

Abstract
FR900482 and the mitomycins are two intriguing classes of alkaloid natural products that have analogous biological mechanisms and obvious structural similarity. Both classes possess potent anti-cancer activity, a feature that has led to their investigation and implementation for the clinical treatment of human cancer. Given the structural similarity between these natural products, we envisioned a common synthetic strategy by which both classes could be targeted through assembling the mitomycin skeleton prior to further oxidative functionalization. Realization of this strategy with respect to FR900482 was accomplished through the synthesis of 7-epi-FR900482, which displayed equal potency relative to the natural product against two human cancer cell lines. With the challenging goal of a synthesis of either mitomycin or FR900482 in mind, several methodologies where explored. While not all of these methods ultimately proved useful for our synthetic goal, a number of them led to intriguing findings that provide a more complete understanding of several methodologies. In particular, amination via π-allyl palladium complexes for the synthesis of tetrahydroquinolines, 8-membered heterocycle formation via carbonylative lactamization, and amination through late-stage C-H insertion via rhodium catalysis all featured prominently in our synthetic studies.
Keywords: alkaloid, asymmetric catalysis, mitomycin, aziridine, DYKAT
FR900482 and the mitomicyns are an important class of bioactive natural products that have been extensively investigated due to their intriguing molecular architecture and potential anti-cancer properties.1 Indeed, numerous synthetic approaches have been reported that are based upon several unique strategies.2,3 While laudable, these methods are hampered by length and multiple exchanges of protecting groups limiting their overall synthetic efficiency.
We hoped to improve on previous efforts by reducing the number of sythetic steps to the desired target. It was also envisioned that a common intermediate could potentially access either the mitomycin or the FR900482 ring system with maximal flexibility for the variation of the aromatic core. Thus potentially either natural product or analogs of both classes could be prepared through a common synthetic strategy. Because no asymmetric route to the mitomycin family has yet been reported, the ability to access either enantiomer of the mitomycin system would represent a significant milestone in synthetic methods toward this family of natural products. Herein we disclose our venture toward that goal.
Results and Discussion
Pd-catalyzed amination
Our initial retrosynthesis of 1 envisioned that the [3.3.1] bicyclic core of FR900482 would be assembled via transannular hemiketalization of the 8-membered hydroxylamine derivative 8. The key transformation involved an 8-endo cyclization of palladium π-allyl complex 11 to generate the benzazocine 10. This intermediate could in turn be derived from allene 12 or the corresponding allylic carbonate 13.
While this retrosynthetic analysis is unconventional, precedent existed for cyclization to yield an 8-membered carbocycle in preference to a possible 6-membered isomer through a palladium π-allyl alkylation.4 For example, both allylic carbonates and allenes were known to favor the formation of the larger 8 or 9 membered rings, with little or none of the smaller 6 or 7 membered ring systems (Eq. 1–3). Although an aniline would be the nucleophile in our case we were hopeful that regioselective formation of the benzazocine would be possible considering that the benzazocine core has minimal transannular interations, which typically disfavor medium sized ring formation.5 This should allow for nucleophilic attack at the sterically more accesssible terminal position of the π–allyl unit.
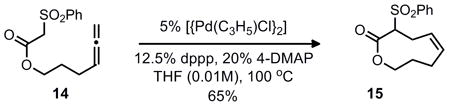 |
Equation 1 |
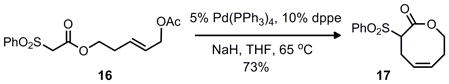 |
Equation 2 |
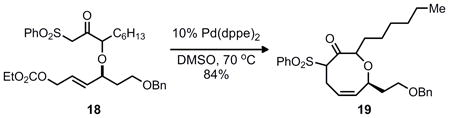 |
Equation 3 |
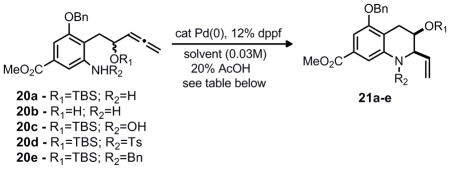
To examine whether this cyclization would be possible, anilines 20a-e were prepared. Hydropalladation of the terminal allene was anticipated to precede 8-endo cyclization onto the palladium π-allyl complex. Under a variety of conditions (Table 1) formation of the desired benzazocine core was not observed and instead the tetrahydroquinoline ring system was formed (21a-e) with good to excellent diastereoselectivity in some cases. Both the yield and the diastereoselectivity of the cyclization varied with the electronic and steric environment of the arylamine nucleophile and allene electrophile.
Table 1.
Selected optimization studies for Pd-π-allyl alkylation of 20
| ||||||
|---|---|---|---|---|---|---|
| Entry | Substrate | Pd Sourcea | Solvent | T(°C) | Yieldb | d.r.c |
| 1 | 20a | Pd2dba3·CHCl3 | THF | 90 | sm | - |
| 2d | 20a | Pd2dba3·CHCl3 | THF | 90 | sm | - |
| 3d | 20a | Pd2dba3·CHCl3 | dioxane | 100 | sm | - |
| 4 | 20a | [{Pd(C3H5)Cl}2] | dioxane | 100 | 94% | 92:8 |
| 5d | 20a | [{Pd(C3H5)Cl}2] | dioxane | 100 | 94% | 76:24 |
| 6 | 20b | Pd2dba3·CHCl3 | THF | 80 | 27% 10% |
62:38 |
| 7 | 20c | [Pd(C3H5)Cl]2 | dioxane | 100 | 21a + 37% 20a | >95:5 |
| 8 | 20d | [Pd(C3H5)Cl]2 | dioxane | 100 | 88% | 85:15 |
| 9 | 20e | [Pd(C3H5)Cl]2 | dioxane | 100 | trace pdt | - |
5% of Pd source.
Isolated yield
Diastereomeric ratios were determined by NMR analysis of the crude reaction mixture.
4% TBAT was used as an additive.
Further experimentation focused on synthesis of the corresponding allylic carbonates (22a and 22b). It was anticipated that some of the entropic barrier to 8-endo cyclization could be overcome by generation of the anti π-allyl complex, which must be the initial intermediate resulting from ionization of 22a or 22b. Furthermore, the 8-endo product must arise from attack on the anti π-allyl, as the syn intermediate would lead to a trans olefin within the newly formed ring. Again good yields and excellent diastereoselectivities could be obtained, but only for the undesired tetrahydroquinolines 23a and 23b. Thus, it appears that Pd-catalyzed cyclizations to form benzazocins cannot be achieved when the formation of a tetrahydroquinoline is a competing process.
While both of these amination methods were ultimately not productive for continuing on with the synthesis, it is important to note that the 1,2,3,4-tetrahydroquinolines 24 are a class of medicinally important compounds in their own right. Among these L-689,5606 (25) and vitantmycin (26)7 are two examples of highly substituted tetrahydroquinolines that might be amenable to synthesis through this method (Figure 2).
Figure 2.
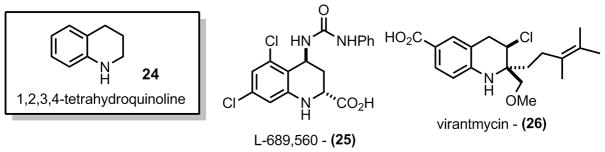
Pharmaceutically important tetrahydroquinolines
Carbonylative Lactamization
Our initial retrosynthetic analysis was modified in favor of a carbonylative lactamization approach (Scheme 2). While carbonylative lactamization had not been reported for the formation of 8-membered heterocycles, this method appeared well suited for formation of the benzazocine core of FR-900482. Preparation of 27 appeared feasible with minimal difficulty and cyclization seemed likely particularly because the olefin and aromatic ring would preclude the transannular interactions and lessen the entropic barrier that causes 8-membered rings to be notoriously difficult to prepare. Synthesis of the requisite lactamization precursor 27 (with incorparation of a handle required for asymmetric synthesis) was accomplished through a Carreira alkynylation of aldehyde 29 with trimethylsilyl acetylene to yield alkyne 31 in good yield and excellent enantioselectivity. The success of this reaction with the highly epimerizable substrate is noteworthy and multiple other alkynylation methods were unsuccessful. While other protocols for this reaction (including Carreria’s catalytic conditions) were attempted, none gave the desired product in acceptable yield. Thus, the synthesis of three lactamization precursors (27a, 27b, and 27c) was accomplished through incorporation of iodine, cis-reduction of the alkyne with diimide, and Sm or Zn reduction of the nitro group to the hydroxylamine oxidation state. It should be noted that samarium(II) iodide functioned with excellent chemoselectivity even in the presence of the vinyl iodide.
Scheme 2.

