

Abstract
α-Crystallin B (cryAB) is the most abundant small heat shock protein in cardiomyocytes (CMs) and has been shown to have potent antiapoptotic properties. Because the mechanism by which cryAB prevents apoptosis has not been fully characterized, we examined its protective effects at the cellular level by silencing cryAB in mouse neonatal CMs using lentivector-mediated transduction of short hairpin RNAs. Subcellular fractionation of whole hearts showed that cryAB is cytosolic under control conditions, and after H2O2 exposure, it translocates to the mitochondria. Phosphorylated cryAB (PcryAB) is mainly associated with the mitochondria, and any residual cytosolic PcryAB translocates to the mitochondria after H2O2 exposure. H2O2 exposure caused increases in cryAB and PcryAB levels, and cryAB silencing resulted in increased levels of apoptosis after exposure to H2O2. Coimmunoprecipitation assays revealed an apparent interaction of both cryAB and PcryAB with mitochondrial voltage-dependent anion channels (VDAC), translocase of outer mitochondrial membranes 20 kDa (TOM 20), caspase 3, and caspase 12 in mouse cardiac tissue. Our results are consistent with the conclusion that the cardioprotective effects of cryAB are mediated by its translocation from the cytosol to the mitochondria under conditions of oxidative stress and that cryAB interactions with VDAC, TOM 20, caspase 3, and caspase 12 may be part of its protective mechanism.
Keywords: mitochondria, reactive oxygen species, phosphorylated α-crystallin B, cardiac cell
α-Crystallin B (cryAB) is a member of the small heat shock protein (HSP) family of molecular chaperones (38). It is the most abundantly expressed small HSP in cardiac muscle, comprising 3–5% of total protein in cardiac myocytes (38). Studies have demonstrated that overexpression of cryAB has potent cardioprotective properties in cultured rat cardiomyocytes (CMs) (28) or in hearts of transgenic mice (32), protecting them from ischemia-reperfusion (I/R) damage. Additionally, using in vitro models, cryAB has been shown to prevent apoptosis induced by various insults, including H2O2 treatment (25). In contrast, silencing of cryAB increases sensitivity to I/R injury and leads to increased cell death in mouse hearts (29). Recently, our group (5) has shown that cryAB is protective against apoptosis in a mouse model of calcineurin hypertrophy, which presents with elevated levels of endoplasmic reticulum (ER) stress (5).
The protective mechanism of cryAB in response to stress has not been fully characterized, but a previous study (3) has indicated that cryAB binds myofilament proteins, thereby preserving contractile protein integrity and myocardial function. CryAB also translocates to the mitochondria (27) and is phosphorylated on Ser59 (18) in response to ischemia and other stresses (17), which is required for limiting myocardial cell apoptosis (28), although the precise interactions mediating this protection have not been elucidated.
Apoptosis in cells occurs by two pathways: the intrinsic pathway involving mitochondria or the extrinsic pathway downstream of death receptors. The mitochondrial and death receptor pathways activate distinct apical caspases (caspase 9 or caspase 8, respectively) that activate the downstream executioner caspase 3 (43). Ischemia or other stresses that mimic ischemia activate the intrinsic apoptotic pathway, leading to the opening of the mitochondrial permeability transition pore (MPTP) and further downstream apoptotic events, culminating in cellular demise (11). Global ischemia as well as reperfusion have been associated with significant increases in ROS, including myocardial H2O2 content (37), which plays a significant role in oxidative stress injury (39). H2O2 also leads to apoptosis in CMs, by activating the intrinsic pathway of apoptosis (41), and thus makes H2O2 a very good in vitro model of I/R injury.
