

Summary
Cardiomyopathies are diseases of the heart resulting in impaired cardiac muscle function, which can lead to heart dilation or overt heart failure. These diseases represent a major cause of global morbidity and death. Innovative preventive and therapeutic measures are urgently needed for early detection, categorization, and treatment of patients at risk of cardiomyopathy. These developments will require a more complete understanding of the molecular effects of impaired cardiac function, even prior to overt disease. The use of gel-free expression proteomics in the detailed analysis of cardiac tissues should yield significant insight into the pathophysiology of these diseases.
Keywords: Cardiac muscle, multidimensional protein identification technology (MudPIT), mass spectrometry
1. Introduction
Cardiomyopathy is a chronic condition of impaired heart function that arises as a result of genetic predisposition and environmental interactions. Because the prognostic outcomes following diagnosis are poor, earlier detection and diagnosis of cardiomyopathy represent a pressing clinical challenge. Although significant progress has been made in identifying genetic, physiological, and environmental factors that predispose individuals to cardiomyopathy, the etiology of this disease has exhibited an unanticipated level of complexity. Additional research into the molecular basis of clinically common forms of cardiomyopathy is needed urgently to speed development of rational prophylactic and therapeutic strategies.
Heart muscle expresses several thousand distinct proteins (1), several hundred of which are likely tissue-specific and critical for proper heart muscle function, performance, and capacity. Although a number of genes/proteins predisposing to cardiomyopathy have been identified (e.g., dystrophin, ABCA1) based on known or suggested physiological function, identification of the full set of gene products associated with this “complex trait” has proven to be a challenge. A nonbiased, comprehensive description of the proteome, or complement of expressed protein products, in healthy and diseased cardiac tissue could provide breakthrough understanding of the pathogenesis of the disease, leading to advanced diagnostic and therapeutic targets.
The proteome is defined as the entire set of proteins that is expressed (produced) in a cell, tissue, or organ at a given time and physiological state (2). The proteome is a dynamic entity dictated by collective rates of gene transcription and pre- and posttranslational controls that serve collectively to regulate protein abundance, subcellular enrichment and turnover in relation to developmental and physiological cues, environmental constraints, and disease perturbations. Tandem mass spectrometry (MS/MS) the study of the structure of gas phase ions as a means to determine the identity of biomolecules—has emerged as the method of choice for large-scale experimental investigation of the proteome (2). Proteins can be “sequenced” after enzymatic digestion with a site-specific protease, typically trypsin. After selecting and fragmenting peptides, the daughter ion spectra are analyzed, usually with the help of a computer-based database search algorithm, to deduce the amino acid sequence of the peptide and, hence, the identity of the corresponding parental protein (3).
The complexity of the mammalian tissue proteome represents a considerable experimental challenge (4–10). Effective pre-fractionation methods are, therefore, required to increase proteome coverage in order to detect low abundance signaling proteins. Historically, two-dimensional (2D)-gel electrophoresis has provided a useful method for high-resolution separation of complex protein samples, including cardiac samples (1). Nonetheless, this technique is biased against the detection of membrane proteins, low-abundance proteins, and proteins with extremes in isoelectric point (pI) and molecular weight (MW). The identification and quantification of gel-separated proteins is also limited by the need to analyze many individual gel spots. To circumvent these problems, several groups have developed protein profiling strategies based on coupling high-efficiency liquid chromatography (LC)-based separation procedures with automated mass spectrometers, allowing for very large-scale “shotgun” sequencing of complex mixtures (5–8,10,11). The archetypal approach, termed “Mud-PIT” (for multidimensional protein identification technology) (11) was pioneered in the laboratory of John Yates, III.
Together with the recent completion of the human and mouse genome sequencing projects (12–14), this proteomic methodology is well-suited to systematic global protein profiling of mammalian tissue and, therefore, offers a powerful means of investigating the effects of disease and therapeutics on mouse tissue. We have begun to apply these methods to examine the biochemical and physiological changes that accompany cardiomyopathy in a comprehensive and unbiased manner. The protocols described in this chapter were adapted and optimized in our laboratory for the analysis of skeletal muscle cell lines (C2C12 cells) (15), microsomal fractions from two different mouse heart muscles (PLN-KO and PLN-I40A [16]), and are currently being expanded to include the detailed analysis of multiple organelle fractions of cardiac tissues obtained from mouse models of dilated cardiomyopathy. A schematic overview of our procedures is outlined in Fig. 1.
Fig. 1.