Modified retrosynthetic analysis of FR-900482
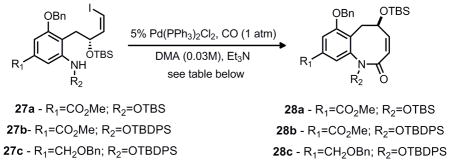
With cyclization substrates 27a-c in hand, a thorough investigation of the palladium-catalyzed cabonylative lactamization was performed (Table 3). Upon refluxing a dilute solution of vinyl iodide 27a, triethylamine, triphenylphosphine, and 5% tetrakis(triphenylphosphine)palladium(0) in THF (0.03M) under an atmosphere of carbon monoxide, a 36% yield of the desired 8-membered ring 28a was obtained, along with some recovered starting material (49% brsm, entry 1). Changing the base (Entry 2) failed to increase the yield, while changing the palladium catalyst and reaction concentration significantly improved the yield (Entries 3 and 4). These optimized reaction conditions proved applicable to modification of the substrate as well. Modification of the protecting group on the hydroxylamine to TBDPS was tolerated, as was employing benzyl ether 27c (Entries 5 and 6).
Table 3.
Selected optimization studies for Pd-catalyzed carbonylative lactamization
| ||||||
|---|---|---|---|---|---|---|
| Entry | Substrate | Pd Source | Solvent | Ligand | Base | Yielda |
| 1 | 27a | Pd[PPh3]4 | THF | PPh3 | Et3N | 36% |
| 2 | 27a | Pd[PPh3]4 | THF | PPh3 | iPr2NEt | 36% |
| 27a | PdCl2[PPh3]2 | DMAb | none | Et3N | 41% | |
| 4 | 27a | PdCl2 [PPh3]2 | DMAc | none | Et3N | 78% |
| 5 | 27b | PdCl2[PPh3]2 | DMAc | none | Et3N | 66% |
| 6 | 27c | PdCl2[PPh3]2 | DMAc | none | Et3N | 64% |
Isolated yield.
0.01M
0.03M
At this stage multiple attempts to install the aziridine functionality on 28 failed. The lactams 28a-c could be selectively reduced (BH3·SMe2, THF, 0 °C, 55%), however, aziridination of either the unsaturated lactam or cis-olefin failed under various nitrene or epoxide based approaches.
In order to circumvent these difficulties, an alternative approach was devised in which the aziridine moiety would be introduced earlier in the synthesis.
Aziridine and Aromatic Fragment Approaches
The revised approach relied on an early and convergent union of an aziridine (39) and aromatic fragment (40) (Scheme 4) and a late stage intramolecular C-C bond formation to complete the 8-membered core (37) of the molecule. Several alternative reactions might accomplish this, but a Nozaki-Hiyama-Kishi or reductive ynal coupling seemed the most likely methods of success.
Scheme 4.
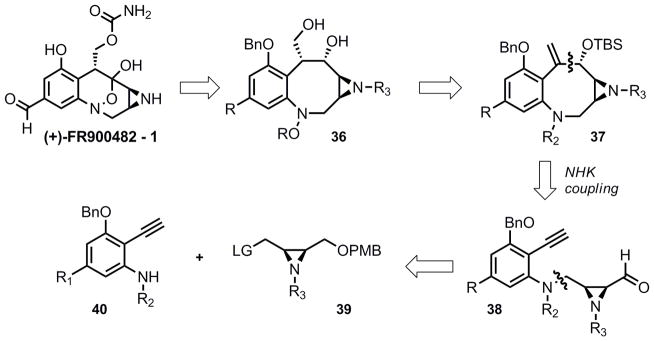
Revised retrosynthesis of FR900482; NHK coupling strategy
As shown in Scheme 5, the aromatic fragment 43 was prepared in three steps from known aldehyde 41.8 A Sonogashira coupling9 of 41 and TMS acetylene was followed by protection of the aldehyde as an acetal. An iron metal reduction of the nitro group gave aniline 43 in a 34% overall yield from 41.
Scheme 5.
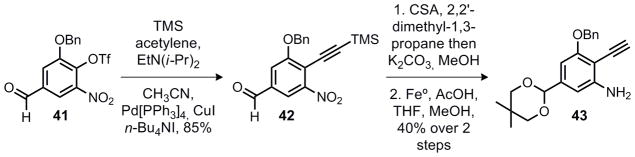
Synthesis of aromatic subunit 45
Synthesis of the aziridine fragment began from the known DYKAT of butadiene monoepoxide (44) and phthalimide (45) outlined in equation 4.10 This reaction yields the protected amino-alcohol 47 in nearly quantitative yield and excellent enantioselectivity.
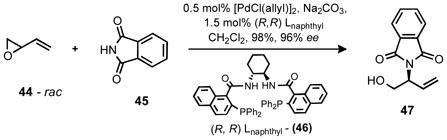 |
Equation 4 |
After conversion of the phthalimide to a Boc carbamate (48), the introduction of the second stereocenter was accomplished in diastereoselective fashion through a syn directed epoxidation11 yielding 49 (Scheme 6). Furthermore, heating the reaction mixture results in selective decomposition of the minor anti epoxide through formation of an oxazolidinone. While the two epoxide diastereomers were chromatographically separable, it proved helpful to heat the reaction for a longer duration on larger scale to simplify the purification process.
Scheme 6.
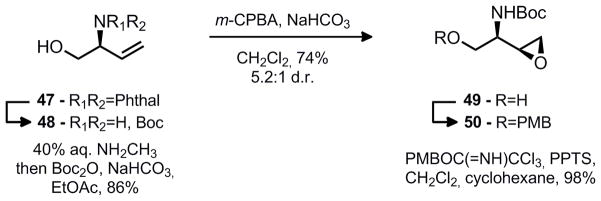
Synthesis of amino-epoxide 50
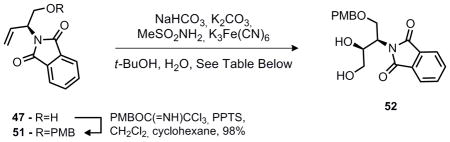
It was anticipated at this stage that the desired aziridine (39) could be prepared via an aza-Payne rearragment12 of 50 if an appropriate base and oxaphilic electrophile is used. Unfortunately, attempts to accomplish this transformation under a variety of conditions proved fruitless and only the oxazolidinone was obtained. One alternative to epoxide opening would be a suitably activated diol, provided reasonable diastereoselectivity could be achieved. Fortunately, under the conditions of Sharpless, good diastereoselectivity and excellent yield were obtained for the formation of diol 52 from 51 (Table 4).
Table 4.
Stereoselective dihydroxylation of 51
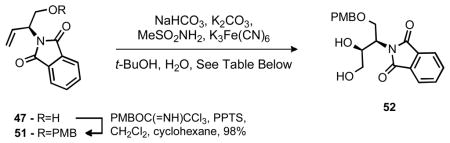
| |||
|---|---|---|---|
| Entry | Catalyst and Ligand | Yield | d.r.a |
| 1 | OsO4 (1 mol%), NMO, THF | 98% | 1.25:1 |
| 2 | K2OsO2(OH)4 (1 mol%), (DHQ)2PHAL (2 mol%) | 71% | 5:1 |
| 3 | K2OsO2(OH)4 (1 mol%), (DHQ)2PYR (2 mol%) | 98% | 8.5:1 |
| 4 | K2OsO2(OH)4 (1 mol%), (DHQ)2AQN (2 mol%) | 90% | 5:1 |
| 5 | K2OsO2(OH)4 (1 mol%), (DHQD)2PHAL (2 mol%) | 85% | 1:1.5 |
Diastereomeric ratio determined by NMR.
With this result in hand, the desired aziridine fragment was quickly completed (Scheme 7). The Sharpless dihydroxylation was sufficiently clean that the immediate product was never purified beyond separatory funnel workup to remove reagents employed in the dihydroxylation. Instead, cleavage of the phthalimide protecting group and in situ protection of the polar amino-diol as a tert-butyl carbamate was accomplished under the conditions described in Scheme 6 to prepare 53.
Scheme 7.

Completion of the aziridine fragment
Chemoselective protection of the primary alcohol with a tert-butyl-diphenylsilyl group and in situ mesylation, followed by warming the crude mesylate in dimethylformamide and cesium carbonate resulted in the formation of aziridine 54. Aziridines 55 and 56 were prepared via desilylation of the TBDPS ether 56 and Dess-Martin oxidation. This entire synthetic sequence requires 8 steps from butadiene monoepoxide and gives 56 in 27% overall yield.
Alternatively, diol 53 could be bis-mesylated and analogous conditions resulted in selective three-membered ring formation providing mesylate 57. Compound 57 could also be prepared by mesylation of 55 (MsCl, Et3N, CH2Cl2, 90%).
While attempts at coupling the fragments 43 and 57 via nucleophilic addition failed, reductive amination of 43 and 56 proved to be a reliable method for accomplishing this transformation (Scheme 8). This reaction was limited to the aniline oxidation state and failed for the corresponding hydroxylamine. Because aldehyde 56 was quickly reduced even in the presence of triacetoxyborohydride, it was essential to drive the dehydration prior to addition of the reducing agent. As a result incorporation of the desired hydroxylamine oxidation state directly was complicated. Similar conditions to yield a coupling product through the intermediacy of a nitrone were not successful.
Scheme 8.