The same stress stimuli that trigger apoptosis, such as oxidative stress (25, 41), induce the synthesis of diverse HSPs, which confer a protective effect against a wide range of cellular stresses. Recent evidence indicates that many HSPs are anti-apoptotic by inhibiting one or more components in the apoptotic cascade (2, 8). In this regard, cryAB has been shown to directly interact with precursors of caspase 3 to suppress its activation in an immortalized rabbit lens epithelial cell line (25) and in breast carcinoma cells (20). In human lens epithelial cells, cryAB has also been shown to interact with the proapoptotic proteins Bax and Bcl-XS, preventing their translocation from the cytosol to the mitochondria, thus leading to decreased apoptosis (26). The antiapoptotic mechanisms of cryAB in CMs, however, have not been fully characterized. Thus, the purpose of this study was to elucidate the mechanisms by which cryAB prevents apoptosis in CMs with a specific focus on the mitochondrial pathway in response to H2O2-induced oxidative stress, an in vitro model that mimics I/R (37).
MATERIALS AND METHODS
Neonatal CM isolation and culture
Neonatal mice were euthanized using isoflurane, in accordance with procedures approved by our institutional Animal Care and Ethics Committee. Hearts were harvested and placed in ice-cold Hanks’ solution [containing 136 mM NaCl, 4.2 mM KCl, 5.6 mM dextrose, 0.44 mM KH2PO4, 0.34 mM NaH2PO4, 4.2 mM NaHCO3, 5 mM HEPES (pH 7.4), and 100 U/ml penicillin-streptomycin (Invitrogen)]. Atria were removed and discarded, and ventricles were cut into small pieces (2– 4 pieces/heart) and washed several times with Hanks’ solution. After washes, tissues were incubated in fresh Hanks’ solution with 1 mg/ml collagenase type II (Worthington Biochemicals) and subjected to gentle rocking for 2 h at room temperature. The suspended cells were pelleted by centrifugation at 800 g for 5 min. Fibroblasts were removed through preplating in 10% FBS-containing media for 1 h.
Lentivector production and transduction of neonatal CMs
Lentivector compatible short hairpin (sh)RNA clones targeting mouse cryAB were obtained from Open-Biosystems (Thermo Scientific). The scrambled shRNA construct used as a negative control was a kind gift from Stephane Angers (University of Toronto, Toronto, ON, Canada). Lentivector production and transduction of neonatal CMs were performed as previously described (5). Plasmids were isolated using Qiagen Maxi preparations according to the manufacturer’s instructions. The packaging plasmid (pCMV-R8.74psPAX2, 2.5 μg), envelope plasmid (VSV-G/pMD2.G, 0.3 μg), and target construct plasmid (pLKO.1, 2.7 μg) expressing either the shRNA or scrambled (Scram) shRNA (as a negative control) were simultaneously transduced into human embryonic kidney (HEK)-293T cells using FuGene (Roche) diluted in Optimem (Invitrogen). Neonatal CMs were incubated with supernatant from transduced HEK-293T cells for 21 h, after which the medium was replaced daily. Transduced CMs were selected by an incubation with 2 μg/ml puromycin for 48 h to remove all nontransduced cells to ensure a homogenous population of transduced cells.
Subcellular fractionation and sucrose gradient separation
Adult mice were euthanized by CO2 asphyxiation. Hearts were harvested, and ventricular tissue was isolated. The tissue was rinsed with ice-cold PBS to remove any remaining blood and placed in ice-cold lysis buffer [containing 250 mM sucrose, 50 mM Tris·HCl (pH 7.4), 5 mM MgCl2, 1 mM DTT, and 1 mM PMSF]. The tissue was dounce homogenized, and differential centrifugation was carried out to isolate nuclear, cytosolic, microsomal, and mitochondrial fractions, as previously described (9). The homogenized sample was also used for sucrose gradient separation, as previously described (34).
CMs from neonatal mice were cultured as described above. Cells were maintained in culture for 5 days, and on the sixth day they were either maintained in culture or stressed with H2O2 (ranging in concentration from 0 to 200 μM) for 24 h. CMs were rinsed with PBS and collected in lysis buffer as described above. CMs in lysis buffer were dounce homogenized, and differential centrifugation was carried out to isolate cytosolic and organellar (including the mitochondria) fractions, as previously described (9).