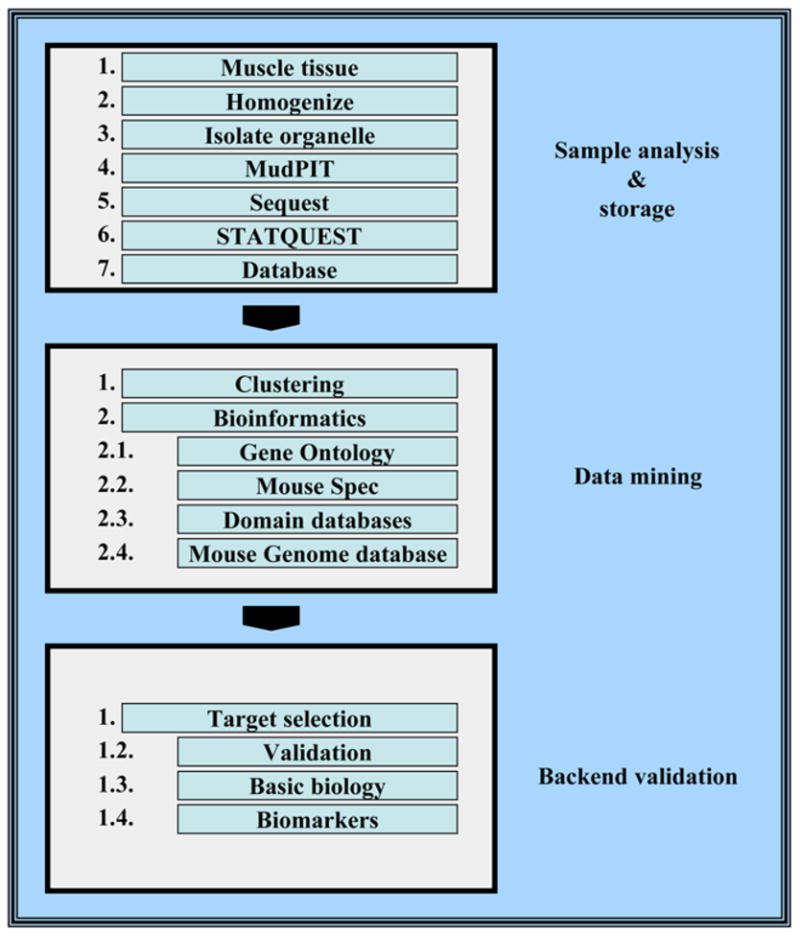
Overview of an integrated heart proteomic profiling methodology. Cardiac muscle tissue is homogenized and subcellular fractions are isolated by differential ultracentrifugation in sucrose gradients. Protein extracts from each organelle are analyzed extensively by multiple independent MudPIT analyses. Generated tandem mass spectra are searched against a protein sequence database by the use of the SEQUEST and STATQUEST algorithms and subsequently filtered to minimize false-positive identifications. High-confidence protein identifications are parsed into an in-house database, and diverse data clustering and mining strategies are used to find interesting patterns of protein expression for biological validation and detailed analysis.
2. Materials
All solid chemicals were from Sigma, whereas HPLC-grade acetonitrile (ACN), methanol, and water were purchased from Fisher Scientific, and heptafluorobutyric acid was obtained from BioLynx (Brockville, Ontario, Canada). Endoproteinase Lys-C was obtained from Roche Diagnostics (Laval, Quebec, Canada).
2.1. Cardiac Muscle Extraction
Buffer for cardiac lysis: 250 mM sucrose, 50 mM Tris-HCl, pH 7.4, 5 mM MgCl2, 1 mM dithiothreitol (DTT), and 1 mM phenylmethylsulfonyl fluoride (PMSF). Store all solutions at 4°C and add DTT and PMSF fresh with each use.
Solution for sucrose cushion 1: 0.9 M sucrose, 50 mM Tris-HCl, pH 7.4, 5 mM MgCl2, 1 mM DTT, and 1 mM PMSF.
Solution for sucrose cushion 2: 2 M sucrose, 50 mM Tris-HCl, pH 7.4, 5 mM MgCl2, 1 mM DTT, and 1 mM PMSF.
Nuclear extraction buffer I: 20 mM HEPES, pH 7.8, 1.5 mM MgCl2, 450 mM NaCl, 0.2 mM EDTA, and 25% glycerol.
Nuclear extraction buffer II: same as nuclear extraction buffer I, with addition of 1% Triton-X 100.
Solution for sucrose cushion: 2 M sucrose, 50 mM Tris-HCl, pH 7.4, 5 mM MgCl2, 1 mM DTT, and 1 mM PMSF.
Mitochondrial extraction buffer I: 10 mM HEPES, pH 7.8.