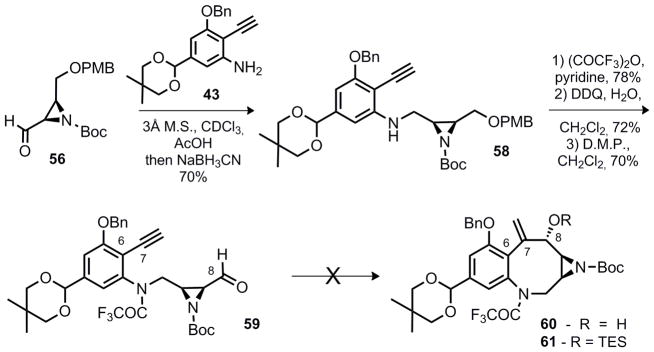
Synthesis of reductive cyclization precursor 61
The desired 8-membered benzazocine core (60 or 61) was anticipated to arise through a reductive cyclization under the conditions of Montgomery13 and Jamison,14 or via prior elaboration of the alkyne (58 or 59) to facilitate an intramolecular N.H.K. coupling reaction (Scheme 8). However, neither approach proved fruitful for the continuation of the synthesis and the synthetic approach needed to be modified prior to continuing forward.
We envisioned that a more established C-C bond forming reaction, such as the Heck coupling,15 would provide the sought after benzazocine core 41. However, in order to prove this hypothesis the parent aromatic and aziridine fragments of the convergent approach in Scheme 4 would have to be modified accordingly (Scheme 9).
Scheme 9.
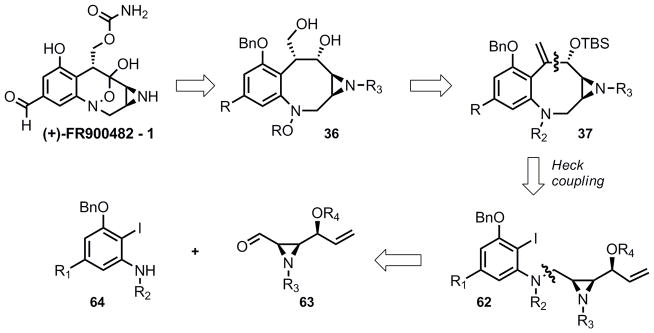
Revised retrosynthesis of FR900482; Heck coupling strategy
The preparation of a suitable aromatic fragment was relatively trivial. Although the Heck reaction could potentially function with the triflate that had been already installed on the aromatic ring 41, it seemed more logical to approach the difficult Heck reaction with the most activated precursor possible. To accomplish this, the triflate was exchanged for an iodine atom via nucleophilc aromatic substitution yielding aryl iodide 65. Furthermore, because the Songashira reaction (in the previous approach; Scheme 5) had proven quite sensitive to the electronic nature of the aromatic ring, it seemed prudent to employ an electron withdrawing group as a surrogate for the C12 aldehyde. As such, the aldehyde was oxidized in a single step to methyl ester 66 by employing N-iodosuccinimide as an oxidizing reagent under basic conditions.16 Finally an iron mediated reduction17 proved to be chemoselective for the conversion of the nitro group to the amine in the presence of the iodide yielding 67 in only six linear steps from 5-nitrovanillin. Furthermore, this route proved to be extremely scaleable and chromatography was only required in the final step to remove the excess iron from the product. In total 20–25 grams of 67 were prepared in the duration of this study.
Modification of the aziridine fragment might appear to be much more challenging. However, during the course of the first generation studies above, Trost and Aponick reported a DYKAT reaction employing divinylcarbonate (68) and phthalimide to produce amino-alcohol 69 as a single diastereomer in very high enantioselectivity.18 To employ this reaction in our synthesis, the subsequent dihydroxylation was required to be chemo and diastereoselective. It has been demonstrated that allylic groups play a pronounced role in the rate of dihydroxylation under Sharpless conditions,19 thus it was anticipated that the required selectivity might be accomplished with the proper choice of protecting group.
To test this possibility, the tert-butylsilyl ether 70 was prepared in three steps from hexa-1,5-diene-3,4-diol (available from TCI-America as a stereorandom mixture). The DYKAT reaction was initially performed under the conditions reported by Trost and Aponick to give the amino-alcohol 69 in 72% yield and 99% ee as a single diastereomer. It was subsequently determined that the catalyst loading could be lowered to 0.5 mol% of [Pd(allyl)Cl]2 and 1.5 mol% of the (R,R) Trost naphthyl ligand without a decrease in yield. Lowering the catalyst loading further resulted in substantially lowered yield due to incomplete conversion, even when the concentration of the reaction medium was increased to maintain the same catalyst concentration. After silyl protection, 70 was submitted to the Sharpless dihydroxylation under conditions identical to those optimized on the first generation system. The reaction was remarkably selective for the desired product. While there was another isomer present in the reaction mixture (~10%), it could not be determined by NMR analysis whether this side-product was a diastereomer, or a product of dihydroxylation of the opposite olefin. Aziridine 74 was accessed in five additional steps via the same sequence that was used for the first generation aziridine synthesis (29% overall yield from 68 in 6 linear steps).
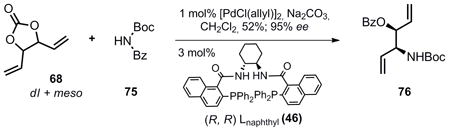
It should be noted that nucleophiles other than phthalimide were successful in the DYKAT reaction. Trost20 recently reported that utilization of N,N-benzoyl-tert-butylcarbamate (75) results in nucleophilic addition of the imide to the π-allyl palladium species, followed by migration of the benzoyl group. With divinyl carbonate this reaction is successful (Equation 5) and results in the direct formation of amino-alcohol 76 in 52% yield (based on the imide or 40% yield based on divinylcarbonate). This reaction was not optimized further. Side-products were obtained indicating that subsequent ionization of the allylic benzoyl group is somewhat competitive with the initial ionization. Unfortunately, the Sharpless dihydroxylation described above was completely ineffective when 76 is employed as a substrate and several diol products were obtained as an inseparable mixture. This strategy would have saved two steps as neither removal of the phthalimide group nor TBS protection would be required. While not useful for the present purpose, 76 may prove to be a versatile synthetic intermediate at some later date and is prepared in only two steps from commercial sources.
 |
Equation 5 |
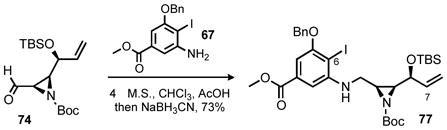
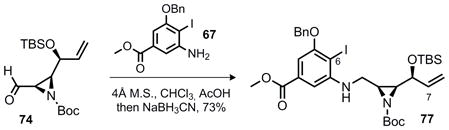
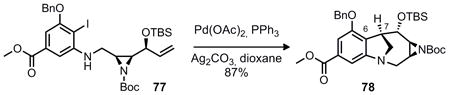
With aziridine 74 and aniline 67 in hand reductive amination proved successful and provided 77 in good yield (Equation 6). Our efforts then turned to the investigation of the crucial C6–C7 bond formation that was planned to proceed via Heck coupling.
 |
Equation 6 |
While conditions analogous to those reported by Danishefsky (Pd[PPh3]4, CH3CN, Et3N, reflux) gave almost no conversion, the conditions of Overman21 gave complete conversion to a single identifiable product (78) which was isolated in 87% yield (Equation 7). However, this product was clearly not the desired exo-cyclic olefin that would be expected from a Heck reaction. Olefin peaks in the δ 5–6 ppm region were not observed; however, mass spectroscopic analysis of the product revealed the anticipated molecular weight ([M]+ = 580.2974 amu). After in depth NMR experiments (COSY, HMBC, HSQC and ROESY) the structure of this new product was determined to be the structure indicated below. Remarkably, transannular amination outcompetes the expected β–hydride elimination in this case, giving rise to 78.22
 |
Equation 7 |
This reaction, while intriguing, presented a substantial obstacle to the synthesis of FR900482. Several changes were made to Heck precursor 77 in an attempt to favor formation of the expected Heck product (37). Removal of the TBS group (n-Bu4NF, THF, quant.) followed by the same Heck reaction conditions led to the formation of two products (presumably diastereomers) neither of which contained olefin signals that would be indicative of an exocyclic double bond. In contrast, protection of the aniline with a trifluoroacetate or acetate gave a product which was unreactive under the same Heck reaction conditions, even after prolonged reflux (dioxane 101 °C). Microwave irradiation of the same reaction mixture resulted in eventual cleavage of the trifluoroacetate (presumably via nucleophilic action of the carbonate base) and immediate formation of the same tetracycle (78) as described above. At this point, the assembly of the 8-membered benzazocine core was abandoned.
It was then anticipated that incorparation of a pyrrolidine ring system prior to the Heck reaction would allow us to reach the desired [3.3.1] bicyclononane ring system present in FR900482. In addition, this strategy would allow us to target either the mitomycin ring system or the FR900482 system through modification of the aromatic ring fragment (Scheme 12).
