

Abstract
The unexpected discovery that somatic cells can be reprogrammed to a pluripotent state yielding induced pluripotent stem cells (iPSCs) has made it possible to produce cardiovascular cells exhibiting inherited traits and disorders. Use of these cells in high throughput analyses should broaden our insight into fundamental disease mechanisms and provide many benefits for patients, including new therapeutics and individually tailored therapies. Here we review recent progress in generating iPSC-based models of cardiovascular disease and their multiple applications in drug development.
Keywords: induced pluripotent stem cells, high throughput screening, functional genomics, chemical biology
Introduction
The reprogramming of somatic cells to a pluripotent state by a cocktail of a few transcription factors will certainly be considered one of the major biological advances of the 21st century. The finding that these induced pluripotent stem cells (iPSCs) can give rise to every cell type in the body quickly triggered an explosion of interest in using patient-specific iPSCs and their differentiated derivatives to model simple and complex clinical presentations of disease.
Remarkable advances have been made in reprogramming technologies beyond the stable integration of four core genes (Oct4, Sox2, Klf4 and c-Myc) originally described by Yamanaka and colleagues in 2006 for mouse cells1 and soon reproduced from human cells2-4. Although the integrated viruses and transgenes become transcriptionally silenced during reprogramming, insertional mutations and residual transgene or virally-induced gene expression may interfere with differentiation or alter subsequent cell phenotypes, leading to questions of whether iPSCs and their derivatives truly reflect a patient's own cells5. To circumvent these issues, newer non-integrating, virus-free and vector-free methods have been developed.6,7 Importantly, the technology for iPSC creation from patient-derived cells has proven robust in that iPSCs can be effectively derived from many tissues, even from elderly and sick individuals, enabling the derivation of a wide range of patient-specific cells over just the past few years8. These advances now set the stage for modeling disease in order to decipher basic mechanisms, predicting toxic drug effects, match a drug therapy to an individual patient, and discover new drugs for a variety of disorders.
Despite such progress, there are significant challenges to implementing large-scale drug screens using iPSC-based models of human disease. The initial problem of directing large-scale production of particular cell types under simplified and defined conditions is being solved for cardiomyocytes, vascular endothelial and smooth muscle cell lineages9-12. Nonetheless, many substantial hurdles must still be overcome. Among these include variability in iPSCs derived even from a single patient, such as could be caused by imperfect reprogramming5, 9. The variability in the disease phenotype among iPSC isolates, even from a single patient, can be so great as to mask differences between iPSCs from affected versus unaffected individuals. Second, in order for a screening campaign to be predictive, any in-dish phenotype must be demonstrated to be relevant to the clinical issues surrounding disease treatment and/or management. It is easy to envisage that the need for highly specialized cells, or a complex cellular context, such as interactions between multiple cell types or three-dimensional architecture, could hamper reproducing the disease phenotype in a culture well. Thus, the correct cell type(s) must be produced and interact appropriately in the cell culture assay. Perhaps less obvious is that many diseases take years to manifest in patients and might not be apparent in the developmentally immature cells derived from patient iPSCs, particularly in the relatively short time interval from pluripotent cell to differentiated derivative used in the laboratory experiment or high throughput screen.
Here we review the derivation of cardiovascular disease-specific iPSCs and progress towards recapitulating clinically relevant disease phenotypes in high throughput formats, with particular focus on how iPSC technology can improve the probability that drugs will be clinically effective through the development of realistic human cardiovascular disease models. In addition, we discuss the challenges to be overcome before the potential of iPSC models can be fully exploited for the elucidation of disease processes, as well as drug toxicity, targets and mechanisms of action.
iPSC-based Cardiovascular Disease Modeling – From Patient to 384-well Plate
In principle, the ability to generate iPSCs from patients will permit the production of unlimited numbers of any cardiovascular cell type needed to model disease. Moreover, natural polymorphisms in the population that confer susceptibility to disease or sensitivity to drugs (e.g. drug-induced Long QT-2) can be isolated, and the differentiated derivatives of the pluripotent cells can, in principle, be used to model the phenotype (Fig. 1). Most disease phenotypes, however, will manifest only in differentiated cells and not in the iPSCs themselves, which are analogous to the pluripotent cells of the peri-implantation embryo. Fortunately, the directed differentiation of cardiovascular cell types is becoming increasingly efficient, at least for heterogeneous populations of cardiomyocytes, vascular endothelial and smooth muscle cells, thanks to protocols that recapitulate normal development, e.g.9-12. Other reviews in this series summarize the generation of patient-specific iPSCs and recent advances in optimizing the production of different cardiovascular cell types.13,14
Figure 1. Utility of iPSC technology for drug discovery.

Drug discovery starts with patient biopsies used for the generation of induced pluripotent stem cells (iPSCs), followed by the directed differentiation to specialized cardiac cell types that are used in disease models for screening. The assays are then screened in high throughput, moderate throughput or small-scale assays that are tailored for a range of activities, from primary drug screens to the design of individualized patient therapies. (Illustration Credit: Ben Smith).
A number of iPSC lines have been isolated from patients with a broad range of genetic cardiovascular diseases (Table 1). In most cases, the relevant cell types - cardiomyocytes and smooth muscle cells – have been produced from these patient iPSCs and reported to recapitulate key aspects of the disease phenotypes. For instance, Long QT and Timothy syndrome patients exhibit prolonged QT intervals on electrocardiography, and cardiomyocytes derived from patient-specific iPSCs showed increased duration of action potentials when measured by single-cell intracellular recording15-18. Similarly, a recent study investigated the phenotype of cardiomyocytes derived from LEOPARD syndrome patient-iPSCs and found the cells to be enlarged, possibly reflecting the hypertrophic cardiomyopathy characteristic of these patients19. In addition, vascular smooth muscle cells from iPSCs generated from Hutchinson-Gilford progeria patients were found to undergo premature senescence, suggesting that the cultures recapitulated aspects of the vascular defects seen in these patients20, 21.
Table 1. Examples of cardiovascular disease-specific human pluripotent stem cell lines.
| Disease name | Genetic cause | Clinical presentation | Disease mechanism | Cell type made | Phenotype displayed in iPSC-derived cells |
Drug testing | Ref. |
|---|---|---|---|---|---|---|---|
| Arrhythmogenic right ventricular cardiomyopathy/dysplasia/(ARVC/D) | Plakophilin-2 (PKP2) L614P | Syncope and sudden death; frequently adolescent/young adult onset; fatty or fatty fibrous replacement of myocardium with thinning of the RV wall |
Desmosomal dysfunction | CMs |
|
|
115 |
| Catecholaminergic polymorphic ventricular tachycardia type 1 (CPVT-1) | RyR2 p.F2483I | Stress-induced ventricular tachyarrhythmia, syncope and sudden cardiac death in children and young adults |
Diastolic Ca2+ leak from the sarcoplasmic reticulum (SR) |
CMs |
|
None | 29 |
| CPVT-1 | RyR2 S406L | Same | Same | CMs |
|
-Dantrolene restored normal Ca2+ spark properties and rescued the arrhythmogenic phenotype | 28 |
| CPVT-1 | RyR2 M4109R | Same | Same | CMs | Similar to above, but also evidence of early after depolarizations (EADs) |
|
31 |
| CPVT-1 | RyR2 P2328S | Same | Same | CMs | Similar to above | None | 30 |
| CPVT-2 | CASQ2 D307H | Ventricular arrhythmias causing sudden death in young individuals |
Intracellular Ca2+ mishandling causing delayed afterdepolarization (DADs) |
CMs |
|
None | 116 |
| Familial dilated cardiomyopathy | TNNT2 R173W | Ventricular dilatation, systolic dysfunction, progressive heart failure and dilated cardiomyopathy. |
Decreased Ca2+ sensitivity and ATPase activity which cause force production impairment |
CMs |
|
-Metoprolol improved sarcomeric organization | 49 |
| Duchenne muscular dystrophy | DMDDeletion of exons 45–52 | Dilated cardiomyopathies and/or cardiac arrythmias |
Cell death caused by extracellular Ca2+influx |
None | 117 | ||
| Hutchinson Gilford Progeria | LMNA C1824T | Premature ageing leading to myocardial infarction |
Disorganization of nuclear lamina and loss of heterochromatin |
VSMCs, ECs, fibroblasts | -Calponin sequestration, premature senescence phenotypes, DNA damage phenotypes associated with vascular ageing |
None | 20, 21 |
| LEOPARD syndrome | PTPN11 T468M | Hypertrophic cardiomyopathy | Pathogenesis unknown | CMs | -CMs are larger, have a higher degree of sarcomeric organization and preferential localization of NFATC4 in the nucleus compared to normal CMs |
None | 19 |
| Long QT-1 | KCNQ1 R190Q | Delayed repolarization; Arrhythmias; Polymorphic ventricular tachycardia |
Prolonged ventricular repolarization due to mutation in repolarizing potassium channel KCNQ1 |
CMs |
|
-Isoproteronol induced EAD was prevented by Propranolol, simulating clinical LQT1 |
15 |
| Long QT-2 | KCNH2 A614V | Same | Significant reduction of the rapid component of the delayed rectifier potassium current (IKr) due to mutation affecting the pore-forming region of the potassium channel KCNH2 |
CMs |
|
|
16 |
| Long QT-2 | KCNH2 G1681A | Same | Same | CMs | Same |
|
18 |
| Long QT-8 (Timothy Syndrome) | Cav1.2 G406R | Same | Mutation causes impairment of voltage-dependent inactivation of Cav1.2 |
CMs |
|
-Roscovitine normalized the Ca2+
defects and improved channel inactivation |
17 |
| SCN5A overlap sydrome | SCN5A 1795insD | ECG features of bradycardia, ventricular and atrial conduction slowing, exibits aspects of both LQT-3, and Brugada Syndrome |
Significant reduction in upstroke velocity of the action potential; and longer action potential duration |
CMs |
|
None | 118 |
| Marfan type 1 | FBN1 1747delC (frameshift) | Aortic aneurysm Mitral valve prolapse Calcification of valve annulus |
Elevated TGFβ signaling; structural weakness of microfibrils in ECM |
Mesen-chymal cells |
|
None | 37 |
| Pompe disease (infantile onset) | GAA C1935A/C1935A; G2040+1T/C1935A; G1062G/C1935A |
Hypotonia and signs of heart failure by the age of 3-5 months of age |
Accumulation of membrane-bound and cytoplasmic glycogen and rupture of lysosomes, aberrant mitochondria, and accumulation of autophagic vesicles leading to cardiomyopathy |
CMs |
|
|
50 |
Abbreviations: cardiomyocytes, CMs; vascular smooth muscle cells, VSMCs; endothelial cells, ECs.