Mitochondrial extraction buffer II: same as nuclear extraction buffer I, with addition of 1.5% Triton-X 100.
Beckman ultraclear centrifuge tubes (14 ↔ 95 mm; Cat. no. 344060).
2.2. Precipitation and Digestion of Cardiac Samples and Solid-Phase Extraction
150 μg Protein in aqueous or detergent solution.
Ice-cold acetone.
8 M Urea, 50 mM Tris-HCl, pH 8.5, 1 mM CaCl2.
50 mM Ammonium bicarbonate.
Endoproteinase Lys-C (Roche Diagnostics).
Poroszyme trypsin beads (Applied Biosystems, Streetsville, Ontario, Canada).
2.3. MudPIT Analyses
SPEC-Plus PT C18 cartridges (Ansys Diagnostics, Lake Forest, CA).
100-μm capillary microcolumn (Polymicro Technologies, Phoenix, AZ).
Zorbax Eclipse XDB-C18 resin (Agilent Technologies, Mississauga, Ontario, Canada).
5 μm Partisphere strong cation exchange resin (Whatman).
Solutions of: Buffer A, 5% ACN, 0.5% acetic acid, and 0.02% heptafluorobutyric acid (HFBA); Buffer B, 100% ACN; Buffer C, 250 mM ammonium acetate in buffer A; and Buffer D, 500 mM ammonium acetate in buffer A.
2.4. Bioinformatics
Cluster 3.0 software (java applet available from http://rana.lbl.gov/).
Sequest database search software (available from Thermo Finnigan).
STATQUEST (developed in-house; see ref. 11).
Swissprot annotation (http://www.expasy.org/sprot/).
Gene Ontology (GO) database (http://www.geneontology.org).
MouseSpec (http://tap.med.utoronto.ca/~posman/mousespec/).
GOminer (http://discover.nci.nih.gov/gominer/).
TreeView (http://rana.lbl.gov/downloads/TreeView/).
3. Methods
3.1. Ventricular Fractionation
Healthy adult mice are euthanized by administration of CO2. The heart is removed, rinsed, and dissected to remove the atria. Ventricular tissues are washed three times in ice-cold phosphate-buffered saline (PBS) and minced finely using a razor blade or scissors. Minced samples are subsequently homogenized carefully using a loose-fitting dounce homogenizer with at least 15 strokes on ice, using ice-cold lysis buffer. All subsequent steps are performed at 4°C. The lysate is centrifuged in a benchtop centrifuge at 800g for 15 min; the supernatant serves as source of cytosol, mitochondria, and microsomes. The pellet, which contains the nuclei, is resuspended in 8 mL lysis buffer and layered onto 0.9 M sucrose, 50 mM Tris-HCl, pH 7.4, 5 mM MgCl2, 1 mM DTT, and 1 mM PMSF, and centrifuged again at 800g for 15 min. The pellet is suspended in 8 mL of 0.9 M sucrose cushion buffer and then carefully applied onto 4 mL of 2 M sucrose cushion buffer in a 13-mL ultracentrifuge tube, and pelleted at 150,000g for 60 min (Beckman SW40.1 rotor). The nuclear pellet is collected, washed once in PBS, suspended in nuclear extraction buffer I, left on ice for 15 min, and centrifuged at 8000g for 20 min. The supernatant is referred to as nuclear extract I. The pellet from this procedure is resuspended in nuclear extraction buffer II, incubated on ice for 30 min, followed by 8000g for 20 min. The resulting supernatant is collected and referred to as nuclear extract II. Following the ultracentrifugation, we also collect the proteins accumulated at the interface of the 250 mM sucrose and 0.9 M sucrose solutions; these proteins are highly enriched in contractile proteins. Proteins are washed twice in PBS, isolated by centrifugation at 14,000g for 10 min, and resuspended in mitochondrial buffer II (see Note 1).
Mitochondria are isolated from the crude cytoplasmic fraction by benchtop centrifugation at 8000g for 20 min. The supernatant is collected and used for microsomal fractions (see Subheading 3.1.3.). The pellet is incubated in 10 mM HEPES for 30 min at 4°C followed by brief sonication pulses at maximum setting. Samples are centrifuged at 8000g for 20 min and the supernatant collected (mitochondria extract I). The pellet is incubated with mitochondrial extraction buffer II for 30 min at 4°C, centrifuged at 8000g for 20 min, and the supernatant collected and referred to as mitochondrial extract II.
