease activity assays were performed as previously described with the following modifications (36, 37). Purified APE1 was incubated with increasing amounts; of E3330 in a total volume of 9 μL assay buffer (5 mM HEPES, 5 mM KCI, 1 mM MgCI2, 0.1% BSA, 0.005% Triton X-100, pH 7.5) at room temperature for 30 min, 1 h or 2 h. One microliter 0.2 pmol 5′-hexachloro-fluorescein phosphoramidite (HEX)-labeled terahydrofuranyl (THF) oligo (the 26 bp oligonucleotide substrate containing a single THF reside in the middle, yielding a HEX-labeled 14-mer fragment (upon repair), was added into the mixture and incubated for 15 min at 37 °C. The reactions were terminated by adding 10 μL formamide. Samples (10 μL) were then applied to a 20% polyacrylamide gel containing 7 M urea in 1X-TBE buffer at 300 volts for 30 min. The amount of 14-mer to 26-mer products detected were quantitated by using the Hitachi FMBio II Fluorescence Imaging System (Hitachi Genetic Systems, South San Francisco, CA) (36, 37).
Fluorescence-based kinetic endonuclease activity assays
The compounds E3330 and E3330-amide were tested with an assay for their ability to inhibit APE1 repair function in a plate-based, high-throughput screening (HTS) assay as previously described(38). A pair of oligonucleotides (Eurogentec Ltd, Belgium), one containing a 6-FAM fluorescein label on its 5′ end and the second containing a 3′ Dabcyl quencher and an internal tetrahydrofuran (THF) as an abasic (AP) site mimic, with complementary bases was annealed as described previously(38). Cleavage at the THF site resulted in release of a 6-FAM-labeled oligonucleotide, which now fluoresced. The increased rate of fluorescence was directly correlated to the amount of APE1 repair activity. Inhibition resulted in a decrease in the rate of fluorescence over time. The assay was performed on an Ultra384 plate reader (Tecan, Durham, North Carolina) in the Chemical Genomics Core Facility at the Indiana University School of Medicine (Indianapolis, Indiana) in a 96-well plate for 3 cycles of 1 min each at 37 °C. A master mix of HTS buffer (50 mM Tris-HCl, pH 8, 50 mM NaCl, 1 mM MgCl2 and 2 mM DTT), water and annealed oligonucleotide (50 nM final concentration) was made, and 100 μL added to each well. A pre-test was performed using 100 μL of the master mix, 50 μL of water and 50 μL of purified human APE1 protein at various concentrations to test for a linear rate of reaction (data not shown). E3330 and E3330-amide were then diluted 20X to the final concentration desired. All dilutions had the vehicle control, DMSO, in equal amounts (0.1% final). Then 10 μL of the 20X compounds were plated with the master mix in triplicate along with 40 μL water. APE1 (50 μL of 0.04 nM) was added to the wells, and the solution immediately assayed. The rate of reaction for each well was normalized to DMSO-only controls and plotted as percent inhibition versus time.
Results
Identification of E3330-interacting regions on APE1 by HDX and MS
In light of the reported low binding (Kd of 390 μM)(30), we performed an HDX analysis with full-length APE1 by using 1.6 mM E3330. This condition should result in approximately 80% complex formation, and indeed we do find detectable differences in the HDX patterns for an experiment done at 4 °C. HDX probes perturbations in the backbone of the protein, quite analogous to the changes monitored by NMR. The backbone perturbations are accompanied by changes in H bonding caused by changes in protein dynamics. The interactions of small molecules such as E3330 with APE1 change the dynamics and allow the interaction sites to be determined. Peptide coverage for the experiment was greater than 90% allowing us to fully assess the folded state of the protein and the effect of added E3330, as is discussed in the following paragraph.
The dynamics of full-length APE1 in the absence of E3330 is in agreement with reported crystal structures (39, 40) (Figure 1): the less structured and exposed regions exhibit relatively fast exchange with deuterium, whereas structured and buried regions of the protein exchange slowly. The most rapidly exchanging regions are represented by peptides 266–271 and 267– 273 in which greater than 70% of the backbone amide hydrogens exchanged with deuterium within 10 s (Figure 1, SI Figure S1). This region is a highly exposed loop on the surface of the protein near the repair active site. On the other hand, regions within the hydrophobic core (63–72, 83–93, 183–191, 208–216, and 285–297) are highly protected from exchange, and they underwent less than 10% exchange even after 1 h of incubation. One region of intermediate exchange contains residues 93–134. These residues make up the terminal strand and half of each of the next two strands on the end of one beta sheet that comprises half of APE1’s beta sandwich fold along with an intervening alpha-helical element. This region also includes two of the three Cys residues, 93 and 99, that are critical for APE1’s redox activity.
Figure 1.

HDX exchange data mapped on the structure of APE1 is consistent with a fully-folded protein. A. The percentage of deuterium uptake for peptides derived from APE1 at different time points are color coded on a ribbon rendering of the crystal structure of human APE1 (PDB:1BIX). The color code explained at the bottom indicates the deuterium uptake level with red indicative of regions that exchange very rapidly and dark blue regions that exchange very slowly. For the most rapidly exchanging peptides, 266–271 and 267– 273, greater than 70% of the backbone amide hydrogens exchanged with deuterium within 10 s. This region was also identified as interacting with E3330.