Impressive as observing disease phenotypes in a dish seems at first blush, it is important to question the extent to which they mirror the patient's clinical presentation so that drugs developed based on the in vitro model will elicit a salutary response in people. In this regard, it is important to bear in mind that hESC and iPSC-derived cardiomyocytes are quite immature electrically, metabolically and mechanically, and do not express the same complement of structural proteins and ion channels nor would be expected to exhibit the same energetics, as their counterparts in the heart of an adult or even a child. As summarized22, 23, hiPSC and hESC-cardiomyocytes are characterized by their small size, poorly organized myofibrils and a punctate and uniform distribution of gap junctions that would influence action potential propagation. In addition, immature pluripotent-derived myocytes show ion channel differences that cause a slower action potential kinetics (typically dV/dt is less than 20 V/s as compared to adult ventricular myocytes with dV/dt of 100-150 V/s), a less well-developed sarcoplasmic reticulum making contractility dependent on Ca2+ entry rather than internal sarcoplasmic reticulum stores, and a relatively depolarized MDP and a slow phase IV depolarization that confers persistent automaticity, and develop a negative, rather than positive, force-frequency relationship. Although not extensively studied to date, the metabolism of pluripotent stem cell-derived cardiomyocytes is expected to resemble that of fetal cardiomyocytes, which rely predominantly on glucose rather than fatty acids as an energy source, as do adult cardiomyocytes24, 25, although increased mitochondrial content and the capacity for oxidative metabolism distinguishes hESC-derived cardiomyocytes from pluripotent progenitors and positively affects their differentiation, e.g.26, 27.
As a consequence of their embryonic nature, iPSC-derived cardiomyocytes as produced currently are not capable of being completely accurate models of diseases that occur only in the physiological context of mature cardiomyocytes, and the same is true for other cardiovascular derivatives albeit less strikingly. Even if the patient iPSC-derived cells exhibit an altered in-dish phenotype relative to normal cells, the phenotype might involve a different set of signaling and genetic pathways than those operating in the patient's own cells, giving rise to a mimic rather than an accurate model of the disease. This presents a serious problem for drug discovery, since the all-important ancillary proteins and pathways that modulate the disease in the patient's cells, and hence comprise high value drug targets, could be quite different. Therefore, in-depth analysis of the disease model is necessary to be certain that it accurately mirrors the clinical presentation of the disease.
Clearly some diseases will be easier to model than others. iPSC-derived channelopathies might represent good candidates for drug development since the mutated proteins themselves are likely to be reasonable drug targets for treating disease, and their cell-autonomous and monogenic nature facilitates assay development. As an example, two recent reports investigated whether the iPSC-derived cardiomyocytes reproduce the phenotype of patients suffering from catecholaminergic polymorphic ventricular tachycardia type 1 (CPVT-1), which is caused by mutations in the cardiac ryanodine receptor type 2 gene (RYR2) and is characterized by stress-induced ventricular arrhythmia that can lead to sudden cardiac death in young individuals with structurally normal hearts. The cardiomyocytes generated from patient iPSCs, with different mutations, showed increased propensity to generate spontaneous Ca2+ sparks in response to catecholaminergic stimulation and increased susceptibility to delayed after depolarizations (DADs)28-31, similar to the physiology of mutant RYR2 receptors in transfected cells and transgenic mice, and the increased propensity for beta-adrenergic triggered arrhythmias in response to exercise and stress32-34.
Although animal models continue to be important for understanding disease mechanisms, they have limitations that might be overcome through the use of human cellular models of disease. In particular, many transgenic mouse models of genetic diseases do not faithfully reflect the human pathophysiology. For example, the spontaneous hematopoietic failure that characterizes human Fanconi anemia patients is not mirrored by mouse models, but does develop in differentiating iPSC cultures35. In the heart, discrepancies between the pathophysiology of transgenic models and the human clinical presentation reflect different ion channel types and distribution, resulting in faster heart rate and shorter action potential duration of the mouse, as well as the differences in cardiac anatomy such as vastly greater wall thickness. Although large animals might pose better disease models, creating transgenic versions of Dolly the sheep with human disease-causing mutations is exceptionally challenging36, and although useful for later stage testing, do not advance target identification or in vitro drug screening. Thus, the early examples of iPSC-based cardiovascular disease models suggest that they are capable of bridging the gap between cell-based studies and animal models with clinical experimentation, which is costly and not generally amenable to mechanistic and target identification studies. Although the recent studies involved monogenic rather than polygenic disorders, presumably because of their relative simplicity, the same strategy and concerns should be relevant to polygenic disease. Regardless of the disease, it is essential that the cellular context, complete with relevant cell type and disease-associated profiles of gene and protein expression, plus the intracellular signaling pathways that influence the disease manifestation, match those in the clinical presentation to ensure that the model comprises the targets for a pharmaceutical therapy.
Modeling the vascular manifestations of Marfan's syndrome (MFS) is a good illustration of contextual relevance. MFS is caused by mutations in the gene encoding fibrillin-1 (FBN1). Although the fibrous connective tissue disorders of MFS were originally attributed to structural weakness of fibrillin-rich extracellular matrix, the pathological manifestations are now thought to involve enhanced bioavailability of TGFβ, explaining the resemblance to MFS-related disorders such as MFS-type II and Loeys-Deitz syndromes caused by elevated signaling through mutated type 1 and type 2 TGFβ receptors. Enhanced TGFβ signaling was recently reported in mesenchymal cells derived from MFS iPSCs and ESCs, and also shown to underlie their spontaneous chondrogenic differentiation, in contrast with normal iPSC/ESC-derived mesenchymal cells that require exogenous TGFβ chondrocytes37. Although consistent with the skeletal manifestations of MFS, they do not model the potentially lethal thoracic aortic aneurysm that is closely associated with smooth muscle dysfunction38. Moreover, the site-specific localization of thoracic aneurysm, as for regional susceptibility to calcification and atherosclerosis, is thought to be linked to the particular developmental origin of vascular smooth muscle cells (VSMCs), which by fate-mapping studies are known to arise from a remarkable diversity of mesenchymal populations in the embryo39. Therefore, relevance of an in-dish phenotype for the vascular MFS phenotype most likely requires recapitulating the VSMC defect. In this regard, the recent paper by Cheung et al.12 is important since it described methods that recapitulate the developmental sequence of events to produce three distinct types of aortic VSMCs from iPSCs: aortic root VSMCs, derived from the lateral plate mesoderm via the secondary heart field, the ascending aorta and arch, which come from neural crest cells that emanate from the lateral border of hindbrain neurectoderm, and the descending aortic VSMCs that originate from somitic mesoderm. Each population exhibits distinct responses to cytokines, including differential activation of matrix metalloproteinase 9 (MMP9) and tissue inhibitor of metalloproteinase 1 (TIMP1), in response to IL-1β that is likely to be relevant to the phenotypic modulation seen prior to aortic aneurysms in animal models40. Given that VSMCs of diverse origin respond differentially to physiological stimuli, the clinical presentation of MFS would be best modeled in neuroectoderm-derived VSMCs since these are analogous to ascending aorta and arch, where the Marfan phenotype is most prominent. Therefore, these cells seem likely to reflect the appropriate cellular context and hence the repertoire of potential drug targets.
Discovery of Disease Mechanisms and Potential Drug Targets
A few groundbreaking studies have probed iPSC-based cardiovascular disease models with small molecules, natural products and biologics (Table 1), clearly demonstrating the feasibility of using disease-in-dish assays to evaluate drug responses. For example, Jung and colleagues found that Dantrolene, which binds to the amino terminal region of RYR2 and restores inter-domain interaction critical for the closed state, alleviated the arrhythmic behavior of CPVT-1 iPSC-derived cardiomyocytes28. Similarly, the application of Ca2+-channel blocker Nifedipine and KATP channel opener Pinacidil shortened the action potential duration and abrogated early after depolarizations in iPSCs-derived Long QT-2 mutant cardiomyocytes16. Although none of the reported iPSC-based studies have evaluated more than a few drugs, these early studies clearly show that the assays can discriminate drug effects, even if translation to clinic will require much more testing than in the immature and monocultured cardiomyocytes. Can iPSC-derived cells be used in high throughput? Moderate throughput screens of mouse and human ESC-derived cells have been effective in identifying compounds that promote cardiogenic mesoderm from stem cells41 and others that target Wnt and TGFβ pathways to control cardiac cell differentiation42, 43, and primary neonatal rat cardiomyocytes have been used in a moderate throughput screen that identified microRNAs that control hypertrophy44. Although encouraging, the studies to date have been low to moderate throughput and therefore have not overcome the formidable hurdles to true high throughput screening (HTS) of hiPSC-based disease models. These include producing the desired and potentially rare cell types for disease modeling reproducibly and in adequate quantity, purity and quality for HTS. Procedures for bioreactor process development, cell banking of primary and differentiated derivatives, and protocols for robotic automation must be devised. Importantly, the assay read-out must have sufficient dynamic range to reliably distinguish disease from control states. Moreover, variability among iPSC line isolates must not be so great as to obliterate a difference between disease and normal states. Implementation of large-scale screening of more than 2 million compounds has been described using mouse ESC-derived neuronal cells, yielding compounds that potentiate a particular glutamate receptor subtype45. Adapting large-scale approaches to iPSC-based human disease models will accelerate their adoption for drug screening.
These issues are addressed during assay development, and ultimately dictate whether the assay can be screened in true high throughput (up to millions of compounds) versus be used in a low or moderate throughput capacity, such as for validating hits or studying their mechanisms of action (typically 1,000 to 50,000 wells). Readouts for cellular assays (Fig. 2) can be whole well measurements, such as by a plate reader (e.g. a gene promoter directing luciferase). This is the most common format and is referred to as HTS. Alternatively, high content screening (HCS) is the analysis of images acquired by automated microscopy, which is capable of providing multiple parameters of features such as cell morphology, protein or gene expression, lipid accumulation, etc. reported for either the whole well or on a cell-by-cell basis. In addition to reading out subcellular details, single cell (cytometric) analysis has the advantage of quantifying the number of cells exhibiting some trait, and this can be a more robust readout than scoring the overall level of activity46. Additionally, the rapid assessment of cells over time enables physiological responses on a whole well basis and is commonly used to measure calcium responses to G-protein coupled receptors (GPCRs) by instruments such as FLIPR (Fluorometric Imaging Plate Reader; Molecular Devices Corp.). Recently, high content imaging and physiological recording are merging with the development of automated means to optically record cellular responses on a cell-by-cell basis at fast (33-100 Hz) rates47.
Figure 2. High throughput assay readouts.
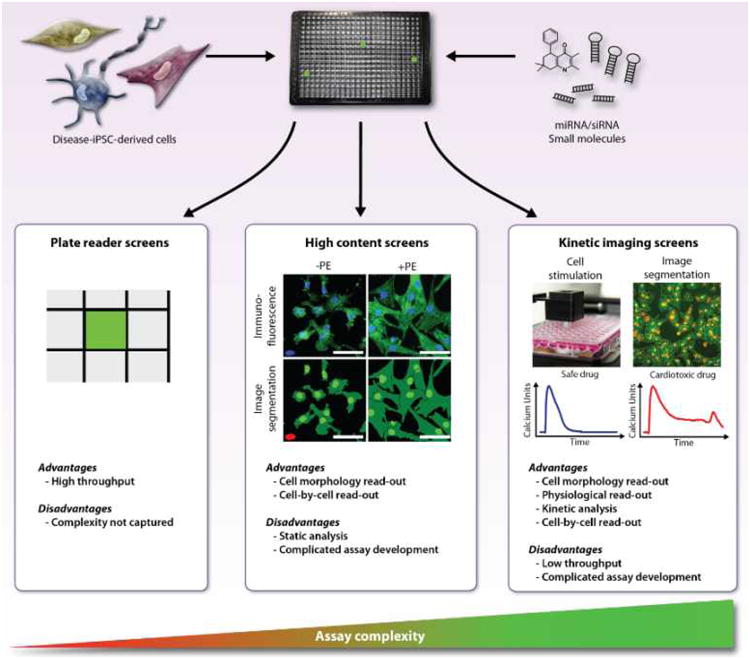
High throughput screening (HTS) readouts measure parameters of entire wells, such as luciferase or fluorescence, and read out by a plate reader (left). Monitoring whole well fluorescence or luminescence over time is also feasible (not shown). In contrast, high content screening (HCS) is based on the quantification of images and has the potential to report multiple parameters on a cell-by-cell (cytometric) basis, such as cardiomyocyte hypertrophy44 (middle panels). Kinetic imaging cytometry is the quantification of fluorescent probes over time and enables cell-by-cell analysis of physiological responses, such as for Ca2+ transients recorded under spontaneous or stimulated conditions, exemplified by the effect of an arrhythmogenic drug47 (right panels). (Illustration Credit: Ben Smith).