Scheme 12.

C-8 oxidation strategy
For this strategy to be feasible, modification to the correct oxidation state at C-8 was essential. One method to accomplish this goal is to perform a C-H insertion reaction directed by the neighboring C-7 hydroxymethyl group in 83 or 84. To set up the C-H insertion, hydroboration of 82 in a diastereoselective manner was crucial. The high number of heteroatoms in such a compact structure makes this novel approach very challenging since there are several activated carbons that could hypothetically react; 1) the two positions adjacent to the aniline nitrogen (C-8 and C-11); 2) the benzylic position (C-7); 3) the aziridine carbons (C-9 and C-10); and 4) the benzylic carbons contiguous to the oxygen atom in the -OBn group (C-13). It was anticipated that a sulfamate ester, such as 83 might favor insertion at the desired C-8 possition over C-7 as has been demonstrated by the studies of Du Bois.23 However, 84 was also explored since the carbamate is present in the natural product and its implementation would lead to a streamlined synthetic approach. Either pyrrolidine diastereomer (81a and 81b) was prepared through inversion (86, Tf2O, pyridine, CH2Cl2) or double inversion ([Ir(COD)Cl]2, 87, DABCO) and the desired Heck reaction proceeded in excellent yield for both substrates (Scheme 13).
Scheme 13.
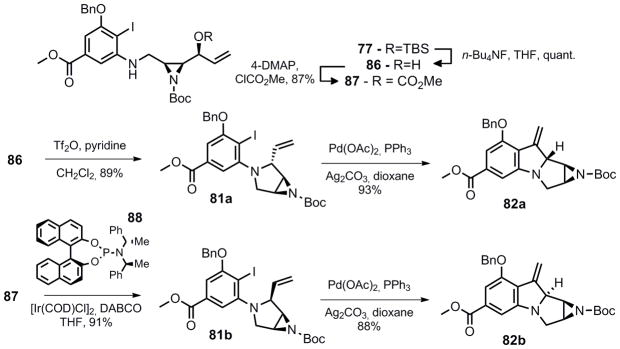
Successful Heck reaction
Hydroboration of 82a with 9-BBN led to a 4:2:1 mixture of 90a, 90b along with the ring opened aziridine (89). Ideally for the C-H insertion the hydroxymethyl and C-8 hydrogen should be cis, thus 90b appeared to be ideally substituted to access the mitomycin ring system. Alternatively, hydroboration of 82b led to exclusive formation of the trans stereochemistry between the C-7 hydroxymethyl and C-8 hydrogen substituents (91).
In order to continue forward, the carbamate 92 and sulfamate ester 94 were prepared (Scheme 15). Although the trans substituted diastereomers (92 and 94) appeared to be a less compelling C-H insertion starting material, it was investigated first since the higher diastereoselectivity in the prior hydroboration allowed access to pure material without tedious separation of diastereomers. Exposure of 92 to the conditions of Du Bois, yielded the five-membered spirocyclic product 93. The sulfamate ester 94 also favored the same regioselectivity rather than the desired C-8 insertion product.
Scheme 15.

Investigation of the C-H Insertion Reaction
Diastereomers 96 and 99 were also attempted in the C-H insertion reaction (Scheme 16). The 1:1 mixture of primary carbamates 96 led to formation of a 1:1 mixture of the undesired 5-membered C-H insertion product 97 (β diastereomer) and 10-membered C-H insertion product 98 (α diastereomer).
Scheme 16.
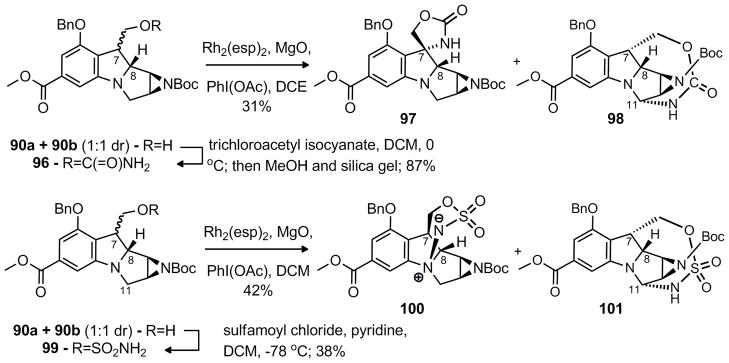
Further investigation of the C-H insertion reaction
The 1:1 diastereomeric mixture of sulfonamides 99 also led to a 1:1 mixture of products. The α diastereomer favored insertion at C-11 to form the 10-membered ring 101 (10% NOE between -CH2OR and H-9). The β diastereomer formed the very unexpected zwiterionic product 100 by attack of the aniline nitrogen (10% NOE between H7-H9 and 6% NOE between -CH2OR and H-8). This intriguing compound proved stable since its NMR was identical after weeks at room temperature on the bench.
The isolation of these large rings was very surprising. Nevertheless, it has been reported by Selkti that structural parameters such as conformational factors play a strong influence and could favor the intramolecular formation of a seven-membered ring in piperidine substrates.24
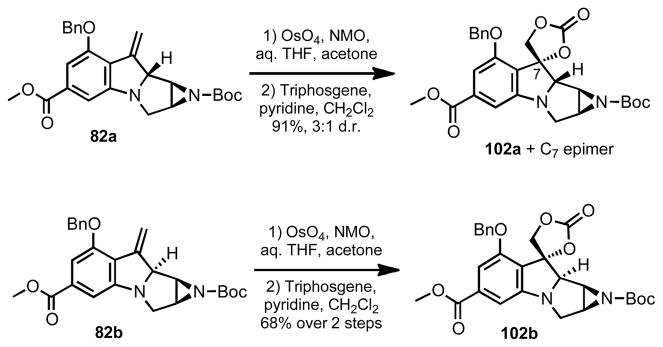
Due to these difficulties, a Polonovski/deoxygenation sequence more reminiscent of Ziegler25 and Dmitrienko’s26 original work was examined. To this end, dihydroxylation of either 82a or 82b and protection of the resulting diol was anticipated to yield material that would serve in the desired C-8 oxidation. Again, 82a was modestly selective for dihydroxylation from the alpha face leading to 102a as the major stereoisomer, which could be readily separated from the minor epimer after protection of the diol as a cyclic carbonate. Olefin 82b led exclusively to a single diastereomer, which was also protected as a cyclic carbonate (102b).
It must be noted that the stereochemistry arising at C-8 for the formation of 102a was inaccurately assigned in our original communication.27 The stereochemistry at C-7, which was assigned by comparison to C-8 was also reversed. This misassignment resulted from the rather large NOE enhancement that was observed in 102a and epi-102a between C-8 and C-9. It was only after synthesis of the C-8 epimer 102b and further NOE studies that the correct assignment of stereochemistry was realized (Figure 3). This finding reversed stereochemistry in several intermediates which had been previously reported, but did not affect the end result as these centers were ultimately irrelavent to the final outcome of the synthesis.
Figure 3.
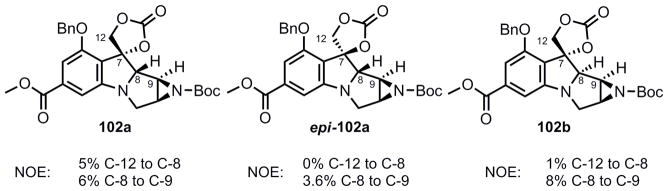
Reassignment of C-8 stereochemistry
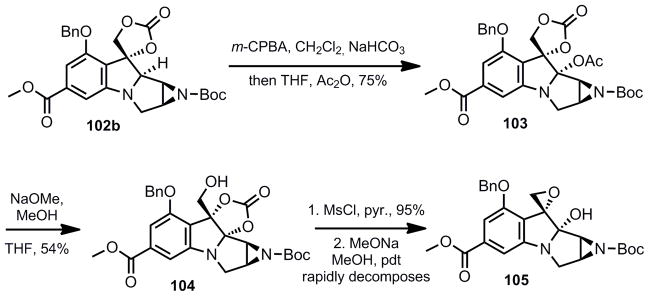
Carbonates 102a, epi-102a and 102b were all subjected to the Polonovski conditions optimized by Ziegler (m-CPBA, CH2Cl2 then Ac2O, THF).28 Compound 102b successfully yielded the C-8 acetate 103. In contrast to Ziegler’s observation that hydrolysis of the acetate occured readily, hydrolysis of 103 did not occur upon exposure to aqueous sodium bicarbonate. Hydrolysis under more forcing conditions (Cs2CO3, MeOH) led to hydrolysis of the carbonate and isolation of the corresponding triol. Hydrolysis with methoxide and methanol led to spontaneous rearrangement to the internal cyclic carbonate 104. None of these products participated successfully in the desired oxidative ring expansion reaction.29 It was anticipated that a benzylic epoxide might prove amenable to deoxygenation following the oxidative expansion reaction. Thus 105 was prepared, but its extreme instability to acid disqualified it as a potential precursor to the desired bicylic system.