The peptides for which changes in deuterium uptake were observed in the presence of E3330 include 68–74 (seen for both the singly and doubly charged peptides), 266–271, and 267–273 representing two distinct regions in the protein (Figure 2, SI Figure S1). For all of these observed peptides, the largest differences in deuterium uptake were observed for the 10 s time point and persisted for at most 10 min. The samples treated with E3330 showed decreased deuterium uptake for a few peptides as compared to the control sample indicative of protection from exchange. The peptides 266–271 and 267–273 include several of the residues identified in the NMR study (M270, M271, N272, and A273) as interacting with E3330. Thus, our results confirm some of the interaction of E3330 as identified in the NMR study. However, the second peptide identified in our HDX studies (68–74) was not identified as a region of interaction by NMR.
Figure 2.


Interaction of E3330 with APE1 as detected by HDX mass spectrometry. (A) HDX data are shown for peptides with slower exchange rates in the presence of 1.6 mM E3330 (open squares) as compared to the exchange rates in the absence of compound (filled squares). (B) The peptides that showed protection from deuterium exchange are shown highlighted on the structure of APE1. Residues 68–74 are shown in orange and 266–273 in magenta. In green is a stick model of E3330 docked in the repair active site with its hydrocarbon tail in close proximity to the region 266–273. Shown as stick models are R73 in orange and R177 in blue, two Arg residues in close proximity to the regions of interaction identified by HDX mass spectrometry.
We also have HDX results when E3330 was incubated with an N-terminally truncated version of APE1 (Δ40 APE1) at a decreased concentration of 25 μM (See Supporting Information). Now we estimate approximately 8% complex formation, considering the equilibrium expression. We might expect that the same peptides, 68–74, 270–284, and 272–284, should show differences in HDX at the lower, 25 μM E3330 concentration, but the changes would be smaller because complex formation is attenuated by a factor of 10 times. We are still able to detect changes over the same regions of the protein as those identified for the experiment at higher (1.6 mM) E3330 concentration (SI Figure S2). This demonstrates that HDX is sufficiently sensitive to report on a small amount (<10%) of complex formation although admittedly this would be difficult to assign in the absence of the results for the 1.6 mM E3330 experiment in which 80% complex formation is predicted. A second finding from this experiment was that the N-terminal region of APE1 does not affect the interactions of E3330 with APE1.
Effect of E3330 on the melting temperature of APE1
To further assess the binding interaction of APE1 with E3330, differential scanning fluorimetry (DSF) and circular dichroism assays were performed. The DSF assay relies on the fluorescence of SYPRO orange to report on the folded state of the protein as the temperature is increased (41). The melting temperature of full-length APE1 as measured in this assay was 52 °C. First, we examined the effect of the catalytically important Mg2+ on the melting temperature of APE1. Titration with Mg2+ increased the melting temperature by 4.3 °C maximally at a concentration of 5 mM with higher concentrations resulting in more modest increases similar to those obtained for much lower concentrations of Mg2+ (SI Figure S3). Surprisingly, titration of APE1 with E3330 decreased the melting temperature in a concentration dependent manner. E3330 was added to a maximum concentration of 1 mM dissolved in either 2% ethanol (data not shown), as used in the NMR study (30), or in 2% DMSO as used in our previous studies and in the HDX study discussed above. In both cases, the melting temperature of APE1 decreased by 5 °C for the 1 mM E3330 sample (Figure 3). The same experiment was repeated in the presence of 1 mM Mg2+, which increases the melting temperature by about 3 °C, with similar results. Although the starting Tm was higher for E3330 and Mg2+, the melting temperature when 1 mM E3330 was added decreased by 4.4 °C (Figure 3).
Figure 3.

Effect of E3330 on APE1’s melting temperature in the presence and absence of Mg2+ as measured by DSF. APE1 was titrated with increasing concentrations of E3330 dissolved in DMSO (10 μM-1 mM) in the presence and absence of 1 mM Mg2+. SYPRO Orange dye was then added and Tm measurements were obtained by DSF. The resulting Tm of APE1 was plotted versus the concentration of E3330 in either the presence or absence of 1 mM Mg2+. Increasing concentrations of E3330 resulted in decreased APE1 melting temperatures in both the presence and absence of Mg2+.
To confirm the results obtained using DSF, we employed circular dichroism to determine the melting temperature of full-length APE1 in the presence and absence of E3330. In reasonable agreement with the DSF experiments, the melting temperature of APE1 was 46 °C (Figure 4A). As noted in the Experimental Procedures, the CD experiments were performed in 10 mM phosphate buffer, pH 7.0 to diminish absorbance contributions from organic buffers. Due to interference in the CD spectrum by DMSO, the solvent used to solubilize E3330, it was not possible to carry out this experiment at concentrations of E3330 above 0.5 mM. Nevertheless, using a concentration of 0.5 mM E3330, the melting temperature of APE1 decreased by 2 °C (Figure 4B). The decrease in melting temperature obtained by DSF was also 2 °C for addition of 0.5 mM E3330. Therefore, the results obtained by both DSF and CD methods are entirely consistent.
Figure 4.