An iPSC-based screening assay, assuming it is capable of finding drug candidates that will translate effectively into clinically meaningful activity in patients, can in principle be used in primary screening to discover compounds with novel targets and/or molecular mechanisms of action (MMOAs). MMOA is defined as the biochemical mechanism through which a drug affects its target(s) to elicit a functional response in the cell – for instance, the precise effects on kinetics, stability/degradation, protein modification or redox processes of the target. Since an iPSC-based disease screen would most likely be a “black box” phenotypic assay, meaning that there is no preconceived hypothesis regarding target or MMOA, it could theoretically be harnessed to reveal novel targets and MMOAs that are unlikely to emerge from target-based screening. However, phenotypic assays have two primary disadvantages that need to be addressed. First, knowledge of the target and MMOA of active compounds aids in the subsequent medicinal chemical optimization of drug leads. Therefore, identifying the relevant macromolecular targets of the small molecule leads will likely become a priority but is often extraordinarily challenging (see below). Second, complex disease model screens are expected to have less throughput and be more costly than comparatively simple target-based approaches, especially if they involve multiple cell types, complicated or prolonged differentiation and culture conditions, and image-based readouts, despite advances in HCS and automated microscopy48. While not precluding using iPSC-based disease assays as primary screens, these limitations prompt the question of whether a disease-in-dish screen should be intended to identify novel targets or drug leads. In other words, should the immediate intention be knowledge or drug discovery?
In order to finely characterize the molecular mechanisms of disease, and bring potential drug targets into focus, pursuing the knowledge discovery path from a primary screen through to target identification and characterization of the drug's MMOA requires satisfying two conditions. First, the cellular context of the assay must contain drug targets relevant to ameliorating human disease. Second, there must be a robust means to identify and validate the cellular macromolecules targeted by the screen hits. The molecular disturbances that are the root cause of the disease phenotype may not necessarily be the same as the cellular macromolecules that could be pharmacologically targeted to alleviate disease symptoms. For instance, effective treatment of long QT caused by hERG mutations might involve small molecule targeting of a current other than IKr. Similarly, in contrast to antisense or RNA interference therapies, the potential targets by which a small molecule might treat cardiomyopathies caused by structural alterations in TNNT2 or Duchenne's muscular dystrophy due to truncated dystrophin are unknown, but might be revealed through studying cardiomyocytes derived from patient-specific iPSCs4, 49. As another example, iPSC models of Pompe disease50 might aid in discovering and characterizing small molecules that target signaling proteins to boost expression of the mutated acid alpha-glucosidase (GAA), as well as alter its bioavailability or trafficking, for those patients who can make protein, in addition to directly targeting GAA (e.g. acting as a small molecule chaperone to restore its shape or function). Thus, the cell type used in the assay must have as close a range of signaling pathways and proteins as the cells affected in the patient. More complex formats, such as involving three-dimensional engineered tissues or hard to obtain cell types that match the clinical presentation might be prohibitively costly or unfeasible for HTS. For these reasons, it is logical to consider pursuing an iterative process that uses smaller scale, moderate throughput screens (MTS) of phenotypic assays against functional and chemical genomics libraries (see below) to unveil disease modifying cellular processes and macromolecular targets. Subsequently, validation of key processes and potential druggable targets can be accomplished through re-screening of additional and selective agents, to eventually yield a refined network of cell signaling proteins and/or gene hierarchies that modulate disease phenotypes (Fig. 3). The resulting cell signaling proteins can then be entered into a conventional target-based drug discovery pipeline.
Figure 3. iPSC-based disease-in-dish modelling to discover novel drug targets.

iPSC technology can contribute the human disease context to multiple points in a pharmaceutical development pipeline (indicated as red in the flow diagram at bottom). Screening disease-in-dish assays against focused libraries of molecules represents a powerful functional and chemical genomics approach to identify new drug targets. The key property of the indicated libraries, except for diversity collections of small molecules, is that the cellular targets can be determined in sufficient throughput and reliability to be used in subsequent pathway analysis and validation steps. In contrast, it remains challenging to identify biologically relevant target of small molecule hits from diversity collections since they are promiscuous and largely uncharacterized. Systems biology tools are employed to create an interaction network consisting of the candidate targets and interacting genes/proteins, the key nodes of which can be validated by re-screening using specific small molecule or siRNA/shRNA agents. Following validation and refinement, the network is analyzed for druggable targets that can be entered into conventional drug discovery at the level of high throughput screening (HTS).
Chemical Genomics
In the target-centric model of drug development, the pipeline begins by screening a biochemical or cell-based assay against a large chemical library to identify compounds that modulate a pre-determined target of interest. Compounds are not tested for efficacy in actual disease-relevant assays until considerably later, and often not against human disease until Phase II clinical trials. Although this approach has been very successful over the past decade in developing drugs against well-characterized targets, it carries the risk that drugs that emerge from satisfying the constraints of target and MMOA may not be effective therapeutically, and this has been proposed to contribute to the high rate of drug candidate attrition, discussed in51, 52. By contrast, it is hoped that iPSC disease-in-dish technology will provide a means to introduce the patient-relevant context at the earliest stages of drug discovery and development (Fig. 3). This would have two main advantages: First, it should be possible to obtain efficacy and toxicity data before the nomination of compounds for costly animal and human testing and, second, the ability to directly identify novel disease relevant targets through a chemical genomics or functional genomics approach.
Chemical genomics is the evaluation of the genome by screening phenotypic assays against libraries of small molecules followed by deconvoluting the cellular targets of the “hits”. Functional genomics is conceptually similar but involves libraries of nucleic acids or proteins (e.g. siRNAs, microRNAs or expressed open reading frames) rather than a chemical library. Chemical and functional genomics are analogous to classical “forward” genetic screening by mutagenesis and have the power to dissect mechanisms of human disease, especially when combined with iPSC technology and a systems biology approach to building a signaling and genetic network among the cellular targets of the screen “hits”53.
Chemical and functional genomics, therefore, are powerful tools to uncover disease mechanisms and reveal novel druggable targets for treatment. The disease-in-dish assay is inherently phenotypic, meaning that the endpoint is an alteration in morphology, behavior or physiology of cells in culture, rather than the direct biochemical activity of the target protein. Unlike target-based assays, which typically pose a single particular protein as a target for screening, phenotypic assays present a plethora of potential cellular macromolecules that could be targeted to elicit a desired cellular response. Phenotypic assays are generally more complex and costlier to develop and execute than target-based assays. Consequently, phenotypic assays are often reserved as secondary assays to evaluate hits from traditional target-based screening, although they are generating considerable interest as a means to cast a broad net for novel classes of therapeutic targets51.
Phenotypic screening of complex bioassays has been successful, and has exploited stem cell assays and even whole organism screens54, 55. Indeed, primary screening of phenotypic assays in the pharmaceutical company setting has led to the initial discovery of a number of new small molecule drugs, in particular first-in-class new molecular entities (NMEs). The calcium receptor allosteric activator Cinacalcet56 and the N-type calcium channel blocker Ziconotide57 are examples of drugs that resulted from phenotypic screens with little or no prior knowledge of the MMOA. In addition, phenotypic screens have also elucidated MMOAs for established targets that in turn led to additional small molecule drugs, reviewed in51. More recently, a phenotypic assay for aberrant mitosis led to the discovery of Monastrol, which targets the motor protein Eg5 and is a promising anti-neoplastic agent58.
The fact that the target of a phenotypic assay is not known beforehand influences the choice of the molecular library for screening (Table 2). Primary HTS drug discovery campaigns often evaluate 300,000-1,000,000 small molecule compounds, typically without replicates, necessitating a high dynamic range, extreme assay robustness and low cost59, 60. On the other hand, screens designed to probe pathways and targets, typical of chemical and functional genomics studies, and well suited to the knowledge discovery mission of academia, are generally moderate throughput, on the order of 1,000 to 50,000 wells. Since the cost in terms of effort and resources is relatively low, such assays can be performed in replicate to accommodate the higher level of assay noise often encountered in phenotypic assays. Whole genome RNA interference, microRNA screens and many small molecule library screens are examples of low or moderate throughput screens.
Table 2. Library design and screening goals.
Different library designs carry distinct advantages and disadvantages, and should be matched to the intent of the screen as well as to considerations of scale and cost.
| Library | Size | Type | Pros | Cons | Suitable for: |
|---|---|---|---|---|---|
| Diversity | 105 - >106 | Small molecule | Immediate drug discovery | High cost Target ID difficult |
Drug discovery |
| Focused | <105 | Small molecule | Restricted target coverage Target ID easier |
Restricted target coverage | Drug discovery Target identification |
| Chemical Genomics | <104 | Small molecule | Target ID easy | Not drug discovery | Knowledge discovery Target identification |
| Functional Genomics | <104 | siRNA, shRNA, miRNA, ORF | Target ID easy | Not drug discovery | Knowledge discovery Target identification Discovery of RNA-based therapeutics? |
Abbreviations: ORF, open reading frame; ID, identification.
Small molecule screen libraries fall broadly into two categories, diversity-oriented and focused. Compounds in diversity collections bind many protein classes, and are often used in phenotypic screens when the target is unknown. The tradeoff in using diversity collections is that any one molecule has a low probability of being active, for an excellent discussion see a recent paper by Stockwell60, and identifying the target remains extraordinarily challenging (see below). Focused libraries, in contrast, are oriented towards specific classes of druggable targets and are constructed based on knowledge of the topology of chemicals that bind particular proteins. For iPSC-based screening assays, these might be focused on structures known to modulate the activity of protein classes hypothesized to result in clinical benefit to patients with a particular disease. Focused libraries can also be assembled to probe the involvement of proteins that mediate particular cell signaling or other cellular processes (e.g. collections of kinase inhibitors and pathway modulators to probe signal transduction pathways), permitting chemical genomic dissection of disease mechanisms. Such libraries have been used to reveal mechanisms of cardiomyocyte differentiation42, but they are constrained by the limited set of proteins they affect in the cell. New strategies are being developed to circumvent the limitations of traditional libraries. Among the most interesting is the idea of “biological activity space”, exemplified by a library of chemical structures tailored to affect tumor proliferation assembled based on the structure-activity relationships of hits from anti-proliferation screens of 60 tumor cell lines61.