Finally, the microsomal fractions are isolated from the supernatant following the first 8000g spin in step 2. Samples are spun at 100,000g for 1 h at 4°C (Beckman SW40.1 rotor). The pellet is extracted using mitochondrial extraction buffer II, left on ice for 30 min, and centrifuged at 8000g for 20 min at 4°C. The supernatant is saved as the “cytosolic” fraction.
A schematic overview of the fractionation methodology is shown in Fig. 2. In addition, we perform conventional biochemical techniques, including immunoblots and enzymatic assays, to examine our fractionation methods. Results of these experiments are shown in Fig. 3. In Fig. 3C, note that we observe substantial contamination of mitochondrial ATP synthase when fractionations are performed using a polytron, compared with a dounce homogenizer, and that the addition of the sucrose cushions further lowers the amount of mitochondrial contamination in other fractions.
Fig. 2.

Fractionation protocol. A summary schematic overview is provided.
Fig. 3.

Analysis of fraction purity. (A) Western blotting of cardiac ventricular subcellular fractions against selected marker proteins. (B) Normalized enzyme activity of creatine kinase and lactate dehydrogenase in subcellular fractions. (C) Western blotting against the mitochondrial membrane protein F1-ATPase β-subunit in fractions isolated under different conditions: (1) use of a polytron homogenize to homogenize cardiac muscle; (2) use of a dounce homogenizer to homogenize the tissues; or (3) use of a dounce homogenizer together with an additional sucrose cushion.
3.2. Digestion of Cell Extracts for MudPIT Analysis
One hundred and fifty micrograms of total protein from each fraction are precipitated overnight with 5 vol of ice-cold acetone followed by centrifugation at 21,000g for 20 min.
The protein pellet is solubilized in 8 M urea, 50 mM Tris-HCl, pH 8.5, at 37°C for 2 h and reduced by the addition of 1 mM DTT for 1 h at room temperature followed by carboxyamidomethylation with 5 mM iodoacetamide for 1 h at 37°C.
The samples are then diluted to 4 M urea with 100 mM ammonium bicarbonate, pH 8.5, and digested with a 1:150 molar ratio of endoproteinase Lys-C at 37°C overnight.
The following day, mixtures are further diluted to 2 M urea with 50 mM ammonium bicarbonate, pH 8.5, and a final concentration of 1 mM CaCl2, and rotated overnight with Poroszyme trypsin beads at 30°C.
The resulting peptide mixtures are solid phase-extracted with SPEC-Plus PT C18 cartridges according to the manufacturer’s instructions and stored at −80°C until further use.
3.3. MudPIT Analysis
A fully automated 12-step, 20-h MudPIT chromatographic procedure is used as described previously (11). A MudPIT consists of 12 independent chromatographic steps each containing a salt bump at the beginning, which aims at moving a subset of peptides from the first dimension of the chromatography column (strong ion exchange) onto the second dimension (reverse phase) of the chromatography column. Here the peptides are separated by a conventional water/ACN gradient and directly sprayed into the mass spectrometer. The 12 steps differ by increasing concentrations in the initial salt bump used to move them onto the reverse-phase material.
An HPLC quaternary pump is interfaced with an LCQ DECA XP ion trap tandem mass spectrometer (ThermoFinnigan, San Jose, CA).
A 100-μm inner diameter-fused silica capillary microcolumn is pulled to a fine tip using a P-2000 laser puller (Sutter Instruments, Novato, CA) and packed first with 10 cm of 5-μm Zorbax Eclipse XDB-C18 resin and then with 6 cm of 5-μm Partisphere strong cation exchange resin.
Peptide samples are loaded manually onto a fresh column using a pressure vessel. The four buffer solutions used for the chromatography are described in Subheading 2.3., item 5. The first 80 min step consists of a gradient from 0 to 80% buffer B for 70 min and a hold at 80% buffer B for 10 min. The next 11 steps are 110 min each with the following profile: 5 min of 100% buffer A, 2 min of x% buffer C/D, 3 min of 100% buffer A, a 10-min gradient from 0 to 10% buffer B, and a 90-min gradient from 10 to 45% buffer B. The 2-min buffer C percentages (x) in steps 2–12 are as follows: 10% C, 20% C, 30% C, 40% C, 50% C, 60% C, 70% C, 80 %C, 90% C, 100% C, and 100% D.