Effect of E3330 on APE1’s melting temperature as measured by CD. A. Overlay of thermal melting profiles of recombinant APE1 with and without E3330, reported as unfolded fraction vs. temperature, as measured through the CD signal at 230 nm. B. First derivative of denaturation sigmoidal curves.
Thus rather than stabilizing the structure of APE1 as previously suggested (30), addition of E3330 results in destabilization or loss of structure as indicated by the lowered melting temperature. We proposed earlier that the binding of E3330 stabilizes a partially unfolded form of APE1, not its low-energy fully-folded structure, and the results thus far are consistent with this hypothesis. That is, both NMR and HDX by MS show that an interaction occurs, and melting point depression shows that the interaction is destabilizing.
Inhibition of APE1’s redox activity by E3330 and E3330-amide
We previously demonstrated that E3330 blocked the redox signaling function of APE1 with AP-1, HIF-1α, NF-κB and other downstream transcription factors as the targets in vitro and in vivo (26, 29, 33, 35, 42). However, we had not established in these studies whether the negatively charged carboxylate of E3330 was required for redox inhibition. Thus, we synthesized a methoxy amide derivative of E3330, (E)-2-((4,5-dimethoxy-2-methyl-3,6-dioxocyclohexa-1,4-dien-1-yl)methylene)-N-methoxy-undecanamide, which we refer to as E3330-amide (Scheme 1). The methoxy amide potentially affords improved solubility as compared to the comparable methyl amide or unsubstituted amide derivative. We then compared the ability of E3330 and E3330-amide, an uncharged derivative of E3330 (Scheme 1), to inhibit the redox activity of APE1 by using a standard EMSA redox assay. In this assay, oxidized c-Jun must be reduced by APE1, resulting in reduced c-Jun that binds DNA and yields a shifted band by EMSA analysis. As demonstrated in Figure 5A, E3330-amide inhibited AP-1-enhanced DNA binding by APE1 in a dose-dependent manner similar to that observed for E3330 albeit with a lower IC50 of 8.5 μM compared to 20 μM for E3330. Subsequent analyses in the cell-based NF-κB luciferase reporter assay performed in Panc1 cells treated with E3330 or E3330-amide resulted in a significant decrease in NF-κB activity. Similar to AP-1, NF-κB is also a target of APE1 redox signaling. In this redox transactivation analysis, E3330 had a higher IC50 (55 μM) than E3330-amide (IC50 of 7 μM) (Figure 5B). These results indicate that E3330-amide is an effective redox inhibitor of APE1, as revealed by using both an in vitro EMSA redox assay and a cell-based redox target transactivation assay. Previous studies have demonstrated that APE1 plays an important role in cell growth and survival through two main functions: DNA repair AP endonuclease activity and redox signaling function (1, 2, 10, 26, 27, 29, 33, 43–50). Inhibition of the redox activity of APE1 by E3330 also prevented tumor cell growth as a cytostatic agent (35, 48, 49). Not surprisingly, E3330-amide also blocked the growth of ovarian cancer cell line SKOV-3X (Figure 5C). The effect was more pronounced with an LD50 of 54 μM compared to E3330, LD50 of 104 μM, in these studies. Thus, E3330-amide has cell-killing capabilities in cancer cells, similar to if not better than E3330.
Figure 5.

Inhibition of APE1’s redox activity by E3330 and E3330-amide. A. Redox EMSA assay. E3330 or its derivative E3330-amide was incubated with 2 μl purified APE1 (reduced with 1.0 mM DTT for 10 min and then diluted to a concentration of 0.06 mM with 0.2 mM DTT in PBS) in EMSA reaction buffer with a total volume 16 μL for 30 min and then the EMSA assay was performed. Final concentrations of APE1 and DTT were 0.006 mM and 0.02 mM, respectively. Controls included reactions with unreduced APE1 and with 0.02 mM DTT, which is carried over in the reduced APE1 samples. E3330-amide inhibited AP-1 DNA-binding enhanced by APE1 in dose-dependent manner as did E3330 albeit at a lower IC50 of 8.5 μM vs 20 μM of E3330. B. Transactivation assay. E3330 or E3330-amide was added to the Panc-1 stable cells with pGreenFire- NF-κB gene. After 40 h treatment, NF-κB stimulated luciferase activity was measured along with a MTT assay to measure cell number. The ratio of luciferase activity over MTT activity was determined as a measure of NF-κB activity. Data are expressed as the mean ± SEM of three independent experiments performed in duplicate and are shown as a percentage of transactivation compared with control of no E3330 or E3330-amide. E3330-amide had a greater effect on NF-κB transactivation with an IC50 of 7 μM compared to an IC50 of 55 μM for E3330. C. Effect of E3330 and E3330-amide on cell growth/ survival in an ovarian cancer cell line. The MTT assay was used to measure the effect of E3330 and E3330-amide on cell growth and survival. Data are expressed as the mean ± SEM of three independent experiments performed in triplicate. E3330-amide caused a decrease in the amount of cell proliferation in dose-dependent manner and was more effective than E3330.