The number of small molecules is cosmologically vast, yet only a fraction are feasible to synthesize and considered likely to yield good drugs59. For this reason, diversity libraries contain compounds from the intersections between chemical properties that historically have defined oral absorption, distribution, metabolism and excretion properties (ADME space and drug-likeness) and particular structural and physicochemical properties that define high affinity, functional interactions with individual target classes. These compounds tend to target proteins with topologically defined drug-binding pockets, such as enzymes, GPCRs, kinases, nuclear receptors and ion channels62, leaving many biologically interesting proteins such as transcription factors, scaffold proteins, and structural proteins largely unexplored60, 63. It is thought that there are over a 1,000,000 total human proteins, including splice variants, post-translational modifications and somatic mutants64, greatly overshadowing the calculated 3,000-10,0000 so-called “druggable” proteins and the approximately 500 proteins targeted by our current pharmacopeia, although few small molecules can be considered completely selective and not all proteins would make good drug targets62, 65. Thus, typical drug discovery libraries, however vast, only scratch the surface of protein diversity within the cell.
A related issue with using a drug discovery (diversity) library for phenotypic screening is that progressing from hit to a novel target remains serendipitous. A common approach for identifying the targets of small molecules is to affinity capture proteins from cell lysates using compound tethered to a solid matrix, followed by mass spectrometry (MS) analysis and subsequent identification of the binding proteins. The challenges of the chemical proteomics approach lies in promiscuity of hits from diversity libraries coupled with poor sensitivity of the pulldowns, although new mass spectroscopic techniques might improve the target identification beyond current capabilities, reviewed in66. Alone, the technology cannot distinguish between proteins that merely bind from those that are relevant for the small molecule's activity in the cell. The problem is that there can be high affinity chemical-protein interactions with proteins that are irrelevant for the activity, and lower affinity interactions with higher abundance proteins that obscure the relevant target.
Because of the difficulties inherent in identifying targets of hits from diversity library screens, we contend that it is more efficient to screen disease-in-dish assays against libraries of molecules for which the targets can be readily identified, and then to move the targets through a process of in vivo validation and ultimately into the conventional drug discovery pipeline, as opposed to directly employ a black box assay in a primary screen of a diversity library (Fig. 3). Libraries for target and pathway characterization comprise signaling pathway modulators (e.g. kinase inhibitors), known drugs and other well-characterized bioactive molecules. These libraries are potent tools for the molecular dissection of disease mechanisms and might aid in revealing novel therapeutic targets, but are not designed for the direct discovery of NMEs. An example of small-scale screening is to test prescribed drugs against patient iPSC-derived cells in order to tailor a therapy to a particular patient class. On a larger scale, chemical and functional genomics screens can be coupled to systems biology to unveil basic disease mechanisms that might ultimately lead to novel drug targets. HTS screens for these novel targets can then be developed to discover new drugs (Fig. 3).
Functional Genomics, RNA interference and microRNA screening
Oligonucleotide libraries offer an alternative to chemical libraries for probing cardiovascular or other disease phenotypes. RNA interference (siRNA or shRNA) technology has proven to be a powerful reverse genetic method to evaluate the function of candidate genes, and even screen entire genomes, for the identification of pathway components that govern a variety of complex processes, such as proteins that sustain pluripotency in ESCs67, 68. RNA interference functions by introducing a double stranded small interfering (siRNA) or short hairpin (shRNA) RNA into the cell in order to target cognate mRNAs for degradation by the RNA-induced Silencing Complex (RISC). siRNA and shRNA libraries are commercially available for human and other genomes and provide a means to predict the physiological and biological consequences of pharmacological target inhibition.
Whereas siRNAs and shRNAs are synthetic tools designed to target single mRNA species (although selectivity is not assured), microRNA are endogenous, ∼22-nucleotide single-stranded RNAs that directly bind and suppress multiple mRNA targets. For instance, miR-223 is estimated by proteomics to affect more than 200 targets in neutrophils alone69, and an estimated 60% of the total proteome is under direct control by microRNAs70. Even more striking, microRNAs often block the expression of multiple proteins that govern the same biological process. miR-486, for instance, blocks the production of multiple proteins that mediate phosphatidylinositol- 3-OH kinase (PI(3)K)–AKT signal transduction71. Similarly miR-133a1 and miR-133a2 were found to negatively regulate multiple smooth muscle genes in cardiomyocytes, consistent with their role as mediators of serum response factor72. microRNAs bind mRNAs through Watson-Crick base pairing of their “seed” sequence to the 3′ untranslated regions (UTRs) or, less commonly, the coding region. By governing translation, they fine-tune nearly every normal and pathological process examined73, 74. In cardiovascular biology, microRNAs control early embryonic development and adult disease, exemplified by the essential roles of miR-1 and miR-133 in heart development 72, 75 and miR-21 and miR-208a in cardiac remodeling after myocardial infarction76, 77 and metabolism78. A large portion of the proteome is regulated by relatively few microRNAs – there are only approximately 1500 human microRNAs in the human genome (www.mirbase.org) - making libraries of oligonucleotide microRNA mimics an efficient means to elucidate disease-modifying mechanisms, regardless of whether or not a particular microRNA identified through such an approach is normally involved in the disease. Indeed, recent screening of oligonucleotide microRNA mimics has led to the discovery of microRNAs that govern formation of cardiogenic progenitors79, cardiomyocyte cell cycle entry80, and cardiomyocyte hypertrophy44.
microRNAs discovered through screening can be matched to candidate targets through computational and biochemical approaches. Computational algorithms are based on the sequence alignment of the microRNA seed sequence to the 3″ UTR of candidate target genes81, 82. The various software packages differ in the methods used to increase specificity, most commonly by exploiting nucleotide composition rules of the putative binding site(s), structural accessibility, or evolutionarily conservation of location of recognition elements within the 3′ UTR. The interactions between microRNAs and their bonafide targets have been hard to model due to sequences surrounding the recognition site that affect recognition, as well as cellular context that can include UTR-binding cofactors that influence site accessibility, reviewed in73, making computational approaches alone too error-prone for use as a sole means of target identification.
To complement these deficiencies, investigators have turned to biochemical strategies, such as cataloguing the proteins or transcripts that are depleted by overexpression of a particular microRNA, or direct determination of mRNAs that are brought into the RISC. For example, immunoprecipitation of RISC using antibodies against Argonaute-2 protein (Ago2), the component of RISC that selects the microRNA strand to be basepaired to the target mRNA, is effective in pulling down target mRNAs in multiple settings, e.g.83-85. When combined with RNAseq, this approach can yield comprehensive insight into context-dependent targets of overexpressed microRNAs, e.g.86, 87. In our laboratory, we use bioinformatics tools to link predicted targets of active microRNAs, along with interacting proteins, into networks that are then tested and refined by evaluating the effects that selective inhibition (by siRNA or selective small molecule inhibitors) of key network nodes have on the original assay readout. In this way, microRNAs and other functional and chemical genomics screen data can be translated into information about signaling and genetic cascades that control or modulate a disease state.
In addition to interrogating pathological mechanisms to identify drug targets, screening of miRs for activity in disease-in-dish assays might provide knowledge of miR function that could lead to the development of therapeutics that mimic or target miR function. Potential RNA-based therapeutics comprise antisense RNA, ribozymes, RNA decoys, aptamers, small interfering RNA (siRNA), short hairpin RNA (shRNA) and miRs that are distinguished not only by their structures and chemical compositions, but also by their targets and mechanisms of action88. Of these, si/shRNAs and miRs exploit RISC-mediated targeting of mRNAs discussed above, and over 20 candidate si/shRNA and miR therapeutics have been evaluated in clinical trials89. These include vascular targets - for instance, siRNAs to VEGFA (Bevasiranib, Opko Health) and VEGFR1 (AGN-745, Allergan) were evaluated in clinical trials for age-related macular degeneration [Opko Health: phase I (NCT00722384) and phase II (NCT00259753); Allergan: Phase I/II (NCT00363714) and phase II (NCT00395057) trials]. Unfortunately, both failed to achieve desirable clinical endpoints for reasons that have been linked to non-selective activation of Toll-like receptors that potentially could be alleviated by chemical modification of the backbone and enhanced delivery90-92. The first anti-miR candidate therapeutic miravirsen, a chemically modified oligonucleotide designed to inhibit miR-122, has recently advanced to a phase II trial (Santaris Pharma: NCT01200420) to treat chronic Hepatitis C virus infection. There have not yet been clinical trials of miRs or anti-miR therapeutics for heart disease, although numerous miRs can be considered as potential therapeutic targets93, 94.
Cardiotoxicity and Arrhythmogenicity Testing
A valuable near-term opportunity for applying iPSC-derived cardiomyocytes in the drug discovery pipeline is to screen for cardiotoxic and arrhythmogenic effects. Drug-induced cardiotoxicity is difficult to predict95 and consequently remains a major factor for drug failure during development and even withdrawal after market launch, adding to the high cost of drug development96, 97. Many recent failures involve fatal ventricular tachyarrhythmias, including the rare Torsade de Pointes (TdP). TdP is commonly related to inhibition of the delayed rectifier potassium current (IKr), which is mediated by the human Ether-à-go-go Related Gene (hERG/KCNH2) and KCNE2 channels that are responsible for action potential repolarization95, 98. Highly predictive assays involving whole heart or slice preparations and in vivo animal testing remain the standard for pre-clinical safety pharmacology, and extensive testing in humans occurs during Phase III clinical trials99. Assays that are currently of sufficient throughput for use in the early stage discovery, when drug candidates emerge, typically focus on single channels such as hERG and use tumor cell lines, such as automated patch clamp recording of hERG-expressing CHO cells99. hESC and hiPSC-derived cardiomyocytes, despite their electrical and mechanical immaturity, nonetheless recapitulate many complexities of human heart muscle cells. Hence, they offer a quantum leap over tumor cell lines and can be used at the earliest stages of drug discovery100, 101. Moreover, it should also be possible to use the pluripotent stem cell-derived cardiomyocytes to test for other cardiotoxic liabilities, such as cardiomyopathies caused by certain anti-cancer drugs102, that would not be readily detected in non-cardiomyocytes.
A limitation of intracellular or patch-clamp recording for safety pharmacology is inadequate throughput for early stage testing. Multiple electrode array (MEA) devices have been explored as an alternative to single cell recording103, yet, although the throughput is greater, it is still restrictive and the electrode arrays are costly. Intracellular calcium ([Ca2+]i) measured by fluorescent probes or by patch-clamp recording of ICa has been reported to be highly predictive of cardiotoxicity and arrhythmogenicity because it integrates the electrophysiological and signaling events leading to muscle contraction104-107. To improve throughput, automated platforms are being developed for high throughput acquisition of Ca2+ and voltage dynamics using fluorescent reporters in contracting cardiomyocytes from hESC and hiPSC sources with sufficient throughput for early stage drug testing and primary screening108, 109. The instrument described in Cerignoli et al.108 records from all cells within each field of view simultaneously, and analyzes results on a cell-by-cell basis, enabling detailed analysis of individual cells or subsets gated by kinetic parameters (Fig. 2).