3.4. Sequest, STATQUEST, and Database Management Systems
Uninterpreted fragmentation (daughter) product ion mass spectra are sequence-mapped against a minimally redundant set of human and mouse protein sequences obtained from the SWISS-PROT and TrEMBL databases using the SEQUEST software algorithm (3) running on a multiprocessor computer cluster. Sequest search results are further validated using an in-house generated, probability-based computer program, termed STATQUEST (5). This program automatically assigns a p-value threshold cut-off corresponding to a defined percentage likelihood of corrected peptide identification. In general, a greater than 95% likelihood of correct identification is used to minimize false-positive identifications and the resulting data are parsed into an in-house SQL type database management system. The use of database management systems is highly advised for large-scale proteomics datasets, because it allows for streamlined, user-defined data queries, which greatly enhance the analysis of genome-wide proteomics projects. Furthermore, the database allows for the storage of a multitude of parameters, such as number of uniquely identified peptides, number of recorded spectra, isoelectric point (pI), and molecular weight of each of the identified proteins, which can be of tremendous value for further in-depth data interpretation (see Note 2).
3.5. Bioinformatics
Computational analysis of large-scale proteomics projects has become the major bottleneck of genome-wide data analysis pipelines. Therefore, a multitude of bioinformatics tools have been developed to speed up the tedious process of data processing and interpretation. In the following section, several essential tools for the analysis of expression proteomics profiles that are in current use in our laboratory will be discussed.
3.5.1. Data Clustering
The sheer size of proteomic datasets make the discovery of meaningful candidates and patterns a very difficult task. Therefore, clustering is often the starting point for grouping a set of expressed proteins based on similarities in their expression patterns. In most cases cluster analysis allows for the discovery of hidden information and regulatory patterns that have yet to be discovered. Several powerful commercial and publicly available software packages are available that are capable of performing most users tasks. A popular publicly available tool is Cluster 3.0, which is based on the original cluster tool developed by the Eisen group (17). First, expression datasets are converted to tab delimited text file containing their experimental conditions (columns), identified proteins (rows) and in the case of proteomics data, some quantitative value of protein expression levels. This file is then opened with the Cluster 3.0 software tool. Next, one or more of the several mathematical models (termed similarity or distance metrics) can be applied to calculate the degree of correlation between the profiles, allowing for subsequent clustering to be performed. A second software tool, termed TreeView, allows users to display the output of the Cluster 3.0 software graphically. Individual clusters can be selected and the protein names/IDs extracted for follow-up analysis. Figure 4 shows a typical clustergram generated for proteins detected in a series of repeat heart subcellular fractionation profiles. As a semi-quantitative estimate of protein abundance, our laboratory typically uses spectral counts (as described in Subheading 3.6.). To optimize the performance of the cluster tool, blanks (i.e., where a protein was not detected in a given fraction) are usually replaced by a non-zero low value (e.g., 0.01) (18).
Fig. 4.

Data visualization. Four individual organelle fractions (cytosol, microsomes, mitochondria, and nuclei) from wild-type heart tissue were analyzed independently by multiple MudPIT analyses. The entire set of proteins identified was clustered using spectral counts as a quantitative estimate of relative protein abundance in each fraction. The profiles are displayed using a “heat map” format. Organelle-specific clusters displaying statistically significantly enriched membership for select functional annotation categories are highlighted by dashed boxes, together with the corresponding GO terms.
3.5.2. Protein Annotation
Protein annotation refers to the known or predicted biological/molecular properties of a protein as extracted from the literature. A large number of highly useful Web pages are available to extract important protein annotations. In particular, the ExPASy molecular biology server (http://ca.expasy.org/) is a highly useful Web tool. This Web page allows biologists to extract extensive information concerning a specific protein (e.g., function, literature links, subcellular localization, etc.). Furthermore, ExPASy serves as a Web portal connecting to diverse, additional knowledge databases, such as Mouse Genome Database (http://www.informatics.jax.org/), domain databases such as (InterPro or Pfam), and Gene Ontology. In addition, the Web portal offers a large number of useful proteomic tools and their corresponding Web pages, which should allow every user to extract a large amount of information on almost every protein.
The Gene Ontology Database (GO; www.geneontology.org) represents another extremely useful database. The GO consortium is aimed at providing the molecular function, biological process, and cellular component of proteins for every major organism in a defined user-friendly vocabulary (19). The GO database consists of three main branches, termed biological process, molecular function, and cellular component. Each main category branches consecutively into a more detailed and complex network of GO terms describing specific functions or properties of particular proteins. By mapping proteins identified in an expression-based proteomics project onto the Gene Ontology database, biologists can accumulate important information about the proteins identified in a particular sample quickly. One of the drawbacks of the GO database is that not every identified protein can be mapped to a defined GO term. Only about 75% of the proteins identified throughout a proteomics project can be linked to one or more GO terms in the GO database.