Effect of E3330 and E3330-amide on APE1 AP endonuclease activity
We and others have demonstrated that E3330 blocks the redox function of APE1 but does not inhibit the apurinic/apyrimidinic (AP) endonuclease activity of APE1 at concentrations lower than 100 μM (23, 29, 33, 35). To determine whether E3330-amide inhibits the endonuclease function of APE1, we used two different assays: a gel-based and a high-throughput screening (HTS) plate-based assay (38, 51). The previously reported analyses were performed by using concentrations of E3330 that inhibited the redox function of APE1 at time points less than 30 minutes. To confirm these previous findings and to ascertain the effect of E3330-amide on APE1 endonuclease activity, we tested inhibition at longer times and higher concentrations. In the gel-based assay (Figure 6A), E3330 did not show any inhibition of APE1 repair activity when reacted with APE1 from 30 min up to 2 h below concentrations of 160 μM. Similarly, in a HTS assay, neither E3330 nor E3330-amide had a significant effect on APE1 repair activity up to 100 μM (Figure 6B, C); APE1 retained at least 85% activity
Figure 6.

Inhibition of APE1’s endonuclease activity by E3330 and E3330-amide. A. Oligonucleotide gel-based APE1 endonuclease activity assays. Oligonucleotide gel-based APE1 endonuclease activity assays were performed as described in Experimental Procedures. The upper band (26-mer) represents uncleaved AP oligonucleotide, whereas the lower band (14-mer) is the cleaved oligonucleotide. No inhibition of APE1’s endonuclease activity was observed for concentrations of E3330 less than 160 μM. B. and C. HTS assay for the inhibition of APE1 endonuclease DNA repair activity. When tested for APE1 DNA repair inhibition, the addition of up to 100 μM E3330 (B) or E3330-amide (C) did not result in a decrease in the reaction rate; an indicator of the lack of inhibition.
Effect of E3330-amide on the melting temperature of APE1
We performed DSF assays using the uncharged derivative E3330-amide to determine whether the negatively charged carboxylate moiety is required for interaction with APE1 in its folded state. In contrast to E3330, no concentration dependent change in melting temperature was observed for E3330-amide (Figure 7A) in the presence or absence of 1 mM Mg2+. Observed differences in melting temperature were 1 ºC or less for different concentrations of E3330-amide. Thus, E3330-amide does not appear to interact with APE1 in its folded state and yet it is a more effective redox inhibitor than E3330
Figure 7.

Effect of E3330-amide on APE1’s melting temperature as measured by DSF. APE1 was titrated with increasing concentrations of E3330-amide dissolved in DMSO (16 μM – 4 mM) in the presence and absence of 1 mM Mg2+. SYPRO Orange dye was then added, and Tm measurements were obtained by DSF. The resulting Tm of APE1 was plotted versus the concentration of E3330-amide in either the presence or absence of 1 mM Mg2+. Increasing concentrations of E3330-amide did not produce a change in the Tm of APE1.
Discussion
Results presented here provide new insights on the mechanism by which E3330 interacts with and inhibits the redox activity of APE1. By using HDX mass spectrometry, we identified two sites of interaction for E3330 that are proximal to the repair active site (Figure 2B). Although these data are consistent with an interaction of E3330 and the repair active site of APE1, the fact that there is no evidence for E3330-amide interacting with the repair active site of APE1 suggests a role for the negatively charged carboxylate in the interaction. There are a number of positively charged residues lining the DNA-binding site of APE1, including R177 and R73, both of which are solvent exposed and would provide favorable electrostatic interactions (Figure 2B) with E3330 localizing it to the regions observed in the HDX experiment to be E3330-interacting. This lack of singular specificity is in accord with the relatively large Kd of 390 μM (30).
The effect of E3330 interacting with APE1 is to lower its melting temperature as measured by DSF and CD. These results suggest that E3330 destabilizes APE1’s structure rather than stabilizing it. Previously, we proposed that even at room temperature APE1 can adopt a partially unfolded state. This idea is supported by APE1’s relatively low melting temperature determined by both DSF and CD (46 °C) in the present studies. We further proposed that the partially unfolded state of APE1 is stabilized through interaction with E3330. In this partially unfolded state, APE1 is susceptible to reaction with NEM, resulting in labeling of all seven of its Cys residues (29). In the absence of E3330, only the two solvent-accessible Cys residues are labeled with NEM (29). Thus, the results presented here strengthen the conclusion from our previous studies (29). Inclusion of Mg2+ increases the melting temperature of APE1consistent with stabilization of the structure. However, addition of E3330 even in the presence of Mg2+ results in a decrease in the melting temperature suggesting that there is no stabilization of APE1’s structure through interaction of the carboxylate group of E3330 and Mg2+, as might have been expected if the compound binds as modeled in the repair active site pocket (30). Our results do not support the proposal that E3330 acts as a redox inhibitor by binding specifically to the repair active site of APE1 and preventing the enzyme from adopting an alternate conformation (30). We further note that inhibition of APE1’s endonuclease activity by E3330 at relatively high concentrations is consistent with destabilization of APE1’s structure observed by DSF and CD at similarly high concentrations.