One of the most appealing aspects of using hiPSC-derived cardiomyocytes in assessing risk is that cells can be prepared from individuals demonstrated to be susceptible to drug induced arrhythmias, such as TdP. In principle, the predictive power of such a panel of cells, at least for detecting cell autonomous disturbances, could outstrip that of clinical testing given the relatively low incidence of susceptible individuals in the general patient population110. Clinically relevant arrhythmia, especially re-entry-based clinical arrhythmia, depends not only on appropriate channel types and distribution, but on complex regional differences in electrophysiological properties within the ventricular wall that can be altered by pathology95, 111. For this reason, absolute QT intervals of isolated myocytes are not very predictive for arrhythmia incidence and multifactorial methods may be more predictive110, 112. Interestingly, Matsa et al.18 observed that cardiomyocytes from a Long QT-2 patient and her asymptomatic carrier (mother) iPSCs showed more evidence of arrhthymias when sparsely plated cells were measured by single cell patch-clamp recording than when field potentials were measured from more dense aggregates by MEA, which revealed arrhythmia in the patient but not the asymptomatic mother, perhaps indicating a stabilizing effect of non-cardiomyocytes and suggesting that assessing complex populations might be better than isolated cardiomyocytes at predicting arrhythmia. Thus, the challenge of how to model complex electrophysiological phenomena using patient-specific iPSC-derived cardiomyocytes remains significant but, if solved, could greatly increase the predictive power of in vitro testing. Peering further into the future, a better understanding of the mechanisms that drive maturation, coupled with the development of materials that reproduce the three-dimensional structure of myocardium, should enormously increase the utility of pluripotent stem cell-derived cardiomyocytes for assessing cardiotoxicity of new drugs.
Conclusions and Prospects
iPSC-based disease models represent a powerful new tool for drug discovery, with promising applications for primary phenotypic screening, elucidation of novel targets, and physiological assays for evaluating cardiotoxicity and optimization of patient specific therapies. Although limited in scope, ground-breaking studies have shown that is possible to elicit disease-relevant phenotypic changes in response to drugs, supporting the view that iPSC models can recapitulate genetically inherited disorders and be used for phenotypic screening. However, a number of major questions regarding the application of the iPSC cardiovascular disease models remain to be answered. Most importantly, to what extent will physiological studies on iPSC derivatives predict a drug's effect on the clinical condition, in particular given that the cells are isolated from other cells in the body, maintained in vitro, and possibly exhibit an immature physiological response to the drugs? For example, the drugs shown by Itzhaki et al.16 to be effective in an iPSC-based Long QT-2 model, Nifedipine and Pinacidil, would not be considered clinically at these doses because they would decrease vascular tone and be unacceptably hypotensive. It would be unrealistic to expect that small molecules found active in iPSC-based disease models in vitro would be immediately and consistently applicable to disease in humans; thus, devising additional disease-relevant assays remains an important challenge of the development process. Nonetheless, early indications suggest that iPSC-based disease models can make predictive primary screens, and, by also enabling more clinically relevant secondary assays, will be an effective entry point to tailoring drug treatments or discovering NMEs.
Phenotypic assays are increasingly implemented in the pharmaceutical company setting not only for secondary assays and in mechanistic studies, but also for primary screening. When used for primary screening they represent a neo-classical alternative to target-based approaches since they mirror practitioner experience as the classical means of discovering compounds. Whether phenotypic approaches can reverse the trend towards fewer innovative new approved drugs per increasing investment in drug research and development and offset the anticipated revenue loss due to patent expirations113 remains uncertain. However, a recent report by Swinney and Anthony51, who analyzed NMEs between 1999-2008, concluded that phenotypic screening outstripped target-based approach for first-in-class small molecule NMEs, 28 to 17, but target-based bested phenotypic for follower small molecule NMEs, 53 to 30. Phenotypic assays also showed a broader range of MMOAs than did target-based assays. This suggests that recapitulating the cellular context in a phenotypic assay increases the likelihood of developing a successful drug against a novel target and/or with optimal MMOA, increasing the efficiency of the drug discovery and development process. Therefore, the ability to generate iPSCs from patients with diseases of characterized as well as uncharacterized etiologies represent an unprecedented and powerful resource since it is now theoretically possible to generate large numbers of organ-specific cell types to produce more realistic disease models.
More research is needed to harness the potential of iPSCs. First, by working out methods for directing the differentiation of disease relevant cell types, including solving the problem of producing cells that recapitulate adult disease without the (possibly many year) latency to overt clinical presentation. Second, it will be important to develop improved methods for recreating the three-dimensional architecture of vascular and myocardial tissue. This will be a tremendous advance that will certainly aid in creating representative physiological models, for example to study substrates for arrhythmia and for measuring physical and other properties relevant to myocardial hypertrophy and heart failure114, and lead to more realistic assays. Finally, improved proteomic methods for the identification of the protein targets of drugs from black box phenotypic assays are needed to help advance compounds through the development pipeline. Pursuing these challenges will introduce the patient context to the earliest stages of the drug pipeline, thereby broadening the scope of target and MMOA diversity and promising to increase the efficiency and productivity of drug discovery.
Acknowledgments
We thank Anne Bang, Vincent Chen, Michael Jackson (Sanford-Burnham Medical Research Institute) and Pilar Ruiz-Lozano (Stanford School of Medicine) for critical discussions and comments on the manuscript.
Sources of Funding: We are grateful for support from California Institute for Regenerative Medicine (CIRM) (RC1-00132), Mathers Charitable Foundation, Fondation Leducq Transatlantic Alliance and NIH (R01 HL113601). AC and EW were both supported by CIRM postdoctoral fellowships and are presently American Heart Association postdoctoral fellows.
Disclosures: The authors acknowledge research grant (MM) and fellowship (AC and EW) support from agencies listed in the Sources of Funding.
Non-standard Abbreviations
- ADME
absorption, distribution, metabolism and excretion
- GAA
acid alpha-glucosidase
- Ago2
Argonaute-2 protein
- CPVT-1
catecholaminergic polymorphic ventricular tachycardia type 1
- DADs
delayed after depolarizations
- FLIPR
Fluorometric Imaging Plate Reader
- GPCRs
G-protein coupled receptors
- HTS
high throughput screening
- hERG
human Ether-à-go-go Related Gene
- iPSCs
induced pluripotent stem cells
- [Ca2+]i
Intracellular calcium
- MFS
Marfan's syndrome
- MS
mass spectrometry
- MMP9
matrix metalloproteinase 9
- MTS
moderate throughput screens
- MMOAs
molecular mechanisms of action
- MEA
multiple electrode array
- NMEs
new molecular entities
- PI(3)K
phosphatidylinositol- 3-OH kinase
- RISC
RNA-induced Silencing Complex
- RYR2
ryanodine receptor type 2
- shRNAs
short hairpin ribonucleic acids
- siRNAs
small interfering ribonucleic acids
- TIMP1
tissue inhibitor of metalloproteinase 1
- TdP
Torsade de Pointes
- UTR
untranslated region
- VSMC
vascular smooth muscle cell
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 2.Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 3.Yu J, Vodyanik MA, Smuga-Otto K, Antosiewicz-Bourget J, Frane JL, Tian S, Nie J, Jonsdottir GA, Ruotti V, Stewart R, Slukvin II, Thomson JA. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007;318:1917–1920. doi: 10.1126/science.1151526. [DOI] [PubMed] [Google Scholar]
- 4.Park IH, Zhao R, West JA, Yabuuchi A, Huo H, Ince TA, Lerou PH, Lensch MW, Daley GQ. Reprogramming of human somatic cells to pluripotency with defined factors. Nature. 2008;451:141–146. doi: 10.1038/nature06534. [DOI] [PubMed] [Google Scholar]
- 5.Saha K, Jaenisch R. Technical challenges in using human induced pluripotent stem cells to model disease. Cell Stem Cell. 2009;5:584–595. doi: 10.1016/j.stem.2009.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Okano H, Nakamura M, Yoshida K, Okada Y, Tsuji O, Nori S, Ikeda E, Yamanaka S, Miura K. Steps toward safe cell therapy using induced pluripotent stem cells. Circ Res. 2013;112 doi: 10.1161/CIRCRESAHA.111.256149. [DOI] [PubMed] [Google Scholar]
- 7.de Almeida PE, Ransohoff JD, Abu N, Wu JC. Immunogenicity of pluripotent stem cell-derived therapeutics. Circ Res. 2013;112 doi: 10.1161/CIRCRESAHA.111.249243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wu SM, Hochedlinger K. Harnessing the potential of induced pluripotent stem cells for regenerative medicine. Nature cell biology. 2011;13:497–505. doi: 10.1038/ncb0511-497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kattman SJ, Witty AD, Gagliardi M, Dubois NC, Niapour M, Hotta A, Ellis J, Keller G. Stage-specific optimization of activin/nodal and bmp signaling promotes cardiac differentiation of mouse and human pluripotent stem cell lines. Cell Stem Cell. 2011;8:228–240. doi: 10.1016/j.stem.2010.12.008. [DOI] [PubMed] [Google Scholar]