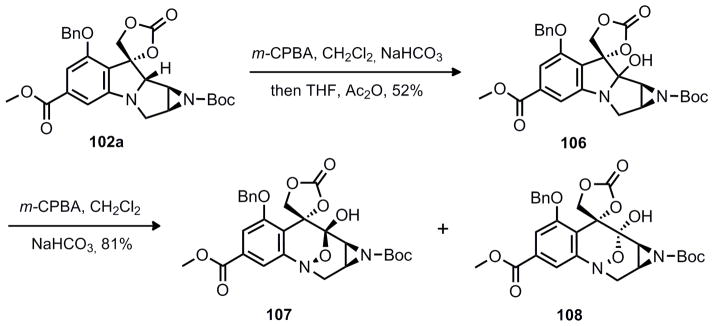
As an alternative, the Polonovski/oxidation sequence was attempted on epi-102a and 102a. While the minor diastereomer failed to give any desired product, 102a led to the anticipated aminal product (106) in moderate yield over two steps. Exposure of this product to a second oxidation led to a mixture of 107 and 108. Surprisingly, the composition of this mixture was dependent on the quality and purification of 106. Exposure of 106 to slightly acidic conditions (SiO2 chromatography) led to intractable mixtures of diastereomers 107 and 108. However, purification of 106 by reversed phase HPLC (NH4OH buffer 0.1%/ACN) led to pure quantities of 107. The most likely explanation for these observations is that the aminal is formed kinetically as a single diastereomer, but exposure to acid results in epimerization of the C8 stereochemistry. Equilibration of these diastereomers after oxidative ring expansion was not observed under acidic or basic conditions. Thus, it is proposed that the oxidative ring expansion is stereospecific and different aminal diastereomers result in opposite diastereomers after the oxidative ring expansion.
Completion of the synthesis rested upon deoxygenation of 107. After a variety of failed attempts to selectively cleave the cyclic carbonate, it was observed that hydrogenolysis of the benzyl ether followed by treatment with sodium borohydride in methanol resulted in not only hydrolysis of the cyclic carbonate, but also led to incorporation of a methoxy group at C-7 (Equation 9).
 |
Equation 9 |
The remarkable transformation outlined in Equation 9 was attributed to facile formation of an ortho-quinone methide intermediate (Scheme 21), which rendered C-7 electrophilic in nature and resulted in the capture of methanol through solvolysis. It was immediately recognized that this reaction might provide access to the desired deoxygenated product if conditions could be found to favor a net reductive termination of the reaction rather than a redox neutral sovolysis. Investigation focused on adjustment of the reaction medium, which might slow or render reversible the undesired solvolysis. While i-PrOH gave only very limited solublity of sodium borohydride, ethanol affected the same reaction yielding 110 as the sole identifiable product (Scheme 20).
Scheme 21.

Mechanism of the deoxygenation
Scheme 20.
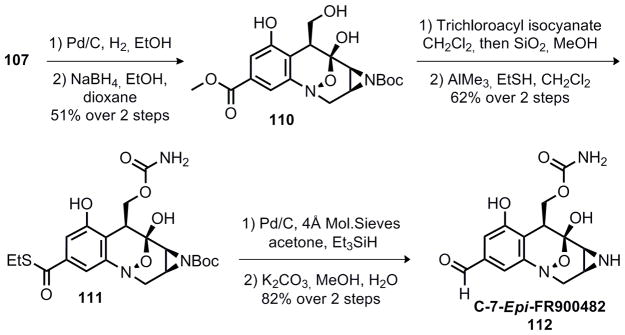
Completion of the synthesis of epi-FR900482
With 110 in hand, but no evidence to confirm the stereochemistry at C-7, further effort focused on completion of the synthetic route.30 The primary alcohol was converted to a primary carbamate via treatment with trichloroacetyl isocyanate followed by in situ hydrolysis of the trichloroacetyl group. Exchange of the methyl ester for an ethyl thioester (111) was followed by Fukuyama reduction with palladium on carbon to yield the corresponding aldehyde.31 The tert-butyl carbamate was removed via hydrolysis with saturated aqueous potassium carbonate in methanol, liberating the free aziridine. The product is isolated as a 3:2 mixture of separable hemiketal isomers, however, comparison to an authentic sample (NMR, TLC) shows that neither isomer matches the natural product, clearly demonstrating our product to be epimeric at C-7.
The likely mechanism which led to the epimeric C-7 stereochemistry is outlined in Scheme 21. Previous experimentation by Danishefsky had generated an ortho-quinone methide intermediate in the absence of a reducing reagent.32 In our case deoxygenation of 107 most likely begins with exchange of the phenol for a phenoxyboronate complex such as 113. Subsequent intramolecular reduction causes chemoselective cleavage of the cyclic carbonate to be possible in the presence of the tert-butyl carbamate protecting group. However, the resulting tetraol (114) is disposed for elimination of its tertiary benzylic alcohol to generate ortho-quinone methide 115. This reactive intermediate is proposed to decompose through a hydride shift yielding aldehyde 116, which is reduced from the less hindered face. Alternatively, the ortho-quinone methide could be directly reduced by sodium borohydride which would presumably lead to the opposite configuration at C-7. The observation that the hydroxymethyl group is formed on the convex face of the molecule seems more consistent with very fast decomposition of the ortho-quinone methide through a hydride shift and then a slower reduction of an aldehyde, which is thermodynamically favored to be on the less hindered face of the molecule.
A substantial amount of effort failed to introduce the C-7 hydroxymethyl substituent in the configuration required for the natural product. While it was disappointing not to have the natural product in hand after so much effort, we were excited by the possibility that the C-7 epimer of FR900482 might have comparable activity based on the established biological mechanism of this natural product.
It is known33 that reduction of the hydroxylamine to the aniline oxidation state is the triggering event that precedes formation of the reactive mitosene intermediate capable of cross-linking DNA (Scheme 22). For FR900482 and epi-FR900482 the triggering event should be the same, generating intermediates 117 and 119 respectively. It is unknown what mechanism the reductive activation proceeds by and it seemed quite possible that the initial N-O bond reduction might be sensitive to the stereochemistry at C-7. Furthermore, dehydration to mitosene intermediate 118 is not instantaneous. Studies on the mitomycins have demonstrated the lifetime of an aminal intermediate such as 117 or 119 is strongly dependent on the substitution of the aromatic ring.[i]34 Thus it also seems likely that the lifetime of such an intermediate could be strongly dependent on the stereochemistry at C-7. However, if these two steps are comparable, the stereochemistry at C-7 would not be expected to have any influence on the biological activity since the eventual mitosene intermediate 118 is identical.
Scheme 22.
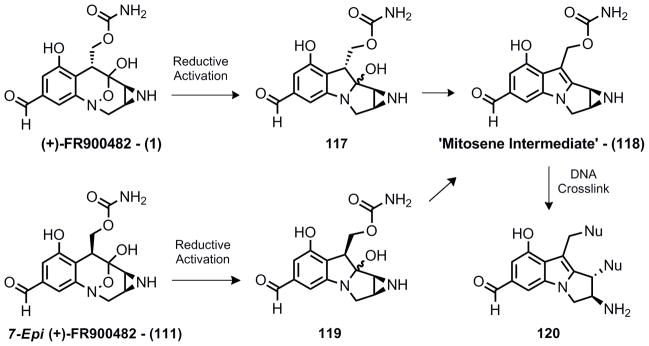
Biological mechanism of action for epi-FR900482 and FR900482
In fact, when FR900482 and epi-FR900482 were tested side by side comparable cyctotoxicity was collected from each within the experimental margin of error (Table 5).
Table 5.
Conclusion
This work highlights several novel and challenging approaches toward FR900482 and mitomycin congeners. Each strategy allowed us to gain a better knowledge in terms of the scope and limitations of the synthetic methodology employed and in terms of the reactivity of the intermediate species involved. For example, we were able to synthesize a challenging 8-membered benzazacine heterocycle, through a carbonylative lactamization. Palladium catalyzed π-allyl alkylation instead afforded highly substituted tetrahydroquinolines which are important pharmacophores. Large N-heterocyclic rings were also produced under Rh-catalized C-H insertion conditions, highlighting the importance of proximity effects in these reactions. Our synthetic efforts ultimately led to 7-epi-FR900482 in 23 linear steps (29 total) and 1.4% overall yield which represents a route that is the shortest yet disclosed for the preparation of close FR900482 analogs. It also demonstrates that the stereochemistry at C-7 is irrelevant to the biological activity of FR900482.
Prior to this study, it was not possible to predict based on what was known about the biological mechanism that a C7 epimer should be equally active compared to the natural product. The question comes down to what is the mechanism by which the compound is activated since the C-7 stereocenter is obliterated. The fact that this stereocenter is irrelevant to activity suggests the activation mode may be non-enzymatic. Because 7-epi-FR900482 has analogous in vitro activity compared to the natural product, its synthesis is functionally equivalent to that of the natural product whose syntheses all are significantly longer. Furthermore, it is also possible that this epimer could have an improved biological profile compared to the natural product, particularly because it may not have all the deleterious side-effects that have caused FK973 to be withdrawn from clinical trials. This is especially true because the convergent synthetic route presented above allows for significant variation of functional groups on epi-FR900482 that would not be possible by semi-synthesis.