The next question is whether the carboxylate of E3330 is required for inhibition of APE1’s redox activity. E3330-amide was found to be a more effective redox inhibitor than E3330 in in vitro redox, cell-based transactivation, and cell-killing assays. Thus, the carboxylate of E3330 is not essential for redox inhibition. In contrast to E3330, E3330-amide does not affect the melting temperature of APE1 suggesting that it does not interact directly with folded APE1. Given that it is a more effective redox inhibitor, we envision the following scenario. Similar to E3330, E3330-amide likely interacts with the redox active form of APE1, which has been proposed to be a locally unfolded state of the protein (29). However, because it lacks the negatively charged carboxylate, E3330-amide has much lower affinity than E3330 for the positively charged repair active site of the fully folded enzyme.
In conclusion, what emerges from this study and previous work is that E3330 is an effective redox inhibitor but is a poor endonuclease inhibitor with in vitro concentrations of greater than 100 μM required for inhibition. E3330 contrasts with a large number of negatively charged endonuclease inhibitors with IC50 values in the 0.1- 10 micromolar range (38, 51–54) and would not have been identified in an experimental screen as an endonuclease inhibitor as most screens are performed using small molecules at a concentration of 10–20 μM. However, at high concentrations, E3330 does interact with the repair active site, albeit without complete specificity, and with low affinity that depends on its negatively charged carboxylate group. We suggest that the properties that make E3330 and its amide derivative effective redox inhibitors are its quinone group, important for inhibiting the redox reaction (35, 42), and its hydrophobic character, allowing it to interact with APE1 in a partially unfolded state (29).
Supplementary Material
Acknowledgments
This work was supported by a grant from the National Institutes of Health, CA114571 to M.M.G., CA121168, CA114571, CA121168S1 and the Riley Children’s Foundation to M.R.K., and a grant from the National Institutes of Health, 8 P41 GM103422, to M.L.G.
We thank Jim Wikel for assistance in designing redox inhibitors, Sandor Vajda and his laboratory at Boston University for assistance in docking of E3330 in the repair active site of APE1, Bruce Pascal from Professor Patrick R. Griffin’s laboratory at Scripps Florida for help with HDX data analysis and helpful discussion, Halesha Basavarajappa and Krishna Mahalingan for assistance with earlier studies related to this work, and members of the Georgiadis and Kelley laboratories for helpful discussion related to this manuscript.
Footnotes
SUPPORTING INFORMATION AVAILABLE
Supporting Information includes HDX peptide data for full-length APE1 and full-length APE1:E3330 (1.6 mM) samples, selected HDX peptide data comparing experiments done at 30 30 μM vs. 1.6 mM E3330, and DSF data for a titration of APE1 with increasing concentrations of Mg2+. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Luo M, He H, Kelley MR, Georgiadis MM. Redox regulation of DNA repair: implications for human health and cancer therapeutic development. Antioxid Redox Signal. 2010;12:1247–1269. doi: 10.1089/ars.2009.2698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tell G, Quadrifoglio F, Tiribelli C, Kelley MR. The Many Functions of APE1/Ref-1: Not Only a DNA Repair Enzyme. Antioxid Redox Signal. 2009;11:601–620. doi: 10.1089/ars.2008.2194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Koukourakis MI, Giatromanolaki A, Kakolyris S, Sivridis E, Georgoulias V, Funtzilas G, Hickson ID, Gatter KC, Harris AL. Nuclear expression of human apurinic/apyrimidinic endonuclease (HAP1/Ref-1) in head-and-neck cancer is associated with resistance to chemoradiotherapy and poor outcome. Int J Radiat Oncol Biol Phys. 2001;50:27–36. doi: 10.1016/s0360-3016(00)01561-3. [DOI] [PubMed] [Google Scholar]
- 4.Kakolyris S, Kaklamanis L, Engels K, Fox SB, Taylor M, Hickson ID, Gatter KC, Harris AL. Human AP endonuclease 1 (HAP1) protein expression in breast cancer correlates with lymph node status and angiogenesis. Br J Cancer. 1998;77:1169–1173. doi: 10.1038/bjc.1998.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Robertson KA, Bullock HA, Xu Y, Tritt R, Zimmerman E, Ulbright TM, Foster RS, Einhorn LH, Kelley MR. Altered expression of Ape1/ref-1 in germ cell tumors and overexpression in NT2 cells confers resistance to bleomycin and radiation. Cancer Res. 2001;61:2220–2225. [PubMed] [Google Scholar]
- 6.Fung H, Demple B. A vital role for Ape1/Ref1 protein in repairing spontaneous DNA damage in human cells. Mol Cell. 2005;17:463–470. doi: 10.1016/j.molcel.2004.12.029. [DOI] [PubMed] [Google Scholar]