Sample preparation for transmission electron microscopy
CM fixation was performed at the Department of Pathology and Laboratory Medicine of Mount Sinai Hospital (Toronto, ON, Canada). Briefly, neonatal CMs transduced with either Scram shRNA or cryAB-targeting shRNA [cryAB knockdown (KD)] for transmission electron microscopy (TEM) were fixed in 2% glutaraldehyde in 0.1 M sodium cacodylate buffer, rinsed in buffer, postfixed in 1% osmium tetroxide in buffer, dehydrated in a graded ethanol series followed by propylene oxide, and embedded in Quetol-Spurr resin. Sections (100 nm thick) were cut on an RMC MT6000 ultramicrotome, stained with uranyl acetate and lead citrate, and viewed in an FEI Tecnai 20 TEM.
Immunogold labeling
Immunogold labeling of cryAB was performed at the Department of Pathology and Laboratory Medicine of Mount Sinai Hospital. Whole hearts from adult mice were fixed in 4% paraformaldehyde and 0.1% glutaraldehyde in 0.1 M sodium cacodylate buffer, rinsed in buffer, dehydrated in a graded ethanol series with progressive lowering of temperature, and embedded in LR white resin. Sections (100 nm thick) were cut on an RMC MT6000 ultra-microtome, labeled with anti-cryAB and anti-phosphorylated cryAB (PcryAB) antibodies followed by 10-nm gold-conjugated secondary antibodies, stained with uranyl acetate and lead citrate, and viewed in an FEI Tecnai 20 TEM. Negative controls consisted of gold-conjugated secondary antibody alone.
Viability assays
Neonatal CMs were subjected to 60 μM H2O2 for 24 h. Viability assays were carried out using a cell counting kit (CCK-8, Dodinjo) according to the manufacturer’s instructions. Early apoptosis was determined based on the dissipation of mitochondrial membrane potential, as measured by JC-1 fluorescent dye (Abcam), a mitochondrial dye, which was added at a concentration of 2.0 μM (10) according to the manufacturer’s instructions. Apoptosis was measured by detecting caspase 3 activity using an assay (R&D Systems) as per the manufacturer’s instructions and by labeling of TUNEL-positive nuclei (PRP-Histology Laboratory, University Health Network, Toronto, ON, Canada). The caspase 3 inhibitor Z-D(OMe)-E(OMe)-V-D(OMe)fluoromethyl ketone (Z-DEVD-FMK; R&D Systems) was used at a concentration of 100 μM (7) according to the manufacturer’s instructions.
ROS detection and inhibition
The presence of ROS in cultured wild-type (WT) and KD neonatal CMs was detected using CellROX deep red reagent (Invitrogen), a fluorogenic probe, according to the manufacturer’s instructions. Briefly, CMs were seeded on 96-well plates and transduced with Scram shRNA or shRNA for cryAB and maintained in culture or stressed with 60 μM H2O2. The dye was then added at a concentration of 5 μM, and fluorescence was measured with a Perkin-Elmer plate reader. The ROS scavengers tiron (41) and sodium pyruvate (42) (Sigma) were used at concentrations of 1.0 mM.
Coimmunoprecipitation
Immunoprecipitations were carried out using protein A/G-agarose beads (Thermo Scientific). Briefly, heart tissue homogenates were obtained from WT control hearts or hearts exposed to 100 μM H2O2 for 1 h (41). The tissue was collected in lysis buffer, as described above. The lysate was cleared by centrifugation for 15 min at 8,000 g at 4°C. To allow antibody-protein complex formation, the cleared lysate was incubated at 4°C under continuous rotation with antibody in binding buffer [140 mM NaCl and 14 mM KCl (pH 7.4)] and 0.1% Triton X-100 with 0.01% BSA for 2 h. Protein A/G-agarose beads were blocked in 0.1% BSA in binding buffer for 2 h. The beads were pelleted, added to the protein sample, and rotated overnight at 4°C. Samples were washed three times and eluted in 0.1 M glycine (pH 2.4).