Thus, the synthesis of 7-epi-FR900482 demonstrates that a substantially shorter route to such structural types is possible and that an unnatural analogue can have equal activity relative to the natural product. We have learned much about the synthesis of such structures and about the biological mechanism that makes these structures valuable. Thus, the main objective of creating a more efficient synthesis of highly active FR analogs for further structure-activity studies in order to find a potent analog devoid of undesired side-effects has indeed been accomplished.
Experimental Section
Reactions were carried out in flame- or oven-dried glassware (overnight, 120 °C) equipped with a Teflon™ coated magnetic stirring bar under a positive stream of argon or nitrogen. All reagents and solvents obtained from commercial sources were used without further purification. Anhydrous toluene, benzene, diethyl ether, hexane, dichloromethane, dimethylformamide and tetrahyrofuran (THF) were obtained from a Solv-Tek solvent purification system by passing through a column of activated alumina. Acetone was distilled from calcium sulfate. Methanol was distilled from magnesium methoxide. Reactions were monitored by thin layer chromatography (TLC) using 0.25 mm pre-coated Silica Gel 60 F254 glass plates (EMD Chemicals Inc.). Normal and flash column chromatography were performed with 32–63 μm standard grade silica gel (Sorben Technologies). Nuclear magnetic resonance (NMR) spectra were recorded on a Varian UI-600 (1H at 600 MHz, 13C at 150 MHz), UI-500 (1H at 500 MHz, 13C at 125 MHz) or Mercury 400 (1H at 400 MHz, 13C at 100 MHz) magnetic resonance spectrometers. Infrared spectra were obtained neat on NaCl plates using a Nicolet IR 100 FT-IR Spectrometer (Thermo Scientific) and reported in wavenumbers (cm−1). High-resolution FAB mass spectrometry (HRMS) data were measured on a Micromass Q-Tof API US mass spectrometer (Waters Corporation, Milford MA) in positive electrospray ionization mode (+ESI). Melting points were determined on a Thomas-Hoover melting point apparatus in open capillaries and are uncorrected. Optical rotation data was obtained with a Jasco DIP-360 digital polarimeter at the sodium D line (589 nm) in the solvent and concentration indicated.
General procedure for carbonylative lactamization: Synthesis of lactame 28a
A solution of vinyl iodide 27a (296 mg, 0.423 mmol) in DMA (4.1 mL) was added at once to a solution of dichlorobis(triphenylphosphine)palladium(II) (14.8 mg, 0.021 mmol), and triethylamine (90.0 mg, 0.124 mL, 0.889 mmol) in DMA (10.0 mL). Carbon monoxide was bubbled through the resulting red solution for 30 min. The reaction mixture was heated to 65°C and stirred at that temperature for 5.5 h. After cooling to room temperature, water was added and the reaction mixture was extracted with EtOAc (x3). The combined organic extracts were washed with brine, dried (MgSO4), filtered, and concentrated under reduced pressure to give 28a as a brown oil. Purification by column chromatography (SiO2, 30% ether/pet ether to 50% ether/pet ether) afforded 198 mg (78%) of the lactame as a white solid, mp = 53–55°C, Rf = 0.37 (50% ether/pet ether). [a]22D = −13.8° (c 5.28, CH2Cl2). IR (CDCl3): 1724, 1700, 1586, 1434, 1325, 1252, 1234 cm−1. 1H NMR (300 MHz, CDCl3): δ 7.81 (s, 1H), 7.56 (d, J = 1.2 Hz, 1H), 7.33 (m, 5H), 6.25 (dd, J = 8.3, 11.7 Hz, 1H), 5.66 (d, J = 11.7 Hz, 1H), 5.55 (m, 1H), 5.09 (s, 2H), 3.89 (s, 3H), 2.88 (m, 2H), 1.01 (s, 9H), 0.77 (s, 9H), 0.15 (d, J = 1.7 Hz, 6H), −0.15 (s, 3H), −0.26 (s, 3H). 13C NMR (75 MHz, CDCl3): δ 169.3, 167.4, 156.3, 154.1, 151.4, 137.1, 129.4, 128.4, 127.8, 127.4, 117.4, 116.5, 108.1, 105.5, 70.1, 69.9, 52.1, 31.1, 30.3, 26.1, 25.7, 18.1, 17.9, −5.24, −5.30, −5.60. HRMS Calc’d. for C32H47NO6Si2: 597.2942. Found: 597.2943.
DYKAT reaction of divinyl carbonate 68: Synthesis of alcohol 69
According to the known procedure of Trost,35 a dry 250 mL flask was charged with sodium carbonate (0.1060 g, 1.0 mmol), phthalimide (2.2364 g, 15.2 mmoles), (R,R)-naphthyl ligand (0.2373 g, 0.30 mmoles) and [(η3-C3H5)PdCl]2 (0.0366 g, 0.10 mmoles). The reaction vessel was evacuated and backfilled with nitrogen several times and then degassed methylene chloride (120 mL) was injected. A separate flask was charged with divinylcarbonate 68, which was dissolved in degassed methylene chloride (40 mL). After 15 minutes the divinylcarbonate solution (20 mmoles, 2.800 g) was transferred via canula into the reaction flask. The reaction was stirred at room temperature for 96h, after which time the solvent was removed under reduced pressure and the remaining crude material was purified by flash chromatography on silica gel (gradient, 10% to 30% EtOAc/PE) to afford the product 69 as a white solid (3.503g, 72%). The enantiomeric excess was determined via evolution on a Chiracell AS column with 5% i-PrOH/heptane at a flow rate of 1 mL/min. Monitoring at 230nm showed the (S,S) enantiomer with a retention of 19.8 min and the (R,R) enantiomer which eluted at 28.3 min. This method showed the enantiomeric excess to be greater than 99%. Rf = 0.35 (4:1 Hexane/EtOAc); [α]D = −86.4 (c = 1.00, CH2Cl2); IR (thin film): 3463, 1707, 1387 cm−1; 1H NMR (400 MHz, CDCl3): δ 7.87-7.81 (m, 2H), 7.76-7.71 (m, 2H), 6.22 (ddd, J = 6.9, 10.4, 17.3 Hz, 1H), 5.83 (ddd, J = 4.7, 10.5, 15.3 Hz, 1H), 5.43-5.15 (m, 4H), 4.89 (t, J = 6.6 Hz, 1H), 4.72-4.65 (m, 1H), 3.52 (d, J = 8.7 Hz, 1H); 13C NMR (100 MHz, CDCl3): δ 168.8, 137.3, 134.2, 132.1, 131.6, 123.5, 118.8, 116.8, 72.7, 58.6 ppm. HRMS (EI+) calc’d for C11H6NO3 (M)+, 243.0895, found 243.0892.
Heck Reaction: Synthesis of exocyclic olefin 82a
The aryl iodide 81a (45.8 mg, 0.080 mmoles), palladium acetate (1.9 mg, 7.9 μmoles), triphenylphosphine (8.9 mg, 31.6 μmoles) and silver carbonate (43.8 mg, 0.16 mmoles) were transferred to a round bottomed flask. The flask was evaculated and refilled with nitrogen three times. Dioxane (1.6 mL, freshly distilled over sodium) was injected and the reaction was heated to 80 °C in an oil bath. After 1 hour the reaction was complete and allowed to cool. The heterogenous suspension was filtered though celite to remove palladium black and the filter cake was washed with ethyl acetate. The filtrate was concentrated under reducted pressure. Flash chromatography (20% EtOAc/hexane) gave compound 82a as a yellow foam (33.0 mg, 93%). Rf = 0.30 (4:1 Hexane/EtOAc); [α]D = +23 (c = 0.90, CDCl3); IR (thin film): 2948, 1716 (sharp), 1566; 1H NMR (500 MHz, CDCl3): δ 7.48-7.33 (m, 5H), 7.18 (d, J = 0.9 Hz, 1H), 7.00 (d, J = 1.0 Hz, 1H), 6.04 (d, J = 3.2 Hz, 1H), 5.38 (d, J = 2.5 Hz, 1H), 5.18 (s, 2H), 4.68 (t, J = 2.7 Hz, 1H), 3.90 (s, 3H), 3.69 (d, J = 11.8 Hz, 1H), 3.38 (dd, J = 4.3, 12.1 Hz, 1H), 3.34 (t, J = 4.4 Hz, 1H), 3.25 (dd, J = 0.4, 4.9 Hz, 1H), 1.49 (s, 9H) ppm; 13C NMR (125 MHz, CDCl3): δ 167.1, 160.6, 159.4, 155.8, 144.0, 136.5, 132.8, 128.9, 128.4, 127.9, 121.5, 110.0, 108.1, 105.8, 82.2, 71.6, 70.6, 55.9, 52.6, 49.2, 45.6, 28.2, ppm; HRMS (EI+) calc’d for C26H28N2O5 (M)+, 448.1998, found 448.1998.