Several tools are available for the biological community to map proteins to GO terms. We developed a Perl-based program termed GOClust. Tab delimited text files of proteins containing a SwissProt/TrEMBL accession number are used in this case. The final output is a series of tables of grouped proteins that share a common annotation to one or more pre-selected GO terms. Alternatively, the Java base program GoMiner (http://discover.nci.nih.gov/gominer/) can be used. Users input a list of SwissProt/TrEMBL accession numbers and the program returns a list of matching GO terms in an attractive pull-down format. An interesting feature of the GoMiner software tool is that two lists—for example, the total list of proteins identified and a subcluster of proteins found to be upregulated in a disease state—can be compared with each other. The program compares the GO terms matched to both lists and provides the user with significantly enriched GO terms in the subcluster, as compared to the total input list. This feature can be extremely useful for finding biological processes or molecular functions of proteins responsible for the development of a disease phenotype, if no hypothesis is readily available. In collaboration with Dr. Tim Hughes (University of Toronto), we have developed a similar software tool, termed MouseSpec. The program accepts protein accession numbers from either the SwissProt/TrEMBL or the IPI databases as an input. The output is a list of statistically enriched GO terms (together with their p value) as compared to a locally stored GO database. A Bonferroni correction factor can be used to correct for multi-hypothesis testing; the p value threshold deemed significant for an individual test is divided by the number of tests conducted, thereby accounting for spurious significance owing to multiple testing over all the categories in the GO database. A cut-off value of 10−3 is used as a final selection criterion to highlight promising, biologically interesting clusters (20).
3.6. Quantitative Nature and Post-Analyses
Confidently identifying as many proteins as possible in a particular sample is an important task. Nevertheless, if comparison of samples, for example, wild-type vs disease, is desired, an estimation of relative protein abundance is also essential. Determination of relative protein abundance on a truly global scale is a very challenging task and several approaches have been published in past few years. The most commonly used approach for quantitative proteomics is the use of isotope labeling, particularly, the isotope coded affinity tag (ICAT) pioneered in the laboratory of Dr. Ruedi Aebersold at the Institute for Systems Biology (21). In this method, two samples are labeled independently with either a light or a heavy isotope containing reagent. Samples are combined, digested enzymatically, and analyzed by LC-MS. Because isotopes possess chemically and physically identical properties, the two differently labeled peptides (light and heavy) will co-elute from the chromatographic column. Nevertheless, because they differ by a defined mass unit, the two peptides will be separated by the mass spectrometer. Integration of the area underneath both peaks permits relative quantification of the proteins identified. The ICAT methodology has been applied successfully to several biologically oriented projects (22–24). However, isotope-labeling reagents, such as the ICAT reagent, are expensive, limiting their application to large-scale projects. Moreover, the quantitative integration of every co-eluting peak requires extensive computation and might only result in the accurate quantification of the higher abundant proteins (21).
An alternative, label-free methodology based on the cumulative number of recorded spectra mapping to an identified protein has been published by the Yates laboratory (25). Although not as accurate as isotope labeling, this technology has bypassed the need for expensive isotope labels and time-consuming back-end analysis tools. From our experience, this methodology generally provides a good first indication of changes in relative protein abundance, especially if a larger number of spectra are recorded for a specific protein and if the changes between two conditions (healthy and disease) are considerable. As a result, this is the method of choice for our cardiac-tissue-profiling projects.
3.7. Verification
As with all large-scale projects, there is the inherent risk of false-positive data being included in the data sets. To minimize this risk, we set our detection stringency limits quite high (i.e., greater than 95% confidence interval for protein determinations). However, in critical experiments, we employ conventional methods of validation of the bioinformatics strategies. This includes, where available, Western blot analyses, assays of enzymatic activities where appropriate, and RT-PCR to measure mRNA levels in cases where antibodies or other assays are not available.
3.8. Conclusion
Advanced high-throughput tandem MS-based shotgun protein profiling techniques and allied computational approaches can be applied to the examination of the effects of cardiomyopathy on global patterns of protein expression and accumulation in heart tissue in a set of well-defined mouse models of cardiac disease. This will provide a unique opportunity for a more complete understanding of the molecular logic that governs cardiac muscle physiology and will provide insights into the biochemical and physiological basis for the development of heart disease. If we understand these progressive processes, we will be able to design therapeutic interventions that will block progression to cardiac disease. We also envision that the information will provide us with the potential for identifying biomarkers of heart disease and even of specific forms of heart disease. If the results are duplicated in large-scale human cohorts, they permit the development of effective clinical methodologies for early intervention and the prevention of progression to heart failure in human patients.
Fig. 5.