- 10.Vazao H, das Neves RP, Graos M, Ferreira L. Towards the maturation and characterization of smooth muscle cells derived from human embryonic stem cells. PLoS ONE. 2011;6:e17771. doi: 10.1371/journal.pone.0017771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kane NM, Meloni M, Spencer HL, Craig MA, Strehl R, Milligan G, Houslay MD, Mountford JC, Emanueli C, Baker AH. Derivation of endothelial cells from human embryonic stem cells by directed differentiation: Analysis of microrna and angiogenesis in vitro and in vivo. Arteriosclerosis, thrombosis, and vascular biology. 2010;30:1389–1397. doi: 10.1161/ATVBAHA.110.204800. [DOI] [PubMed] [Google Scholar]
- 12.Cheung C, Bernardo AS, Trotter MW, Pedersen RA, Sinha S. Generation of human vascular smooth muscle subtypes provides insight into embryological origin-dependent disease susceptibility. Nat Biotechnol. 2012;30:165–173. doi: 10.1038/nbt.2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li Qian, Srivastava D. Direct cardiac reprogramming: From developmental biology to cardiac regeneration. Circ Res. 2013;112 doi: 10.1161/CIRCRESAHA.112.300625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ma T, Xie M, Laurent T, Ding S. Progress in the reprogramming of somatic cells. Circ Res. 2013;112:xxx–xxx. doi: 10.1161/CIRCRESAHA.111.249235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moretti A, Bellin M, Welling A, Jung CB, Lam JT, Bott-Flugel L, Dorn T, Goedel A, Hohnke C, Hofmann F, Seyfarth M, Sinnecker D, Schomig A, Laugwitz KL. Patient-specific induced pluripotent stem-cell models for long-qt syndrome. N Engl J Med. 2010;363:1397–1409. doi: 10.1056/NEJMoa0908679. [DOI] [PubMed] [Google Scholar]
- 16.Itzhaki I, Maizels L, Huber I, Zwi-Dantsis L, Caspi O, Winterstern A, Feldman O, Gepstein A, Arbel G, Hammerman H, Boulos M, Gepstein L. Modelling the long qt syndrome with induced pluripotent stem cells. Nature. 2011;471:225–229. doi: 10.1038/nature09747. [DOI] [PubMed] [Google Scholar]
- 17.Yazawa M, Hsueh B, Jia X, Pasca AM, Bernstein JA, Hallmayer J, Dolmetsch RE. Using induced pluripotent stem cells to investigate cardiac phenotypes in timothy syndrome. Nature. 2011;471:230–234. doi: 10.1038/nature09855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Matsa E, Rajamohan D, Dick E, Young L, Mellor I, Staniforth A, Denning C. Drug evaluation in cardiomyocytes derived from human induced pluripotent stem cells carrying a long qt syndrome type 2 mutation. European heart journal. 2011;32:952–962. doi: 10.1093/eurheartj/ehr073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Carvajal-Vergara X, Sevilla A, D'Souza SL, Ang YS, Schaniel C, Lee DF, Yang L, Kaplan AD, Adler ED, Rozov R, Ge Y, Cohen N, Edelmann LJ, Chang B, Waghray A, Su J, Pardo S, Lichtenbelt KD, Tartaglia M, Gelb BD, Lemischka IR. Patient-specific induced pluripotent stem-cell-derived models of leopard syndrome. Nature. 2010;465:808–812. doi: 10.1038/nature09005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu GH, Barkho BZ, Ruiz S, Diep D, Qu J, Yang SL, Panopoulos AD, Suzuki K, Kurian L, Walsh C, Thompson J, Boue S, Fung HL, Sancho-Martinez I, Zhang K, Yates J, 3rd, Izpisua Belmonte JC. Recapitulation of premature ageing with ipscs from hutchinson-gilford progeria syndrome. Nature. 2011;472:221–225. doi: 10.1038/nature09879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang J, Lian Q, Zhu G, Zhou F, Sui L, Tan C, Mutalif RA, Navasankari R, Zhang Y, Tse HF, Stewart CL, Colman A. A human ipsc model of hutchinson gilford progeria reveals vascular smooth muscle and mesenchymal stem cell defects. Cell Stem Cell. 2011;8:31–45. doi: 10.1016/j.stem.2010.12.002. [DOI] [PubMed] [Google Scholar]
- 22.Chen HS, Kim C, Mercola M. Electrophysiological challenges of cell-based myocardial repair. Circulation. 2010;120:2496–2508. doi: 10.1161/CIRCULATIONAHA.107.751412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zeevi-Levin N, Itskovitz-Eldor J, Binah O. Functional properties of human embryonic stem cell-derived cardiomyocytes. Critical reviews in eukaryotic gene expression. 2010;20:51–59. doi: 10.1615/critreveukargeneexpr.v20.i1.40. [DOI] [PubMed] [Google Scholar]
- 24.Cox SJ, Gunberg DL. Metabolite utilization by isolated embryonic rat hearts in vitro. Journal of embryology and experimental morphology. 1972;28:235–245. [PubMed] [Google Scholar]
- 25.Fisher DJ, Heymann MA, Rudolph AM. Myocardial oxygen and carbohydrate consumption in fetal lambs in utero and in adult sheep. The American journal of physiology. 1980;238:H399–405. doi: 10.1152/ajpheart.1980.238.3.H399. [DOI] [PubMed] [Google Scholar]
- 26.Chung S, Dzeja PP, Faustino RS, Perez-Terzic C, Behfar A, Terzic A. Mitochondrial oxidative metabolism is required for the cardiac differentiation of stem cells. Nature clinical practice Cardiovascular medicine. 2007;4(1):S60–67. doi: 10.1038/ncpcardio0766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.San Martin N, Cervera AM, Cordova C, Covarello D, McCreath KJ, Galvez BG. Mitochondria determine the differentiation potential of cardiac mesoangioblasts. Stem Cells. 2011;29:1064–1074. doi: 10.1002/stem.654. [DOI] [PubMed] [Google Scholar]
- 28.Jung CB, Moretti A, Mederos YSM, Iop L, Storch U, Bellin M, Dorn T, Ruppenthal S, Pfeiffer S, Goedel A, Dirschinger RJ, Seyfarth M, Lam JT, Sinnecker D, Gudermann T, Lipp P, Laugwitz KL. Dantrolene rescues arrhythmogenic ryr2 defect in a patient-specific stem cell model of catecholaminergic polymorphic ventricular tachycardia. EMBO molecular medicine. 2012;4:180–191. doi: 10.1002/emmm.201100194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fatima A, Xu G, Shao K, Papadopoulos S, Lehmann M, Arnaiz-Cot JJ, Rosa AO, Nguemo F, Matzkies M, Dittmann S, Stone SL, Linke M, Zechner U, Beyer V, Hennies HC, Rosenkranz S, Klauke B, Parwani AS, Haverkamp W, Pfitzer G, Farr M, Cleemann L, Morad M, Milting H, Hescheler J, Saric T. In vitro modeling of ryanodine receptor 2 dysfunction using human induced pluripotent stem cells. Cellular physiology and biochemistry: international journal of experimental cellular physiology, biochemistry, and pharmacology. 2011;28:579–592. doi: 10.1159/000335753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kujala K, Paavola J, Lahti A, Larsson K, Pekkanen-Mattila M, Viitasalo M, Lahtinen AM, Toivonen L, Kontula K, Swan H, Laine M, Silvennoinen O, Aalto-Setala K. Cell model of catecholaminergic polymorphic ventricular tachycardia reveals early and delayed afterdepolarizations. PLoS ONE. 2012;7:e44660. doi: 10.1371/journal.pone.0044660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Itzhaki I, Maizels L, Huber I, Gepstein A, Arbel G, Caspi O, Miller L, Belhassen B, Nof E, Glikson M, Gepstein L. Modeling of catecholaminergic polymorphic ventricular tachycardia with patient-specific human-induced pluripotent stem cells. Journal of the American College of Cardiology. 2012;60:990–1000. doi: 10.1016/j.jacc.2012.02.066. [DOI] [PubMed] [Google Scholar]
- 32.Sedej S, Heinzel FR, Walther S, Dybkova N, Wakula P, Groborz J, Gronau P, Maier LS, Vos MA, Lai FA, Napolitano C, Priori SG, Kockskamper J, Pieske B. Na+-dependent sr ca2+ overload induces arrhythmogenic events in mouse cardiomyocytes with a human cpvt mutation. Cardiovasc Res. 2010;87:50–59. doi: 10.1093/cvr/cvq007. [DOI] [PubMed] [Google Scholar]
- 33.Paavola J, Viitasalo M, Laitinen-Forsblom PJ, Pasternack M, Swan H, Tikkanen I, Toivonen L, Kontula K, Laine M. Mutant ryanodine receptors in catecholaminergic polymorphic ventricular tachycardia generate delayed afterdepolarizations due to increased propensity to ca2+ waves. European heart journal. 2007;28:1135–1142. doi: 10.1093/eurheartj/ehl543. [DOI] [PubMed] [Google Scholar]
- 34.Fernandez-Velasco M, Rueda A, Rizzi N, Benitah JP, Colombi B, Napolitano C, Priori SG, Richard S, Gomez AM. Increased ca2+ sensitivity of the ryanodine receptor mutant ryr2r4496c underlies catecholaminergic polymorphic ventricular tachycardia. Circ Res. 2009;104:201–209. doi: 10.1161/CIRCRESAHA.108.177493. 212p following 209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Raya A, Rodriguez-Piza I, Guenechea G, Vassena R, Navarro S, Barrero MJ, Consiglio A, Castella M, Rio P, Sleep E, Gonzalez F, Tiscornia G, Garreta E, Aasen T, Veiga A, Verma IM, Surralles J, Bueren J, Izpisua Belmonte JC. Disease-corrected haematopoietic progenitors from fanconi anaemia induced pluripotent stem cells. Nature. 2009;460:53–59. doi: 10.1038/nature08129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jacobsen JC, Bawden CS, Rudiger SR, McLaughlan CJ, Reid SJ, Waldvogel HJ, MacDonald ME, Gusella JF, Walker SK, Kelly JM, Webb GC, Faull RL, Rees MI, Snell RG. An ovine transgenic huntington's disease model. Human molecular genetics. 2010;19:1873–1882. doi: 10.1093/hmg/ddq063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Quarto N, Leonard B, Li S, Marchand M, Anderson E, Behr B, Francke U, Reijo-Pera R, Chiao E, Longaker MT. Skeletogenic phenotype of human marfan embryonic stem cells faithfully phenocopied by patient-specific induced-pluripotent stem cells. Proc Natl Acad Sci U S A. 2012;109:215–220. doi: 10.1073/pnas.1113442109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Milewicz DM, Guo DC, Tran-Fadulu V, Lafont AL, Papke CL, Inamoto S, Kwartler CS, Pannu H. Genetic basis of thoracic aortic aneurysms and dissections: Focus on smooth muscle cell contractile dysfunction. Annu Rev Genomics Hum Genet. 2008;9:283–302. doi: 10.1146/annurev.genom.8.080706.092303. [DOI] [PubMed] [Google Scholar]
- 39.Ruddy JM, Jones JA, Spinale FG, Ikonomidis JS. Regional heterogeneity within the aorta: Relevance to aneurysm disease. The Journal of thoracic and cardiovascular surgery. 2008;136:1123–1130. doi: 10.1016/j.jtcvs.2008.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Owens GK, Kumar MS, Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev. 2004;84:767–801. doi: 10.1152/physrev.00041.2003. [DOI] [PubMed] [Google Scholar]