- 7.Tell G, Crivellato E, Pines A, Paron I, Pucillo C, Manzini G, Bandiera A, Kelley MR, Di Loreto C, Damante G. Mitochondrial localization of APE/Ref-1 in thyroid cells. Mutat Res. 2001;485:143–152. doi: 10.1016/s0921-8777(00)00068-9. [DOI] [PubMed] [Google Scholar]
- 8.Chattopadhyay R, Wiederhold L, Szczesny B, Boldogh I, Hazra TK, Izumi T, Mitra S. Identification and characterization of mitochondrial abasic (AP)-endonuclease in mammalian cells. Nucleic Acids Res. 2006;34:2067–2076. doi: 10.1093/nar/gkl177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tell G, Wilson DM., 3rd Targeting DNA repair proteins for cancer treatment. Cell Mol Life Sci. 2010;67:3569–3572. doi: 10.1007/s00018-010-0484-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kelley M, Georgiadis M, Fishel M. APE1/Ref-1Role in Redox Signaling: Translational Applications of Targeting the Redox Function of the DNA Repair/Redox Protein APE1/Ref-1. Current Molecular Pharmacology. 2012;5:36–53. doi: 10.2174/1874467211205010036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Demple B, Herman T, Chen DS. Cloning and expression of APE, the cDNA encoding the major human apurinic endonuclease: definition of a family of DNA repair enzymes. Proc Natl Acad Sci U S A. 1991;88:11450–11454. doi: 10.1073/pnas.88.24.11450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xanthoudakis S, Curran T. Identification and characterization of Ref-1, a nuclear protein that facilitates AP-1 DNA-binding activity. EMBO J. 1992;11:653–665. doi: 10.1002/j.1460-2075.1992.tb05097.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bhakat KK, Yang SH, Mitra S. Acetylation of human AP-endonuclease 1, a critical enzyme in DNA repair and transcription regulation. Methods Enzymol. 2003;371:292–300. doi: 10.1016/S0076-6879(03)71022-2. [DOI] [PubMed] [Google Scholar]
- 14.Vascotto C, Fantini D, Romanello M, Cesaratto L, Deganuto M, Leonardi A, Radicella JP, Kelley MR, D’Ambrosio C, Scaloni A, Quadrifoglio F, Tell G. APE1/Ref-1 interacts with NPM1 within nucleoli and plays a role in the rRNA quality control process. Mol Cell Biol. 2009;29:1834–1854. doi: 10.1128/MCB.01337-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hirota K, Matsui M, Iwata S, Nishiyama A, Mori K, Yodoi J. AP-1 transcriptional activity is regulated by a direct association between thioredoxin and Ref-1. Proc Natl Acad Sci U S A. 1997;94:3633–3638. doi: 10.1073/pnas.94.8.3633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wei SJ, Botero A, Hirota K, Bradbury CM, Markovina S, Laszlo A, Spitz DR, Goswami PC, Yodoi J, Gius D. Thioredoxin nuclear translocation and interaction with redox factor-1 activates the activator protein-1 transcription factor in response to ionizing radiation. Cancer Res. 2000;60:6688–6695. [PubMed] [Google Scholar]
- 17.Ziel KA, Campbell CC, Wilson GL, Gillespie MN. Ref-1/Ape is critical for formation of the hypoxia-inducible transcriptional complex on the hypoxic resonse element of the rat pulmonary artery endothelial cell VEGF. FASEB J Epub. 2004 doi: 10.1096/fj.03-1160fje. [DOI] [PubMed] [Google Scholar]
- 18.Gray MJ, Zhang J, Ellis LM, Semenza GL, Evans DB, Watowich SS, Gallick GE. HIF-1alpha, STAT3, CBP/p300 and Ref-1/APE are components of a transcriptional complex that regulates Src-dependent hypoxia-induced expression of VEGF in pancreatic and prostate carcinomas. Oncogene. 2005;24:3110–3120. doi: 10.1038/sj.onc.1208513. [DOI] [PubMed] [Google Scholar]
- 19.Pines A, Perrone L, Bivi N, Romanello M, Damante G, Gulisano M, Kelley MR, Quadrifoglio F, Tell G. Activation of APE1/Ref-1 is dependent on reactive oxygen species generated after purinergic receptor stimulation by ATP. Nucleic Acids Res. 2005;33:4379–4394. doi: 10.1093/nar/gki751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pines A, Bivi N, Romanello M, Damante G, Kelley MR, Adamson ED, D’Andrea P, Quadrifoglio F, Moro L, Tell G. Cross-regulation between Egr-1 and APE/Ref-1 during early response to oxidative stress in the human osteoblastic HOBIT cell line: evidence for an autoregulatory loop. Free Radic Res. 2005;39:269–281. doi: 10.1080/10715760400028423. [DOI] [PubMed] [Google Scholar]
- 21.Jayaraman L, Murthy KG, Zhu C, Curran T, Xanthoudakis S, Prives C. Identification of redox/repair protein Ref-1 as a potent activator of p53. Genes Dev. 1997;11:558–570. doi: 10.1101/gad.11.5.558. [DOI] [PubMed] [Google Scholar]
- 22.Madhusudan S, Hickson ID. DNA repair inhibition: a selective tumour targeting strategy. Trends Mol Med. 2005;11:503–511. doi: 10.1016/j.molmed.2005.09.004. [DOI] [PubMed] [Google Scholar]