Immunoblot analysis
Total cellular protein was harvested from control cardiac ventricular homogenates from WT animals or from cardiac ventricular homogenates stressed with 100 μM H2O2 for 1 h and was subjected to standard immunoblot analysis. Protein concentrations were determined by Bradford assay, and equal protein levels were loaded. The following antibodies were used to target specific proteins: rabbit polyclonal to cryAB (Stressgen, 1:1,000), Ser59 PcryAB (Stressgen, 1:1,000), rabbit polyclonal to caspase 3 (Abcam, 1:1,000), rabbit polyclonal to caspase 12 (Abcam, 1:1,000), mouse monoclonal to caspase 9 (Cell Signalling Technologies, 1:1,000), rabbit polyclonal to cytochrome c (Cell Signalling Technologies, 1:1,000), goat polyclonal to voltage-dependent anion channel (VDAC; Santa Cruz Biotechnology, 1:100), mouse monoclonal to translocase of outer mitochondrial membranes 20 kDa (TOM 20; Santa Cruz Biotechnology, 1:1,000), and mouse monoclonal to cryAB (Santa Cruz Biotechnology, 1:100).
Immunofluorescence
Adult CMs were isolated as previously described (33) and fixed in ice-cold 90% methanol. Staining was performed as previously described (34). Briefly, cells were dissociated in modified Hanks’ solution (Hanks’ solution containing 10 mM taurine, 0.1 mM EGTA, 10 mM 2,3-butanedione monoxime, 1 mg/ml BSA, and 1 mg/ml collagenase) with a magnetic stir bar added to the tube and rotated very gently at 37°C for 5 min to dissociate cells. Once dissociated, cells were fixed in 90% ice-cold methanol. For immuno-fluorescence, nonspecific interactions were first suppressed with 5% horse serum in permeabilization buffer [0.2% Tween 20 and 0.5% Triton X-100 in PBS (pH 7.0)] for 30 min, and samples were then incubated with primary antibodies in permeabilization buffer overnight at 4°C. Samples were then washed three times with PBS and incubated with either Alexa fluor 488 or Alexa fluor 633. Two-dimensional images were collected using a Leica DM IRBE inverted microscope equipped with a Leica TCS SP laser scanning confocal system. Spectra for Alexa fluor 488 were collected by excitation at 488 nm and emission between 490 and 510 nm; images for Alexa fluor 633 were collected by excitation at 633 nm and emission between 640 and 670 nm. Three-dimensional (3-D) images were collected using a Quorum Angstrom Grid Axiovert 200M inverted structured illumination microscope system. For 3-D region of interest analyses, sequential sections of stained cells were acquired for 3-D image reconstruction and colocalization measurements. A 3-D volume was constructed from sequential z-sections of cells using Imaris software (version 7.4.2, Bitplane, Zurich, Switzerland). Colocalization statistics were calculated using Imaris software.
Statistical analysis
Statistical differences were determined by ANOVA and an unpaired Student’s t-test. Post hoc Tukey tests were performed when ANOVA was significant. Two-way ANOVA was used to determine interactions between independent variables, cryAB KD and viability, or cryAB KD and apoptosis. Results were considered significant at P < 0.05.