- 41.Sadek H, Hannack B, Choe E, Wang J, Latif S, Garry MG, Garry DJ, Longgood J, Frantz DE, Olson EN, Hsieh J, Schneider JW. Cardiogenic small molecules that enhance myocardial repair by stem cells. Proc Natl Acad Sci U S A. 2008;105:6063–6068. doi: 10.1073/pnas.0711507105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Willems E, Spiering S, Davidovics H, Lanier M, Xia Z, Dawson M, Cashman J, Mercola M. Small-molecule inhibitors of the wnt pathway potently promote cardiomyocytes from human embryonic stem cell-derived mesoderm. Circ Res. 2011;109:360–364. doi: 10.1161/CIRCRESAHA.111.249540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Willems E, Teixeira JC, Schade D, Cai W, Bushway PJ, Lanier M, Walsh CT, Reeves P, Kirchausen T, Izpisua-Belmonte JC, Cashman J, Mercola M. Small-molecule mediated type ii tgfβ receptor degradation promotes cardiomyogenesis in embryonic stem cells. Cell Stem Cell. 2012 doi: 10.1016/j.stem.2012.04.025. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jentzsch C, Leierseder S, Loyer X, Flohrschütz I, Sassi Y, Hartmann D, Thum T, Laggerbauer B, Engelhardt S. A phenotypic screen to identify hypertrophy-modulating micrornas in primary cardiomyocytes. Journal of Molecular and Cellular Cardiology. 2012;52:13–20. doi: 10.1016/j.yjmcc.2011.07.010. [DOI] [PubMed] [Google Scholar]
- 45.McNeish J, Roach M, Hambor J, Mather RJ, Weibley L, Lazzaro J, Gazard J, Schwarz J, Volkmann R, Machacek D, Stice S, Zawadzke L, O'Donnell C, Hurst R. High-throughput screening in embryonic stem cell-derived neurons identifies potentiators of alpha-amino-3-hydroxyl-5-methyl-4-isoxazolepropionate-type glutamate receptors. J Biol Chem. 2010;285:17209–17217. doi: 10.1074/jbc.M109.098814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bushway PJ, Mercola M, Price JH. A comparative analysis of standard microtiter plate reading versus imaging in cellular assays. Assay Drug Dev Technol. 2008;6:557–567. doi: 10.1089/adt.2008.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cerignoli F, Charlot D, Ingermanson R, Gehalot P, Price JH, McDonough PM, Mercola M. High throughput drug risk assessment in human cardiomyocytes by kinetic image cytometry. Submitted (J Pharm Toxicol Methods) 2012 doi: 10.1016/j.vascn.2012.08.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Conrad C, Gerlich DW. Automated microscopy for high-content rnai screening. J Cell Biol. 2010;188:453–461. doi: 10.1083/jcb.200910105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sun N, Yazawa M, Liu J, Han L, Sanchez-Freire V, Abilez OJ, Navarrete EG, Hu S, Wang L, Lee A, Pavlovic A, Lin S, Chen R, Hajjar RJ, Snyder MP, Dolmetsch RE, Butte MJ, Ashley EA, Longaker MT, Robbins RC, Wu JC. Patient-specific induced pluripotent stem cells as a model for familial dilated cardiomyopathy. Science Translational Medicine. 2012;4:130ra147. doi: 10.1126/scitranslmed.3003552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Huang HP, Chen PH, Hwu WL, Chuang CY, Chien YH, Stone L, Chien CL, Li LT, Chiang SC, Chen HF, Ho HN, Chen CH, Kuo HC. Human pompe disease-induced pluripotent stem cells for pathogenesis modeling, drug testing and disease marker identification. Human molecular genetics. 2011;20:4851–4864. doi: 10.1093/hmg/ddr424. [DOI] [PubMed] [Google Scholar]
- 51.Swinney DC, Anthony J. How were new medicines discovered? Nature reviews Drug discovery. 2011;10:507–519. doi: 10.1038/nrd3480. [DOI] [PubMed] [Google Scholar]
- 52.Kola I, Landis J. Can the pharmaceutical industry reduce attrition rates? Nature reviews Drug discovery. 2004;3:711–715. doi: 10.1038/nrd1470. [DOI] [PubMed] [Google Scholar]
- 53.Stockwell BR. Chemical genetics: Ligand-based discovery of gene function. Nature reviews Genetics. 2000;1:116–125. doi: 10.1038/35038557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sachinidis A, Sotiriadou I, Seelig B, Berkessel A, Hescheler J. A chemical genetics approach for specific differentiation of stem cells to somatic cells: A new promising therapeutical approach. Combinatorial chemistry & high throughput screening. 2008;11:70–82. doi: 10.2174/138620708783398322. [DOI] [PubMed] [Google Scholar]
- 55.Wheeler GN, Brandli AW. Simple vertebrate models for chemical genetics and drug discovery screens: Lessons from zebrafish and xenopus. Developmental dynamics: an official publication of the American Association of Anatomists. 2009;238:1287–1308. doi: 10.1002/dvdy.21967. [DOI] [PubMed] [Google Scholar]
- 56.Nemeth EF. Misconceptions about calcimimetics. Annals of the New York Academy of Sciences. 2006;1068:471–476. doi: 10.1196/annals.1346.044. [DOI] [PubMed] [Google Scholar]
- 57.Valentino K, Newcomb R, Gadbois T, Singh T, Bowersox S, Bitner S, Justice A, Yamashiro D, Hoffman BB, Ciaranello R, et al. A selective n-type calcium channel antagonist protects against neuronal loss after global cerebral ischemia. Proc Natl Acad Sci U S A. 1993;90:7894–7897. doi: 10.1073/pnas.90.16.7894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mayer TU, Kapoor TM, Haggarty SJ, King RW, Schreiber SL, Mitchison TJ. Small molecule inhibitor of mitotic spindle bipolarity identified in a phenotype-based screen. Science. 1999;286:971–974. doi: 10.1126/science.286.5441.971. [DOI] [PubMed] [Google Scholar]
- 59.Lipinski C, Hopkins A. Navigating chemical space for biology and medicine. Nature. 2004;432:855–861. doi: 10.1038/nature03193. [DOI] [PubMed] [Google Scholar]
- 60.Stockwell BR. Exploring biology with small organic molecules. Nature. 2004;432:846–854. doi: 10.1038/nature03196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Weinstein JN, Myers TG, O'Connor PM, Friend SH, Fornace AJ, Jr, Kohn KW, Fojo T, Bates SE, Rubinstein LV, Anderson NL, Buolamwini JK, van Osdol WW, Monks AP, Scudiero DA, Sausville EA, Zaharevitz DW, Bunow B, Viswanadhan VN, Johnson GS, Wittes RE, Paull KD. An information-intensive approach to the molecular pharmacology of cancer. Science. 1997;275:343–349. doi: 10.1126/science.275.5298.343. [DOI] [PubMed] [Google Scholar]
- 62.Overington JP, Al-Lazikani B, Hopkins AL. How many drug targets are there? Nature reviews Drug discovery. 2006;5:993–996. doi: 10.1038/nrd2199. [DOI] [PubMed] [Google Scholar]
- 63.Crews CM. Targeting the undruggable proteome: The small molecules of my dreams. Chem Biol. 2010;17:551–555. doi: 10.1016/j.chembiol.2010.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jensen ON. Modification-specific proteomics: Characterization of post-translational modifications by mass spectrometry. Current opinion in chemical biology. 2004;8:33–41. doi: 10.1016/j.cbpa.2003.12.009. [DOI] [PubMed] [Google Scholar]
- 65.Meisner NC, Hintersteiner M, Uhl V, Weidemann T, Schmied M, Gstach H, Auer M. The chemical hunt for the identification of drugable targets. Current opinion in chemical biology. 2004;8:424–431. doi: 10.1016/j.cbpa.2004.06.011. [DOI] [PubMed] [Google Scholar]
- 66.Rix U, Superti-Furga G. Target profiling of small molecules by chemical proteomics. Nat Chem Biol. 2009;5:616–624. doi: 10.1038/nchembio.216. [DOI] [PubMed] [Google Scholar]
- 67.Ding L, Paszkowski-Rogacz M, Nitzsche A, Slabicki MM, Heninger AK, de Vries I, Kittler R, Junqueira M, Shevchenko A, Schulz H, Hubner N, Doss MX, Sachinidis A, Hescheler J, Iacone R, Anastassiadis K, Stewart AF, Pisabarro MT, Caldarelli A, Poser I, Theis M, Buchholz F. A genome-scale rnai screen for oct4 modulators defines a role of the paf1 complex for embryonic stem cell identity. Cell Stem Cell. 2009;4:403–415. doi: 10.1016/j.stem.2009.03.009. [DOI] [PubMed] [Google Scholar]
- 68.Hu G, Kim J, Xu Q, Leng Y, Orkin SH, Elledge SJ. A genome-wide rnai screen identifies a new transcriptional module required for self-renewal. Genes Dev. 2009;23:837–848. doi: 10.1101/gad.1769609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Baek D, Villen J, Shin C, Camargo FD, Gygi SP, Bartel DP. The impact of micrornas on protein output. Nature. 2008;455:64–71. doi: 10.1038/nature07242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Friedman RC, Farh KK, Burge CB, Bartel DP. Most mammalian mrnas are conserved targets of micrornas. Genome research. 2009;19:92–105. doi: 10.1101/gr.082701.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Small EM, O'Rourke JR, Moresi V, Sutherland LB, McAnally J, Gerard RD, Richardson JA, Olson EN. Regulation of pi3-kinase/akt signaling by muscle-enriched microrna-486. Proc Natl Acad Sci U S A. 2010;107:4218–4223. doi: 10.1073/pnas.1000300107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Liu N, Bezprozvannaya S, Williams AH, Qi X, Richardson JA, Bassel-Duby R, Olson EN. Microrna-133a regulates cardiomyocyte proliferation and suppresses smooth muscle gene expression in the heart. Genes Dev. 2008;22:3242–3254. doi: 10.1101/gad.1738708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bartel DP. Micrornas: Target recognition and regulatory functions. Cell. 2009;136:215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Filipowicz W, Bhattacharyya SN, Sonenberg N. Mechanisms of post-transcriptional regulation by micrornas: Are the answers in sight? Nature reviews Genetics. 2008;9:102–114. doi: 10.1038/nrg2290. [DOI] [PubMed] [Google Scholar]
- 75.Zhao Y, Ransom JF, Li A, Vedantham V, von Drehle M, Muth AN, Tsuchihashi T, McManus MT, Schwartz RJ, Srivastava D. Dysregulation of cardiogenesis, cardiac conduction, and cell cycle in mice lacking mirna-1-2. Cell. 2007;129:303–317. doi: 10.1016/j.cell.2007.03.030. [DOI] [PubMed] [Google Scholar]
- 76.Thum T, Gross C, Fiedler J, Fischer T, Kissler S, Bussen M, Galuppo P, Just S, Rottbauer W, Frantz S, Castoldi M, Soutschek J, Koteliansky V, Rosenwald A, Basson MA, Licht JD, Pena JT, Rouhanifard SH, Muckenthaler MU, Tuschl T, Martin GR, Bauersachs J, Engelhardt S. Microrna-21 contributes to myocardial disease by stimulating map kinase signalling in fibroblasts. Nature. 2008;456:980–984. doi: 10.1038/nature07511. [DOI] [PubMed] [Google Scholar]
- 77.van Rooij E, Sutherland LB, Qi X, Richardson JA, Hill J, Olson EN. Control of stress-dependent cardiac growth and gene expression by a microrna. Science. 2007;316:575–579. doi: 10.1126/science.1139089. [DOI] [PubMed] [Google Scholar]
- 78.Grueter CE, van†Rooij E, Johnson BA, DeLeon SM, Sutherland LB, Qi X, Gautron L, Elmquist JK, Bassel-Duby R, Olson EN. A cardiac microrna governs systemic energy homeostasis by regulation of med13. Cell. 2012;149:671–683. doi: 10.1016/j.cell.2012.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Colas AR, McKeithan WL, Cunningham TJ, Bushway PJ, Garmire LX, Duester G, Subramaniam S, Mercola M. Whole-genome microrna screening identifies let-7 and mir-18 as regulators of germ layer formation during early embryogenesis. Genes Dev. 2012;26:2567–2579. doi: 10.1101/gad.200758.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Eulalio A, Mano M, Ferro MD, Zentilin L, Sinagra G, Zacchigna S, Giacca M. Functional screening identifies mirnas inducing cardiac regeneration. Nature. 2012 doi: 10.1038/nature11739. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 81.Alexiou P, Maragkakis M, Papadopoulos GL, Reczko M, Hatzigeorgiou AG. Lost in translation: An assessment and perspective for computational microrna target identification. Bioinformatics. 2009;25:3049–3055. doi: 10.1093/bioinformatics/btp565. [DOI] [PubMed] [Google Scholar]