- 23.Luo M, Zhang J, He H, Su D, Chen Q, Gross ML, Kelley MR, Georgiadis MM. Characterization of the redox activity and disulfide bond formation in apurinic/apyrimidinic endonuclease. Biochemistry. 2012;51:695–705. doi: 10.1021/bi201034z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim YJ, Kim D, Illuzzi JL, Delaplane S, Su D, Bernier M, Gross ML, Georgiadis MM, Wilson DM., 3rd S-glutathionylation of cysteine 99 in the APE1 protein impairs abasic endonuclease activity. J Mol Biol. 2011;414:313–326. doi: 10.1016/j.jmb.2011.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Walker LJ, Robson CN, Black E, Gillespie D, Hickson ID. Identification of residues in the human DNA repair enzyme HAP1 (Ref-1) that are essential for redox regulation of Jun DNA binding. Mol Cell Biol. 1993;13:5370–5376. doi: 10.1128/mcb.13.9.5370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Georgiadis M, Luo M, Gaur R, Delaplane S, Li X, Kelley M. Evolution of the redox function in mammalian apurinic/apyrimidinic endonuclease. Mutat Res. 2008;643:54–63. doi: 10.1016/j.mrfmmm.2008.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vascotto C, Bisetto E, Li M, Zeef LA, D’Ambrosio C, Domenis R, Comelli M, Delneri D, Scaloni A, Altieri F, Mavelli I, Quadrifoglio F, Kelley MR, Tell G. Knock-in reconstitution studies reveal an unexpected role of Cys-65 in regulating APE1/Ref-1 subcellular trafficking and function. Mol Biol Cell. 2011;22:3887–3901. doi: 10.1091/mbc.E11-05-0391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shimizu N, Sugimoto K, Tang J, Nishi T, Sato I, Hiramoto M, Aizawa S, Hatakeyama M, Ohba R, Hatori H, Yoshikawa T, Suzuki F, Oomori A, Tanaka H, Kawaguchi H, Watanabe H, Handa H. High-performance affinity beads for identifying drug receptors. Nat Biotechnol. 2000;18:877–881. doi: 10.1038/78496. [DOI] [PubMed] [Google Scholar]
- 29.Su D, Delaplane S, Luo M, Rempel DL, Vu B, Kelley MR, Gross ML, Georgiadis MM. Interactions of Apurinic/Apyrimidinic Endonuclease with a Redox Inhibitor: Evidence for an Alternate Conformation of the Enzyme. Biochemistry. 2011;50:82–92. doi: 10.1021/bi101248s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Manvilla BA, Wauchope O, Seley-Radtke KL, Drohat AC. NMR studies reveal an unexpected binding site for a redox inhibitor of AP endonuclease 1. Biochemistry. 2011;50:10540–10549. doi: 10.1021/bi201071g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li M, Zhong Z, Zhu J, Xiang D, Dai N, Cao X, Qing Y, Yang Z, Xie J, Li Z, Baugh L, Wang G, Wang D. Identification and characterization of mitochondrial targeting sequence of human apurinic/apyrimidinic endonuclease 1. J Biol Chem. 2010;285:14871–14881. doi: 10.1074/jbc.M109.069591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang J, Chalmers MJ, Stayrook KR, Burris LL, Garcia-Ordonez RD, Pascal BD, Burris TP, Dodge JA, Griffin PR. Hydrogen/deuterium exchange reveals distinct agonist/partial agonist receptor dynamics within vitamin D receptor/retinoid X receptor heterodimer. Structure. 2010;18:1332–1341. doi: 10.1016/j.str.2010.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Luo M, Delaplane S, Jiang A, Reed A, He Y, Fishel M, Nyland RL, II, Borch RF, Qiao X, Georgiadis MM, Kelley MR. Role of the multifunctional DNA repair and redox signaling protein Ape1/Ref-1 in cancer and endothelial cells: Small molecule inhibition of Ape1’s redox function. Antioxid Redox Signal. 2008;10:1853–1867. doi: 10.1089/ars.2008.2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang D, Luo M, Kelley MR. Human apurinic endonuclease 1 (APE1) expression and prognostic significance in osteosarcoma: enhanced sensitivity of osteosarcoma to DNA damaging agents using silencing RNA APE1 expression inhibition. Mol Cancer Ther. 2004;3:679–686. [PubMed] [Google Scholar]
- 35.Kelley MR, Luo M, Reed A, Su D, Delaplane S, Borch RF, Nyland RL, Gross ML, Georgiadis MM. Functional Analysis of Novel Analogues of E3330 That Block the Redox Signaling Activity of the Multifunctional AP Endonuclease/Redox Signaling Enzyme APE1/Ref-1. Antioxid Redox Signal. 2011;14:1387–1401. doi: 10.1089/ars.2010.3410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kreklau EL, Limp-Foster M, Liu N, Xu Y, Kelley MR, Erickson LC. A novel fluorometric oligonucleotide assay to measure O(6)- methylguanine DNA methyltransferase, methylpurine DNA glycosylase, 8- oxoguanine DNA glycosylase and abasic endonuclease activities: DNA repair status in human breast carcinoma cells overexpressing methylpurine DNA glycosylase. Nucleic Acids Res. 2001;29:2558–2566. doi: 10.1093/nar/29.12.2558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Luo M, Kelley MR. Inhibition of the human apurinic/apyrimidinic endonuclease (APE1) repair activity and sensitization of breast cancer cells to DNA alkylating agents with lucanthone. Anticancer Res. 2004;24:2127–2134. [PubMed] [Google Scholar]