RESULTS
Expression pattern of cryAB in mouse cardiac muscle
CryAB protein is diffusely present in the cytosol under normal physiological conditions in CMs (23). To determine the endogenous distribution of cryAB protein among different cellular compartments, subcellular fractionation of adult mouse hearts was performed by differential centrifugation and sucrose gradient separation. Differential centrifugation was used to generate nuclear, cell organellar (including the mitochondria), microsomal, and cytosolic fractions. The positive fraction markers used were GAPDH for the cytosol, histone H3 for the nucleus, Na+-K+-ATPase for the microsomes, and VDAC for the organellar/mitochondrial fraction. As shown in Fig. 1A, fractions were relatively pure, as GAPDH was exclusively found in the cytosolic fraction, VDAC was found restricted to the mitochondrial fraction, histone H3 was preferentially found in the nuclear fraction, and Na+-K+-ATPase was predominately found in the microsomal fraction with some expression in the organellar/mitochondrial fraction. Immunoblot analysis for the expression of cryAB in each subcellular fractions showed the highest levels (83% of total levels) seen in the cytosol, in agreement with the literature (23). The remaining levels were 4% of total levels in the microsomal fraction, 12% of total levels in the nuclear fraction, and negligible levels (<1%) in the nuclear fraction (Fig. 1A). PcryAB, on the other hand, was predominately found in the mitochondrial fraction (75% of total levels) and at lower levels in the cytosol (25% of total levels). The cytosolic localization of cryAB was further confirmed by sucrose gradient separation of whole heart lysates. As shown in Fig. 1B, cryAB was restricted to fractions 4 – 6, with some expression in fraction 7. These data are consistent with GAPDH, a known cytosolic protein, which was also detected in fractions 4 – 6. VDAC, a mitochondrial marker, was mainly isolated with fractions 2– 4 and showed some expression in fractions 5 and 6. PcryAB was mainly isolated in fractions 2 and 3, consistent with VDAC, and to a lower extent in fraction 4, consistent with GAPDH, suggesting a mainly mitochondrial localization of PcryAB. Na+-K+-ATPase, a plasma membrane protein normally found within the microsome biochemical fraction (22), was isolated within the light fractions (with some expression in fractions 8 –12). Finally, electron microscopy images showed the distribution of cryAB in the cytosol and PcryAB predominantly in the mitochondria in adult mouse cardiac muscle. The representative images shown in Fig. 1C demonstrate that ~75% of gold-labeled cryAB (111 of 149 particles in 10 separate images) were found in the cytosol, 11% in the mitochondria, and 14% associated with the sarcomeres. When similar experiments were performed using gold-labeled PcryAB antibody, 75% of the gold particles were found in the mitochondria, whereas 10% were found in the cytosol and 15% were in the sarcomeres (73 total particles were observed in 10 separate images). Negative controls were labeled with gold-conjugated secondary antibody alone.
Fig. 1.

α-Crystallin B (cryAB) and phosphorylated cryAB (PcryAB) distribution in cardiac cells. A: immunoblot of cryAB and PcryAB in different subcellular fractions isolated from adult mouse hearts. The distribution of GAPDH as a marker for the cytosolic fraction, histone H3 for the nuclear fraction, and Na+-K+-ATPase for the microsomal fraction was assessed. A minimum of three experiments was performed for each fractionation. B: sub-cellular fractionation of wild-type (WT) adult mouse heart lysates run on a continuous 20–60% sucrose gradient. Fractions 1–12 contained the highest to lowest sucrose concentrations, respectively. The mitochondrial marker voltage-dependent anion channel (VDAC), the plasma membrane protein marker Na+-K+-ATPase, and GAPDH as a cytosol protein were detected as markers to monitor the fractionation procedure. A minimum of 3 experiments was performed for each gradient. C: electron microscopy images showing immunogold staining of cryAB and PcryAB under control conditions along with negative controls for cryAB and PcryAB staining. *Presence of gold particles. Scale bar = 100 nm. Images are representative of a minimum of 50 separate microscopy fields.
CryAB silencing induces loss of viability in CMs
To determine whether cryAB expression in mouse CMs affects cell viability, we silenced cryAB using lentivector-mediated transduction of shRNA targeting cryAB expression. Figure 2A shows the reduced levels of cryAB expression after the transduction of cryAB-specific shRNA lentivirus relative to a Scram shRNA (control vector). The transduction resulted in reduced cryAB expression to 28 ± 2% in KD CMs compared with Scram shRNA CMs (P < 0.05; Fig. 2, A and B).
Fig. 2.