Analysis of MudPIT protein detection. (A) Protein abundance and dynamic range. Throughout the analysis of four organelle fractions in wild-type heart, a total of 1652 proteins with a total of 79,197 spectra were detected. Displayed are the fraction (%) of proteins and spectra for specific subsets of the total identifications. These results show that although high-abundance proteins make up only a small fraction of the proteins identified, they are nevertheless preferentially detected with a large number of corresponding spectra, potentially masking proteins of lower abundance. (B) Random sampling and detection saturation. The total number of high-confidence proteins detected in the heart cytosol after a certain number of individual MudPIT analyses is presented. Repeated analysis results in an apparent saturation in the total cumulative number of proteins that can be identified.
Acknowledgments
Work in our laboratory is supported by grants from the Natural Science and Engineering Research Council of Canada (to AE), the Ontario Genomics Institute and Genome Canada (to AE and DHM), by Heart and Stroke Foundation of Ontario Grant T-5042 and CIHR Grants MT-12545 and MOP-49493 and the Neuromuscular Research Partnership Program (to DHM), and by the Muscular Dystrophy Association (to AOG). AOG is supported by a fellowship from the Heart and Stroke Foundation of Canada; TK was supported by a fellowship from the Josef Schormuller Gedachtnisstiftung.
Footnotes
The major difficulty in applying high-resolution, global protein analysis to muscle tissues is the presence of high concentrations of sarcomeric, mitochondrial, and cytoskeletal proteins that are not present in other tissue types. We have addressed this issue by including a gentle dounce homogenization, which minimizes the rupture of mitochondria during our fractionation, thereby diminishing the contamination of other fractions with mitochondrial proteins. In addition, to remove a substantial amount of contractile and cytoskeletal proteins, we include two sucrose cushions in our fractionation protocol. These cushions allow for a cleaner nuclei preparation with little contractile protein contamination; unfortunately, the total yield of nuclear protein is very low (see Fig. 3).
The extreme complexity of mammalian cardiac tissue is problematic even for high-resolution separation methodologies such as the MudPIT technique. Heart tissue contains an overwhelming number of expressed proteins (in the range of at least several thousand). Enzymatic digestion further increases this complexity to tens of thousands of peptides. Identification of proteins in MudPIT-based studies is based on the isolation and fragmentation of individual peptides eluting from the HPLC column into the mass spectrometer. However, not all peptide ions can be successfully analyzed by the mass spectrometer owing to limitations in the speed of scanning or overall duty-cycle (25). This confounding issue is complicated further by the wide range in protein concentrations typically found in mammalian tissue. As a result, peptides from lower abundance proteins often elute from the chromatographic columns without ever being detected by the mass spectrometer, because they can be masked by a few very high-abundance peptide peaks generated by higher-abundance proteins (see Fig. 5A).
Several partial solutions have been devised to surmount this problem. For instance, simplification of the protein mixture, e.g., by subcellular pre-fractionation, provides a simple, yet powerful, approach for improving the odds of detection of lower-abundance proteins. Nevertheless, organellar extracts still contain a highly complex mixture of proteins, resulting in the missed identification of many expressed proteins. Organelle extracts can be further simplified using conventional fractionation methodologies aimed at protein level fractionation, such as ion-exchange or size-exclusion chromatography. However, because most proteomic profiling projects are aimed at the comparison of particular samples (e.g., healthy vs disease state), a simple comparison can result in misleading interpretations. Therefore, repeat analyses of the same sample (i.e., running multiple LC-MS analyses), can improve the overall detection coverage (total number of protein identifications) and markedly increase detection of lower-abundance proteins (see Fig. 5B).