Supplementary Material
Figure 1.

FR00482 and mitomycin family of compounds
Scheme 1.
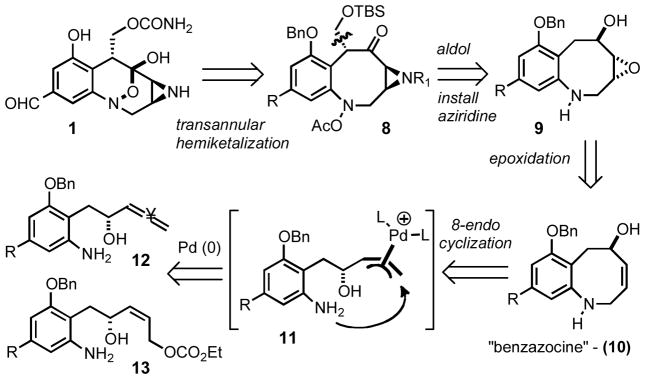
Original retrosynthetic analysis of FR-900482.
Scheme 3.

Synthesis of vinyl iodides 27a, 27b, and 27c
a. cat AgNO3, NIS, acetone, 87%; b. DIBAL-H, THF; c. K2CO3, MeOH; d. NaH, BnBr, Bu4NI, THF; e. 35, pyr., AcOH, MeOH; f. SmI2(4 eq), THF, MeOH; g. Zn (2.2 eq), THF, MeOH, H2O; h. TBDPSCl or TBSCl, imid., CH2Cl2
Scheme 10.
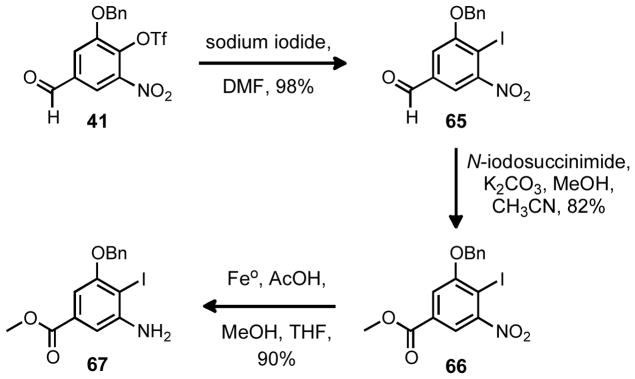
Synthesis of a modified aromatic fragment
Scheme 11.

Synthesis of a modified aziridine fragment
Scheme 14.

Hydroboration of 82a and 82b
Scheme 17.
Dihydroxylation of 82a and 82b
Scheme 18.
Polonovski and oxidation/ring expansion of 102
Scheme 19.
Successful Polonovski/oxidative ring expansion
Table 2.
Selected optimization studies for Pd-π-allyl alkylation of 22.
| |||||
|---|---|---|---|---|---|
| Entry | Substrate | Pd Sourcea | Solvent | Ligand | Yieldc |
| 1 | 22a | Pd2dba3·CHCl3 | THF | PPh3 | 23% |
| 2 | 22a | Pd2dba3·CHCl3 | THF | dppp | 51% |
| 3 | 22a | Pd2dba3·CHCl3 | THF | P(OPh)3 | 67% |
| 4 | 22a | [{Pd(C3H5)Cl}2] | THF | P(OPh)3 | 69% |
| 5 | 22a | Pd2dba3·CHCl3 | THF | -- | 23% |
| 6 | 22b | Pd2dba3·CHCl3 | THF | P(OPh)3 | 73% |
| 7 | 22b | Pd2dba3·CHCl3 | DMSO | P(OPh)3 | 75% |
| 8 | 22b | Pd2dba3·CHCl3 | PhH | P(OPh)3 | 23% |
| 9 | 22b | Pd2dba3·CHCl3 | THF | P(O-iPr)3 | 41% |
| 10 | 22b | Pd(OAc)2b | THF | P(OPh)3 | 86% |
| 11 | 22b | Pd(OAc)2 | DMSO | dppf | 85% |
| 12 | 22b | Pd(dppe)2b | DMSO | -- | 82% |
10% of Pd source unless stated otherwise.
20% of Pd source.
All yields are isolated yields after silica gel chromatography.
Acknowledgments
We gratefully acknowledge D. Barrett and Astellas Pharmaceuticals for providing an authentic sample of FR900482 and Dr. Yong Li and Hugu Menzella for biological testing of FR900482 and its epimer. We also acknowledge S. Lynch for his invaluable help in the acquisition of 2-D NMR data. We thank the Stanford Graduate Fellowship Program and NIH (GM 33049) for their financial support of our program and Johnson Matthey for their supply of precious metal salts.
Contributor Information
Prof. Barry M. Trost, Email: bmtrost@stanford.edu.
Dr. Brendan M. O’Boyle, Email: brendan_oboyle@merck.com.
Dr. Wildeliz Torres, Email: wildeliz@hotmail.com.
Dr. Michael K. Ameriks, Email: mameriks@its.jnj.com.
References
- 1.Bradner WT. Cancer Treatment Reviews. 2001;27:35. doi: 10.1053/ctrv.2000.0202. [DOI] [PubMed] [Google Scholar]; (b) Pazdur R, Ho DH, Daugherty K, Bradner WT, Krakoff IH, Rbner MN. Invest New Drugs. 1991;9:337. doi: 10.1007/BF00183587. [DOI] [PubMed] [Google Scholar]; (c) Shimomura K, Manda T, Mukumoto S, Masuda K, Nakamura T, Mizota T, Matsumoto S, Nishigaki F, Oku T, Mori J, Shibayama F. Cancer Res. 1988;48:1166. [PubMed] [Google Scholar]
- 2.For a review on the topic see: Coleman RS. Current Opinion in Drug Discovery and Development. 2001;4:435.Successful approaches: Fukuyama T, Goto S. Tetrahedron Lett. 1989;30:6491.Fukuyama T, Xu L, Goto S. J Am Chem Soc. 1992;114:383.McClure KF, Danishefsky SJ, Schulte GK. J Org Chem. 1994;59:355.Schkeryantz JM, Danishefsky SJ. J Am Chem Soc. 1995;117:4722.Danishefsky SJ, Schkeryantz JM. Synlett. 1995:475.Katoh T, Itoh E, Yoshino T, Terashima S. Tetrahedron Lett. 1996;37:3471.Yoshino T, Nagata Y, Itoh E, Hashimoto M, Katoh T, Terashima S. Tetrahedron Lett. 1996;37:3475.Katoh T, Yoshino T, Nagata Y, Nakatani S, Terashima S. Tetrahedron Lett. 1996;37:3479.Katoh T, Itoh E, Yoshino T, Terashima S. Tetrahedron. 1997;53:10229.Yoshino T, Nagata T, Itoh E, Hashimoto M, Katoh T, Terashima S. Tetrahedron. 1997;53:10239.Katoh T, Nagata Y, Yoshino T, Nakatani S, Terashima S. Tetrahedron. 1997;53:10253.Suzuki M, Kambe M, Tokuyama H, Fukuyama T. Angew Chem, Int Ed. 2002;41:4686. doi: 10.1002/anie.200290016.Suzuki M, Kambe M, Tokuyama H, Fukuyama T. J Org Chem. 2004;69:2831. doi: 10.1021/jo049862z.Judd TC, Williams RM. Angew Chem, Int Ed. 2002;41:4683. doi: 10.1002/anie.200290015.Judd TC, Williams RM. J Org Chem. 2004;69:2825. doi: 10.1021/jo035828t.
- 3.Formal syntheses: Fellows IM, Kaelin DE, Martin SF. J Am Chem Soc. 2000;122:10781.Paleo MR, Aurrecoechea N, Jung KY, Rapoport HJ. Org Chem. 2003;68:130. doi: 10.1021/jo0206521.Shaw KJ, Luly JR, Rapoport H. J Org Chem. 1985;50:4515.Jones RJ, Rapoport H. J Org Chem. 1990;55:1144.Synthesis of analogues or related natural products: Kambe M, Arai E, Suzuki M, Tokuyama H, Fukuyama T. Org Lett. 2001;3:2575. doi: 10.1021/ol016243t.Trost BM, O’Boyle BM. Org Lett. 2008;10:1369. doi: 10.1021/ol800127a.Incomplete studies: Dmitrienko GI, Denhart D, Mithani S, Prasad G, Taylor NJ. Tetrahedron Lett. 1992;33:5705.Mithani S, Drew DM, Rydberg EH, Taylor NJ, Mooibroek S, Dmitrienko GI. J Am Chem Soc. 1997;119:1159.Trost BM, Ameriks MK. Org Lett. 2004;6:1745. doi: 10.1021/ol049583y.Ziegler FE, Belema M. J Org Chem. 1994;59:7962.Ziegler FE, Belema M. Org Chem. 1997;62:1083.Zhang W, Wang C, Jimenez LS. Synth Commun. 2000;30:351.Greshock TJ, Funk RL. J Am Chem Soc. 2002;124:754. doi: 10.1021/ja0123554.Young IS, Kerr MA. Angew Chem Int Ed. 2003;42:3023. doi: 10.1002/anie.200351573.Lim HJ, Sulikowski GA. Tetrahedron Lett. 1996;37:5243.Colandrea VJ, Rajaraman S, Jimenez LS. Org Lett. 2003;5:785. doi: 10.1021/ol026738y.