Viability in cryAB-silenced cardiomyocytes (CMs). A: immunoblot for cryAB and actin in CMs either transduced with scrambled short hairpin (sh)RNAs (Scram shRNA) or cryAB targeting shRNA virus [cryAB knockdown (KD)]. In cryAB KD cells, cryAB levels were ~28% of the expression levels in CMs transduced with the Scram shRNA control plasmid. B: quantification of cryAB expression levels in CMs transduced with cryAB-targeting shRNA (cryAB KD) compared with CMs transduced with Scram shRNA. C: quantification of viability in CMs transduced with cryAB-targeting shRNA (cryAB KD) compared with Scram shRNA at increasing H2O2 concentrations. *P < 0.05 vs. Scram shRNA control CMs and **P < 0.05 vs. KD control; #P < 0.05, differences between groups (Scram shRNA vs. KD). Values are reported as means ± SE; n = 3/treatment. D: quantification of caspase 3 activity in Scram shRNA (*P < 0.05) and cryAB KD (**P < 0.05) CMs after exposure to 60 μM H2O2 compared with control conditions. #P < 0.05, differences between groups (Scram shRNA vs. KD). Values are reported as means ± SE; n = 6/treatment. E: quantification of TUNEL-positive nuclei in Scram shRNA (*P < 0.05) and KD (**P < 0.05) CMs after exposure to 60 μM H2O2 compared with control conditions. #P < 0.05, differences between groups (Scram shRNA vs. KD). Values are reported as means ± SE; n = ~1,000 cells/treatment.
Since cryAB has antiapoptotic properties (25, 28, 32) and a previous study (25) has shown that it prevents apoptosis induced by oxidative insult, neonatal CMs transduced with either cryAB-targeting lentivectors or Scram shRNA control lentivectors were analyzed after the administration of 60, 100, or 200 μM H2O2 for 24 h (41) to explore the role of cryAB on the viability of CMs exposed to H2O2. CryAB KD neonatal CMs showed a significant reduction in viability compared with neonatal CMs transduced with the Scram shRNA control construct at all H2O2 concentrations used (Fig. 2C). As expected, in Scram shRNA CMs, there was a progressive decrease in viability with increasing H2O2 concentrations relative to reference levels at baseline. In the absence of H2O2, viability in cryAB KD CMs was 50 ± 6% compared with levels seen in Scram shRNA CMs (P < 0.05). Decreases in viability were observed in KD CMs with increasing H2O2 concentrations, and these were significantly more pronounced at all H2O2 concentrations tested. Altogether, cryAB KD appears to promote cell death at baseline and after exposure to H2O2, and exposure to H2O2 seems to cause reductions in viability in Scram shRNA and cryAB KD CMs, which are more pronounced in KD CMs. Since cryAB KD alone induced significant cell death and treatment with 60 μM H2O2 led to significant reductions in viability in both Scram shRNA and cryAB KD CMs, subsequent experiments were performed using 60 μM H2O2 to exacerbate the cell death initially observed at baseline in KD CMs.
To confirm the activation of apoptosis at baseline and after exposure to H2O2, we measured caspase 3 activity in control and cryAB KD CMs under resting conditions and after exposure to H2O2 and in the absence or presence of caspase 3 inhibitors (Fig. 2D). Caspase 3 activity levels in Scram shRNA CMs under baseline conditions were set as a reference level of 1 arbitrary unit (AU). In cryAB KD CMs, caspase 3 activity levels were significantly higher than in Scram shRNA control CMs (130 ± 2%) at rest (P < 0.05). The addition of caspase 3 inhibitors had negligible effects on caspase 3 activity levels in Scram shRNA CMs under basal conditions, bringing the levels down to 90 ± 2% compared with baseline conditions. In KD CMs, however, caspase 3 activity levels were significantly lowered to 70 ± 2% of their baseline levels after the addition of caspase 3 inhibitors (P < 0.05). As expected, after exposure to H2O2, caspase 3 activity levels increased to 240 ± 4% in control CMs (P < 0.05), whereas in KD CMs, they increased approximately fivefold with the addition of H2O2 (460 ± 5% relative to Scram shRNA CM baseline levels). The addition of H2O2 together with caspase 3 inhibitors resulted in caspase 3 activity levels close to those observed at basal conditions (110 ± 3%) in Scram shRNA CMs and KD CMs (120 ± 3% of reference levels). We confirmed the levels of apoptosis in Scram shRNA CMs and cryAB KD CMs at baseline and after exposure to 60 μM H2O2 by TUNEL staining. Analysis of TUNEL-positive nuclei (Fig. 2E) showed a significantly higher percentage of TUNEL-positive nuclei in cryAB KD CMs compared with Scram shRNA CMs at baseline (30 ± 3% vs. 9 ± 2%, P < 0.05) and after exposure to H2O2 (67 ± 6% vs. 30 ± 5%, P < 0.05). These results suggest that KD CMs underwent apoptosis by activation of caspase 3 more readily compared with Scram shRNA CMs at baseline, an effect that was exacerbated after exposure to H2O2, suggestive of a protective role for cryAB against apoptosis.