References
- 1.Dos Remedios CG, Liew CC, Allen PD, et al. Genomics, proteomics and bioinformatics of human heart failure. J Muscle Res Cell Motil. 2003;24:251–260. doi: 10.1023/a:1025433721505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aebersold R, Mann M. Mass spectrometry-based proteomics. Nature. 2003;422:198–207. doi: 10.1038/nature01511. [DOI] [PubMed] [Google Scholar]
- 3.Eng JK, McCormack AL, Yates JR., 3rd An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J Am Soc Mass Spectrom. 1994;5:976–989. doi: 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- 4.Cagney G, Emili A. De novo peptide sequencing and quantitative profiling of complex protein mixtures using mass-coded abundance tagging. Nat Biotechnol. 2002;20:163–170. doi: 10.1038/nbt0202-163. [DOI] [PubMed] [Google Scholar]
- 5.Kislinger T, Emili A. Going global: protein expression profiling using shotgun mass spectrometry. Curr Opin Mol Ther. 2003;5:285–293. [PubMed] [Google Scholar]
- 6.Andersen JS, Lam YW, Leung AK, et al. Nucleolar proteome dynamics. Nature. 2005;433:77–83. doi: 10.1038/nature03207. [DOI] [PubMed] [Google Scholar]
- 7.Andersen JS, Wilkinson CJ, Mayor T, et al. Proteomic characterization of the human centrosome by protein correlation profiling. Nature. 2003;426:570–574. doi: 10.1038/nature02166. [DOI] [PubMed] [Google Scholar]
- 8.Mootha VK, Bunkenborg J, Olsen JV, et al. Integrated analysis of protein composition, tissue diversity, and gene regulation in mouse mitochondria. Cell. 2003;115:629–640. doi: 10.1016/s0092-8674(03)00926-7. [DOI] [PubMed] [Google Scholar]
- 9.Durr E, Yu J, Krasinska KM, et al. Direct proteomic mapping of the lung microvascular endothelial cell surface in vivo and in cell culture. Nat Biotechnol. 2004;22:985–992. doi: 10.1038/nbt993. [DOI] [PubMed] [Google Scholar]
- 10.Schirmer EC, Florens L, Guan T, et al. Nuclear membrane proteins with potential disease links found by subtractive proteomics. Science. 2003;301:1380–1382. doi: 10.1126/science.1088176. [DOI] [PubMed] [Google Scholar]
- 11.Washburn MP, Wolters D, Yates JR., 3rd Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nat Biotechnol. 2001;19:242–247. doi: 10.1038/85686. [DOI] [PubMed] [Google Scholar]
- 12.Waterston RH, Lindblad-Toh K, Birney E, et al. Initial sequencing and comparative analysis of the mouse genome. Nature. 2002;420:520–562. doi: 10.1038/nature01262. [DOI] [PubMed] [Google Scholar]
- 13.Venter JC, Adams MD, Myers EW, et al. The sequence of the human genome. Science. 2001;291:1304–1351. doi: 10.1126/science.1058040. [DOI] [PubMed] [Google Scholar]
- 14.Lander ES, Linton LM, Birren B, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 15.Kislinger T, Gramolini AO, Pan Y, et al. Proteome dynamics during C2C12 myoblast differentiation. Mol Cell Proteomics. 2005;4:887–901. doi: 10.1074/mcp.M400182-MCP200. [DOI] [PubMed] [Google Scholar]
- 16.Pan Y, Kislinger T, Gramolini AO, et al. Identification of biochemical adaptations in hyper- or hypocontractile hearts from phospholamban mutant mice by expression proteomics. Proc Natl Acad Sci USA. 2004;101:2241–2246. doi: 10.1073/pnas.0308174101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eisen MB, Spellman PT, Brown PO, et al. Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci USA. 1998;95:14863–14868. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cox B, Kislinger T, Emili A. Integrating gene and protein expression data: pattern analysis and profile mining. Methods. 2005;35:303–314. doi: 10.1016/j.ymeth.2004.08.021. [DOI] [PubMed] [Google Scholar]
- 19.Ashburner M, Ball CA, Blake JA, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Robinson MD, Grigull J, Mohammad N, et al. FunSpec: a web-based cluster interpreter for yeast. BMC Bioinformatics. 2002;3:35. doi: 10.1186/1471-2105-3-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gygi SP, Rist B, Gerber SA, et al. Quantitative analysis of complex protein mixtures using isotope-coded affinity tags. Nat Biotechnol. 1999;17:994–999. doi: 10.1038/13690. [DOI] [PubMed] [Google Scholar]
- 22.Brand M, Ranish JA, Kummer NT, et al. Dynamic changes in transcription factor complexes during erythroid differentiation revealed by quantitative proteomics. Nat Struct Mol Biol. 2004;11:73–80. doi: 10.1038/nsmb713. [DOI] [PubMed] [Google Scholar]
- 23.Han DK, Eng J, Zhou H, et al. Quantitative profiling of differentiation-induced microsomal proteins using isotope-coded affinity tags and mass spectrometry. Nat Biotechnol. 2001;19:946–951. doi: 10.1038/nbt1001-946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shiio Y, Donohoe S, Yi EC, et al. Quantitative proteomic analysis of Myc oncoprotein function. EMBO J. 2002;21:5088–5096. doi: 10.1093/emboj/cdf525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu H, Sadygov RG, Yates JR., 3rd A model for random sampling and estimation of relative protein abundance in shotgun proteomics. Anal Chem. 2004;76:4193–4201. doi: 10.1021/ac0498563. [DOI] [PubMed] [Google Scholar]