- 4.Trost BM, Vos BA, Brzezowski CM, Martina DP. Tetrahedron Lett. 1992;33:717. [Google Scholar]
- 5.Transannular interactions are minimized by unsaturation in the forming ring.
- 6.(a) Omura S, Nakagawa A. Tetrahedron Lett. 1981;22:2199. [Google Scholar]; (b) Steinhagen H, Corey EJ. Org Lett. 1999;1:823. doi: 10.1021/ol9908575. [DOI] [PubMed] [Google Scholar]; (c) Hill ML, Raphael RA. Tetrahedron Lett. 1986;27:1293. [Google Scholar]; (d) Morimoto Y, Matsuda F, Shirahama H. Tetrahedron. 1996;52:10609. [Google Scholar]; (e) Morimoto Y, Shirahama H. Tetrahedron. 1996;52:10631. [Google Scholar]; (f) Morimoto Y, Matsuda F, Shirahama H. Synlett. 1991:202. [Google Scholar]
- 7.(a) Leeson PD, Carling RW, Moore KW, Moseley AM, Smith JD, Stevenson G, Chan T, Baker R, Foster AC, Grimwood S, Kemp JA, Marshall GR, Hoogsteen KJ. Med Chem. 1992;35:1954. doi: 10.1021/jm00089a004. [DOI] [PubMed] [Google Scholar]; (b) Carling RW, Leeson PD, Moseley AM, Smith JD, Saywell K, Tricklebank MD, Kemp JA, Marshall GR, Foster AC, Grimwood S. Bioorg Med Chem Lett. 1993;3:65. [Google Scholar]; (c) Takamura M, Funabashi K, Kanai M, Shibasaki MJ. J Am Chem Soc. 2001;123:6801. doi: 10.1021/ja010654n. [DOI] [PubMed] [Google Scholar]
- 8.Fellows IM, Kaelin DE, Martin SF. J Am Chem Soc. 2000;122:10781. [Google Scholar]
- 9.Negishi E, Anastasia L. Chem Rev. 2003;103:1979. doi: 10.1021/cr020377i. [DOI] [PubMed] [Google Scholar]
- 10.(a) Trost BM, Van Vranken DL, Bingel C. J Am Chem Soc. 1992;114:9327. [Google Scholar]; (b) Trost BM, Bunt RC, Lemoine RC, Calkins TL. J Am Chem Soc. 2000;122:5986. [Google Scholar]
- 11.Ohfune Y, Kurokawa N. Tetrahedron Lett. 1984;25:1587. [Google Scholar]
- 12.Najime R, Pilard S, Vaultier M. Tetrahedron Lett. 1992;33:5351. [Google Scholar]
- 13.(a) Montgomery J. Angew Chem Int Ed. 2004;43:3890. doi: 10.1002/anie.200300634. [DOI] [PubMed] [Google Scholar]; (b) Montgomery J, Tang XT. J Am Chem Soc. 1999;121:6098. [Google Scholar]; (c) Knapp-Reed B, Mahandru GM, Montgomery J. J Am Chem Soc. 2005;127:13156. doi: 10.1021/ja054590i. [DOI] [PubMed] [Google Scholar]; (d) Tang X, Montgomery J. J Am Chem Soc. 2000;122:6950. [Google Scholar]
- 14.(a) Chan J, Jamison TF. J Am Chem Soc. 2003;125:11514. doi: 10.1021/ja0373925. [DOI] [PubMed] [Google Scholar]; (b) Colby EA, O’Brien KC, Jamison TF. J Am Chem Soc. 2005;127:4297. doi: 10.1021/ja042733f. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Colby EA, O’Brien KC, Jamison TF. J Am Chem Soc. 2004;126:998. doi: 10.1021/ja039716v. [DOI] [PubMed] [Google Scholar]; (d) Chan J, Jamison TF. J Am Chem Soc. 2004;126:10682. doi: 10.1021/ja0470968. [DOI] [PubMed] [Google Scholar]
- 15.Abelman MM, Overman LE, Tran VD. J Am Chem Soc. 1990;112:6959. [Google Scholar]
- 16.McDonald C, Holcomb H, Kennedy K, Kirkpatrick E, Leathers T, Vanemon P. J Org Chem. 1989;54:1213. [Google Scholar]
- 17.Fonseca T, Gigante B, Marques MM, Gilchrist TL, De Clercq E. Bioorganic and Medicinal Chemistry. 2004;12:103. doi: 10.1016/j.bmc.2003.10.013. [DOI] [PubMed] [Google Scholar]
- 18.Trost BM, Aponick A. J Am Chem Soc. 2006;128:3931. doi: 10.1021/ja0578348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Corey EJ, Noe MC. J Am Chem Soc. 1996;118:319. [Google Scholar]
- 20.Trost BM, Fandrick DR, Brodmann T, Stiles DT. Ang Chem Int Ed. 2007;46:6123. doi: 10.1002/anie.200700835. [DOI] [PubMed] [Google Scholar]
- 21.Abelman MM, Overman LE, Tran VD. J Am Chem Soc. 1990;112:6959. [Google Scholar]
- 22.For a more complete discussion see: Trost BM, O’Boyle BM, Hund D. Chem Eur J. 2010;16:9772. doi: 10.1002/chem.201001689.
- 23.(a) Espino CG, Du Bois J. Angew Chem. 2001;113:618. doi: 10.1002/1521-3773(20010202)40:3<598::AID-ANIE598>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2001;40:598. [Google Scholar]; (b) Espino CG, Wehn PM, Chow J, Du Bois J. J Am Chem Soc. 2001;123:6935. [Google Scholar]
- 24.Toumieux S, Compain P, Martin OR, Selkti M. Org Lett. 2006;8:4493. doi: 10.1021/ol061649x. [DOI] [PubMed] [Google Scholar]
- 25.(a) Ziegler FE, Belema M. J Org Chem. 1994;59:7962. [Google Scholar]; (b) Ziegler FE, Belema M. J Org Chem. 1997;62:1083. [Google Scholar]
- 26.Dmitrienko GI, Denhart D, Mithani S, Prasad GKB, Taylor NJ. Tetrahedron Lett. 1992;33:5705. [Google Scholar]; (b) Mithani S, Drew DM, Rydberg EH, Taylor NJ, Mooibroek S, Dmitrienko GI. J Am Chem Soc. 1997;119:1159. [Google Scholar]
- 27.See reference 3f.
- 28.It should be noted that the Polonovski oxidation was unsuccessful with other protecting groups for the 1,2 diol (acetonide). An electron withdrawing protecting group appeared to be required for reasonable efficiency in the Polonovski reaction.
- 29.Only starting material was recovered when 103 and 104 were treated with m-CPBA and NaHCO3 in CH2Cl2 followed by Ac2O in THF.
- 30.The connectivity of 110 was confirmed by NMR analysis (gCOSY, gHMBC, gHSQC), but the configuration at C7 remained uncertain.
- 31.Fukuyama T, Tokuyama H. Aldrichim Acta. 2004;37:87. [Google Scholar]
- 32.See reference 2e.
- 33.(a) Woo J, Sigurdsson ST, Hopkins PB. J Am Chem Soc. 1993;115:1199. [Google Scholar]; (b) Huang H, Pratum TK, Hopkins PB. J Am Chem Soc. 1994;116:2703. [Google Scholar]; (c) Paz MM, Hopkins PB. Tetrahedron Lett. 1997;38:343. [Google Scholar]; (d) Paz MM, Hopkins PB. J Am Chem Soc. 1997;119:5999. [Google Scholar]; (e) Williams RM, Rajski SR, Rollins SB. Chemistry and Biology. 1997;4:127. doi: 10.1016/s1074-5521(97)90256-8. [DOI] [PubMed] [Google Scholar]; (f) Beckerbauer L, Tepe JJ, Cullison J, Reeves R, Williams RM. Chemistry and Biology. 2000;7:805. doi: 10.1016/s1074-5521(00)00028-4. [DOI] [PubMed] [Google Scholar]; (g) Williams RM, Ducept P. Biochemistry. 2003;42:14696. doi: 10.1021/bi035202x. [DOI] [PubMed] [Google Scholar]
- 34.Gruschow S, Chang LC, Mao Y, Sherman DH. J Am Chem Soc. 2007;129:6470. doi: 10.1021/ja0700193. [DOI] [PubMed] [Google Scholar]
- 35.Trost BM, Aponick A. J Am Chem Soc. 2006;128:3931. doi: 10.1021/ja0578348. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.