To determine whether cryAB KD and exposure to H2O2 are interacting variables affecting CM viability, we performed two-way ANOVA and found that, although cryAB KD had significant effects on lowering viability (P < 0.05) and exposure to H2O2 also significantly lowered viability at all concentrations tested in Scram shRNA and cryAB KD CMs (P < 0.05), the effects of cryAB KD and H2O2 exposure combined on CM viability seemed to be additive. In other words, the interaction of cryAB KD and H2O2 exposure only approached statistical significance (P < 0.1). However, as shown in Fig. 2, A and B, cryAB silencing was not 100% efficient, which could account for the higher than expected viability that was observed in cryAB KD CMs in the presence of extrinsic oxidative stress. We performed two-way ANOVA to determine whether cryAB KD and exposure to H2O2 were interacting variables affecting CM apoptosis. CryAB KD and H2O2 were interacting variables (P < 0.05) that had synergistic effects on caspase 3 activity levels as well as on TUNEL staining (P < 0.05). These results suggest that although cryAB KD does not seem to have a significant effect on CM viability after H2O2, it does, however, have a significant effect on CM apoptosis, as measured by caspase 3 activity levels and TUNEL staining.
Upregulation and translocation of cryAB to the mitochondria under stress conditions
Expression levels of cryAB have been shown to increase in response to H2O2 exposure (35). To determine whether similar upregulation occurs in our CM model, CMs isolated from adult mice were treated with 100 μM H2O2, based on the literature (41), and cryAB levels were determined. Compared with untreated conditions, after exposure to H2O2, total cryAB levels were significantly increased (2.1-fold over control levels, P < 0.05). PcryAB levels were also significantly increased under H2O2 treatment, with a 1.8-fold increase (P < 0.05) in protein levels (Fig. 3, A and B).
Fig. 3.

Upregulation and translocation of cryAB and PcryAB to the mitochondria under oxidative stress conditions. A: immunoblots of cryAB and PcryAB along with actin standards in WT mouse adult CM controls or CMs exposed to 100 μM H2O2 for 1 h. B: quantification of cryAB and PcryAB levels in adult mouse CMs after exposure to 100 μM H2O2 relative to control levels (*P < 0.05). C: immunoblots of cytosolic and organellar subcellular fractions for cryAB and PcryAB in cultured neonatal CMs under control conditions and after exposure to 60 μM H2O2. The distribution of VDAC, a mitochondrial marker, and GAPDH, a cytosolic marker, was assessed to monitor the purity of the fractions. D: quantification of the mitochondrial and cytosolic distribution of cryAB and PcryAB in cultured neonatal CMs after exposure to 60 μM H2O2 relative to control conditions (#P < 0.05). The band intensities of cryAB organellar levels are expressed in arbitrary units (AU). Values are reported as means ± SE; n = 3. *P < 0.05, mitochondria vs. control cytosol conditions; **P < 0.05, mitochondria vs. stress cytosol conditions.