- 82.Selbach M, Schwanhausser B, Thierfelder N, Fang Z, Khanin R, Rajewsky N. Widespread changes in protein synthesis induced by micrornas. Nature. 2008;455:58–63. doi: 10.1038/nature07228. [DOI] [PubMed] [Google Scholar]
- 83.Beitzinger M, Peters L, Zhu JY, Kremmer E, Meister G. Identification of human microrna targets from isolated argonaute protein complexes. RNA biology. 2007;4:76–84. doi: 10.4161/rna.4.2.4640. [DOI] [PubMed] [Google Scholar]
- 84.Easow G, Teleman AA, Cohen SM. Isolation of microrna targets by mirnp immunopurification. RNA. 2007;13:1198–1204. doi: 10.1261/rna.563707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zhang L, Ding L, Cheung TH, Dong MQ, Chen J, Sewell AK, Liu X, Yates JR, 3rd, Han M. Systematic identification of c. Elegans mirisc proteins, mirnas, and mrna targets by their interactions with gw182 proteins ain-1 and ain-2. Molecular cell. 2007;28:598–613. doi: 10.1016/j.molcel.2007.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Goff LA, Davila J, Swerdel MR, Moore JC, Cohen RI, Wu H, Sun YE, Hart RP. Ago2 immunoprecipitation identifies predicted micrornas in human embryonic stem cells and neural precursors. PLoS ONE. 2009;4:e7192. doi: 10.1371/journal.pone.0007192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Matkovich SJ, Van Booven DJ, Eschenbacher WH, Dorn GW., 2nd Risc rna sequencing for context-specific identification of in vivo microrna targets. Circ Res. 2011;108:18–26. doi: 10.1161/CIRCRESAHA.110.233528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Melnikova I. Rna-based therapies. Nat rev drug discovery. 2007;6:863–864. doi: 10.1038/nrd2314. [DOI] [PubMed] [Google Scholar]
- 89.Burnett JC, Rossi JJ. Rna-based therapeutics: Current progress and future prospects. Chem Biol. 2012;19:60–71. doi: 10.1016/j.chembiol.2011.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Cho WG, Albuquerque RJ, Kleinman ME, Tarallo V, Greco A, Nozaki M, Green MG, Baffi JZ, Ambati BK, De Falco M, Alexander JS, Brunetti A, De Falco S, Ambati J. Small interfering rna-induced tlr3 activation inhibits blood and lymphatic vessel growth. Proc Natl Acad Sci U S A. 2009;106:7137–7142. doi: 10.1073/pnas.0812317106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kleinman ME, Yamada K, Takeda A, Chandrasekaran V, Nozaki M, Baffi JZ, Albuquerque RJ, Yamasaki S, Itaya M, Pan Y, Appukuttan B, Gibbs D, Yang Z, Kariko K, Ambati BK, Wilgus TA, DiPietro LA, Sakurai E, Zhang K, Smith JR, Taylor EW, Ambati J. Sequence- and target-independent angiogenesis suppression by sirna via tlr3. Nature. 2008;452:591–597. doi: 10.1038/nature06765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Samuel-Abraham S, Leonard JN. Staying on message: Design principles for controlling nonspecific responses to sirna. The FEBS journal. 2010;277:4828–4836. doi: 10.1111/j.1742-4658.2010.07905.x. [DOI] [PubMed] [Google Scholar]
- 93.Condorelli G, Latronico MV, Dorn GW., 2nd Micrornas in heart disease: Putative novel therapeutic targets? European heart journal. 2010;31:649–658. doi: 10.1093/eurheartj/ehp573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Divakaran V, Mann DL. The emerging role of micrornas in cardiac remodeling and heart failure. Circ Res. 2008;103:1072–1083. doi: 10.1161/CIRCRESAHA.108.183087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Hoffmann P, Warner B. Are herg channel inhibition and qt interval prolongation all there is in drug-induced torsadogenesis? A review of emerging trends. J Pharmacol Toxicol Methods. 2006;53:87–105. doi: 10.1016/j.vascn.2005.07.003. [DOI] [PubMed] [Google Scholar]
- 96.Qian JY, Guo L. Altered cytosolic ca2+ dynamics in cultured guinea pig cardiomyocytes as an in vitro model to identify potential cardiotoxicants. Toxicology in Vitro. 2010;24:960–972. doi: 10.1016/j.tiv.2009.12.027. [DOI] [PubMed] [Google Scholar]
- 97.Denning C, Anderson D. Cardiomyocytes from human embryonic stem cells as predictors of cardiotoxicity. Drug Discovery Today: Therapeutic Strategies. 2008;5:223–232. [Google Scholar]
- 98.Redfern WS, Carlsson L, Davis AS, Lynch WG, MacKenzie I, Palethorpe S, Siegl PK, Strang I, Sullivan AT, Wallis R, Camm AJ, Hammond TG. Relationships between preclinical cardiac electrophysiology, clinical qt interval prolongation and torsade de pointes for a broad range of drugs: Evidence for a provisional safety margin in drug development. Cardiovasc Res. 2003;58:32–45. doi: 10.1016/s0008-6363(02)00846-5. [DOI] [PubMed] [Google Scholar]
- 99.Guth BD. Preclinical cardiovascular risk assessment in modern drug development. Toxicological Sciences. 2007;97:4–20. doi: 10.1093/toxsci/kfm026. [DOI] [PubMed] [Google Scholar]
- 100.Jonsson MK, Duker G, Tropp C, Andersson B, Sartipy P, Vos MA, van Veen TA. Quantified proarrhythmic potential of selected human embryonic stem cell-derived cardiomyocytes. Stem cell research. 2010;4:189–200. doi: 10.1016/j.scr.2010.02.001. [DOI] [PubMed] [Google Scholar]
- 101.Otsuji TG, Minami I, Kurose Y, Yamauchi K, Tada M, Nakatsuji N. Progressive maturation in contracting cardiomyocytes derived from human embryonic stem cells: Qualitative effects on electrophysiological responses to drugs. Stem cell research. 2010;4:201–213. doi: 10.1016/j.scr.2010.01.002. [DOI] [PubMed] [Google Scholar]
- 102.Menna P, Salvatorelli E, Minotti G. Cardiotoxicity of antitumor drugs. Chemical Research in Toxicology. 2008;21:978–989. doi: 10.1021/tx800002r. [DOI] [PubMed] [Google Scholar]
- 103.Braam SR, Tertoolen L, van de Stolpe A, Meyer T, Passier R, Mummery CL. Prediction of drug-induced cardiotoxicity using human embryonic stem cell-derived cardiomyocytes. Stem cell research. 2010;4:107–116. doi: 10.1016/j.scr.2009.11.004. [DOI] [PubMed] [Google Scholar]
- 104.Takahashi A, Camacho P, Lechleiter JD, Herman B. Measurement of intracellular calcium. Physiol Rev. 1999;79:1089–1125. doi: 10.1152/physrev.1999.79.4.1089. [DOI] [PubMed] [Google Scholar]
- 105.Mattheakis LC, Ohler LD. Seeing the light: Calcium imaging in cells for drug discovery. Drug Discovery Today. 2000;5:15–19. [Google Scholar]
- 106.Monteith GR, Bird GSJ. Techniques: High-throughput measurement of intracellular ca2+ back to basics. Trends in pharmacological sciences. 2005;26:218–223. doi: 10.1016/j.tips.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 107.Komukai K, O-Uchi J, Morimoto S, Kawai M, Hongo K, Yoshimura M, Kurihara S. Role of ca2+/calmodulin-dependent protein kinase ii in the regulation of the cardiac l-type ca2+ current during endothelin-1 stimulation. Am J Physiol Heart Circ Physiol. 2010;298:1902–1907. doi: 10.1152/ajpheart.01141.2009. [DOI] [PubMed] [Google Scholar]
- 108.Cerignoli F, Charlot D, Whittaker R, Ingermanson R, Gehalot P, Savtchenko A, Gallacher DJ, Towart R, Price JH, McDonough PM, Mercola M. High throughput measurement of ca(2+) dynamics for drug risk assessment in human stem cell-derived cardiomyocytes by kinetic image cytometry. Journal of pharmacological and toxicological methods. 2012;66:246–256. doi: 10.1016/j.vascn.2012.08.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Lee P, Klos M, Bollensdorff C, Hou L, Ewart P, Kamp TJ, Zhang J, Bizy A, Guerrero-Serna G, Kohl P, Jalife J, Herron TJ. Simultaneous voltage and calcium mapping of genetically purified human induced pluripotent stem cell-derived cardiac myocyte monolayers. Circ Res. 2012;110:1556–1563. doi: 10.1161/CIRCRESAHA.111.262535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kannankeril P, Roden DM, Darbar D. Drug-induced long qt syndrome. Pharmacological reviews. 2010;62:760–781. doi: 10.1124/pr.110.003723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Antzelevitch C. Arrhythmogenic mechanisms of qt prolonging drugs: Is qt prolongation really the problem? J Electrocardiol. 2004;37(Suppl):15–24. doi: 10.1016/j.jelectrocard.2004.08.004. [DOI] [PubMed] [Google Scholar]
- 112.Valentin JP, Hoffmann P, De Clerck F, Hammond TG, Hondeghem L. Review of the predictive value of the langendorff heart model (screenit system) in assessing the proarrhythmic potential of drugs. Journal of pharmacological and toxicological methods. 2004;49:171–181. doi: 10.1016/j.vascn.2004.03.008. [DOI] [PubMed] [Google Scholar]
- 113.Paul SM, Mytelka DS, Dunwiddie CT, Persinger CC, Munos BH, Lindborg SR, Schacht AL. How to improve r&d productivity: The pharmaceutical industry's grand challenge. Nature reviews Drug discovery. 2010;9:203–214. doi: 10.1038/nrd3078. [DOI] [PubMed] [Google Scholar]
- 114.Eschenhagen T, Zimmermann WH. Engineering myocardial tissue. Circ Res. 2005;97:1220–1231. doi: 10.1161/01.RES.0000196562.73231.7d. [DOI] [PubMed] [Google Scholar]
- 115.Ma D, Wei H, Lu J, Ho S, Zhang G, Sun X, Oh Y, Tan SH, Ng ML, Shim W, Wong P, Liew R. Generation of patient-specific induced pluripotent stem cell-derived cardiomyocytes as a cellular model of arrhythmogenic right ventricular cardiomyopathy. European heart journal. 2012 doi: 10.1093/eurheartj/ehs226. [DOI] [PubMed] [Google Scholar]
- 116.Novak A, Barad L, Zeevi-Levin N, Shick R, Shtrichman R, Lorber A, Itskovitz-Eldor J, Binah O. Cardiomyocytes generated from cpvtd307h patients are arrhythmogenic in response to beta-adrenergic stimulation. Journal of cellular and molecular medicine. 2012;16:468–482. doi: 10.1111/j.1582-4934.2011.01476.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Park IH, Arora N, Huo H, Maherali N, Ahfeldt T, Shimamura A, Lensch MW, Cowan C, Hochedlinger K, Daley GQ. Disease-specific induced pluripotent stem cells. Cell. 2008;134:877–886. doi: 10.1016/j.cell.2008.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Davis RP, Casini S, van den Berg CW, Hoekstra M, Remme CA, Dambrot C, Salvatori D, Oostwaard DW, Wilde AA, Bezzina CR, Verkerk AO, Freund C, Mummery CL. Cardiomyocytes derived from pluripotent stem cells recapitulate electrophysiological characteristics of an overlap syndrome of cardiac sodium channel disease. Circulation. 2012;125:3079–3091. doi: 10.1161/CIRCULATIONAHA.111.066092. [DOI] [PubMed] [Google Scholar]