- 38.Bapat A, Glass LS, Luo M, Fishel ML, Long EC, Georgiadis MM, Kelley MR. Novel small molecule inhibitor of Ape1 endonuclease blocks proliferation and reduces viability of glioblastoma cells. J Pharmacol Exp Ther. 2010;334:988–998. doi: 10.1124/jpet.110.169128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gorman MA, Morera S, Rothwell DG, La Fortelle E, Mol CD, Tainer JA, Hickson ID, Freemont PS. The crystal structure of the human DNA repair endonuclease HAP1 suggests the recognition of extra-helical deoxyribose at DNA abasic sites. EMBO J. 1997;16:6548–6558. doi: 10.1093/emboj/16.21.6548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Beernink PT, Segelke BW, Hadi MZ, Erzberger JP, Wilson DM, 3rd, Rupp B. Two divalent metal ions in the active site of a new crystal form of human apurinic/apyrimidinic endonuclease, Ape1: implications for the catalytic mechanism. J Mol Biol. 2001;307:1023–1034. doi: 10.1006/jmbi.2001.4529. [DOI] [PubMed] [Google Scholar]
- 41.Niesen FH, Berglund H, Vedadi M. The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nature protocols. 2007;2:2212–2221. doi: 10.1038/nprot.2007.321. [DOI] [PubMed] [Google Scholar]
- 42.Nyland RL, Luo M, Kelley MR, Borch RF. Design and synthesis of novel quinone inhibitors targeted to the redox function of apurinic/apyrimidinic endonuclease 1/redox enhancing factor-1 (Ape1/ref-1) J Med Chem. 2010;53:1200–1210. doi: 10.1021/jm9014857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jiang A, Gao H, Kelley MR, Qiao X. Inhibition of APE1/Ref-1 Redox Activity with APX3330 Blocks Retinal Angiogenesis in vitro and in vivo. Vision Res. 2011;51:93–100. doi: 10.1016/j.visres.2010.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jedinak A, Dudhgaonkar S, Kelley MR, Sliva D. Apurinic/Apyrimidinic endonuclease 1 regulates inflammatory response in macrophages. Anticancer Res. 2011;31:379–385. [PMC free article] [PubMed] [Google Scholar]
- 45.Jiang Y, Zhou S, Sandusky GE, Kelley MR, Fishel ML. Reduced expression of DNA repair and redox signaling protein APE1/Ref-1 impairs human pancreatic cancer cell survival, proliferation, and cell cycle progression. Cancer Invest. 2010;28:885–895. doi: 10.3109/07357907.2010.512816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fishel ML, Colvin ES, Luo M, Kelley MR, Robertson KA. Inhibition of the redox function of APE1/Ref-1 in myeloid leukemia cell lines results in a hypersensitive response to retinoic acid-induced differentiation and apoptosis. Exp Hematol. 2010;38:1178–1188. doi: 10.1016/j.exphem.2010.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jiang Y, Guo C, Fishel ML, Wang Z-Y, Vasko MR, Kelley MR. Role of APE1 in differentiated neuroblastoma SH-SY5Y cells in response to oxidative stress: Use of APE1 small molecule inhibitors to delineate APE1 functions. DNA Repair. 2009;8:1273–1282. doi: 10.1016/j.dnarep.2009.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jiang Y, Guo C, Vasko MR, Kelley MR. Implications of Apurinic/Apyrimidinic Endonuclease in Reactive Oxygen Signaling Response after Cisplatin Treatment of Dorsal Root Ganglion Neurons. Cancer Res. 2008;68:6425–6434. doi: 10.1158/0008-5472.CAN-08-1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fishel ML, He Y, Reed AM, Chin-Sinex H, Hutchins GD, Mendonca MS, Kelley MR. Knockdown of the DNA repair and redox signaling protein Ape1/Ref-1 blocks ovarian cancer cell and tumor growth. DNA Repair (Amst) 2008;7:177–186. doi: 10.1016/j.dnarep.2007.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zou GM, Luo MH, Reed A, Kelley MR, Yoder MC. Ape1 regulates hematopoietic differentiation of embryonic stem cells through its redox functional domain. Blood. 2007;109:1917–1922. doi: 10.1182/blood-2006-08-044172. [DOI] [PubMed] [Google Scholar]
- 51.Madhusudan S, Smart F, Shrimpton P, Parsons JL, Gardiner L, Houlbrook S, Talbot DC, Hammonds T, Freemont PA, Sternberg MJ, Dianov GL, Hickson ID. Isolation of a small molecule inhibitor of DNA base excision repair. Nucleic Acids Res. 2005;33:4711–4724. doi: 10.1093/nar/gki781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Seiple LA, Cardellina JH, 2nd, Akee R, Stivers JT. Potent inhibition of human apurinic/apyrimidinic endonuclease 1 by arylstibonic acids. Mol Pharmacol. 2008;73:669–677. doi: 10.1124/mol.107.042622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zawahir Z, Dayam R, Deng J, Pereira C, Neamati N. Pharmacophore guided discovery of small-molecule human apurinic/apyrimidinic endonuclease 1 inhibitors. J Med Chem. 2009;52:20–32. doi: 10.1021/jm800739m. [DOI] [PubMed] [Google Scholar]
- 54.Simeonov A, Kulkarni A, Dorjsuren D, Jadhav A, Shen M, McNeill DR, Austin CP, Wilson DM., 3rd Identification and characterization of inhibitors of human apurinic/apyrimidinic endonuclease APE1. PLoS One. 2009;4:e5740. doi: 10.1371/journal.pone.0005740. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.