Under conditions of I/R, cryAB translocates to the mitochondria and contractile units (19). After I/R, phosphorylation of cryAB on the Ser59 residue enhances protection against apoptosis, and it has been suggested that cryAB and PcryAB may have different protective effects in CMs (19). To determine the distribution of cryAB and PcryAB in H2O2-induced oxidative stress, subcellular fractionation of cultured control neonatal mouse CMs and CMs exposed to 60 μM H2O2 for 24 h was performed. Figure 3, C and D, shows higher levels of cryAB expression in the cytosol and lower levels in the mitochondria. Under normal culture conditions, cryAB levels observed in the cytosol were 4.8-fold higher than the levels observed in the mitochondria. In contrast, upon exposure to 60 μM H2O2, cryAB translocated to the mitochondria, resulting in a 2.1-fold increase in cryAB levels associated with the mitochondria compared with control conditions (P < 0.05). PcryAB, on the other hand, was found at very low levels in the cytosol compared with the levels observed in mitochondria. Expression ratio levels of PcryAB were calculated to be 1:5 cytosolic to mitochondrial. Under stress conditions, PcryAB was almost exclusively associated with the mitochondria, with only < 5 ± 0.2% of total PcryAB detected in the cytosol (P < 0.05; Fig. 3, C and D). GAPDH and VDAC were used as fraction markers for the cytosol and mitochondria, respectively. Under control conditions, GAPDH and VDAC were exclusively found in the cytosolic and mitochondrial fraction, respectively. Taken together, these findings suggest that part of the protective mechanism of cryAB after exposure to oxidative stress may involve its translocation to the mitochondria.
CryAB silencing induces mitochondrial dysfunction
Since CM viability was significantly decreased under control conditions in cryAB KD CMs, it was of interest to determine the cause for the increased cell death in the absence of a stressor. The association of cryAB and PcryAB with the mitochondria after exposure to oxidative stress suggested potential mitochondrial involvement in cryAB KD-induced apoptosis. Furthermore, previously, cryAB R120G transgenic mice were demonstrated to exhibit desmin-related cardiomyopathy where mitochondrial respiration was compromised, leading to alterations in the permeability transition pore, compromised inner membrane potential, and elevated levels of apoptosis (24). To test whether reduced levels of cryAB might affect the mitochondria by the induction of oxidative stress, the levels of ROS in cryAB KD and Scram shRNA CMs were determined at baseline, after exposure to 60 μM H2O2, and in the presence of ROS scavengers (Fig. 4A). At baseline, ROS levels in cryAB KD CMs were 1.9-fold higher than levels observed in Scram shRNA CMs (P < 0.05). With the addition of ROS scavengers, ROS levels were decreased in Scram shRNA CMs to 75 ± 2% of the levels observed in Scram shRNA cells without ROS inhibition. In cryAB KD CMs treated with ROS scavengers, ROS levels were decreased to near control levels of 110 ± 3% relative to baseline levels observed in Scram shRNA CMs (significantly different compared with untreated cryAB KD CMs, P < 0.05). These results suggest that some oxidative stress was associated with the culture process but that there was higher production of ROS by the mitochondria in cryAB KD CMs with or without scavengers. After the addition of H2O2, there was a 2.4-fold increase in ROS levels in Scram shRNA CMs (P < 0.05) and much greater 5.8-fold increase in cryAB KD CMs (P < 0.05) relative to reference levels in Scram shRNA CMs. In the presence of H2O2 and ROS scavengers, cells with Scram shRNA showed a 54 ± 7%, decrease in ROS levels compared with H2O2 treatment alone, whereas in KD CMs, ROS levels were decreased to 55 ± 4% compared with levels observed with H2O2 treatment alone but still remained elevated compared with Scram shRNA CMs (P < 0.05; Fig. 4A). These results suggest that the oxidative stress observed at baseline in Scram shRNA and KD CMs is exacerbated by the addition of exogenous H2O2.
Fig. 4.
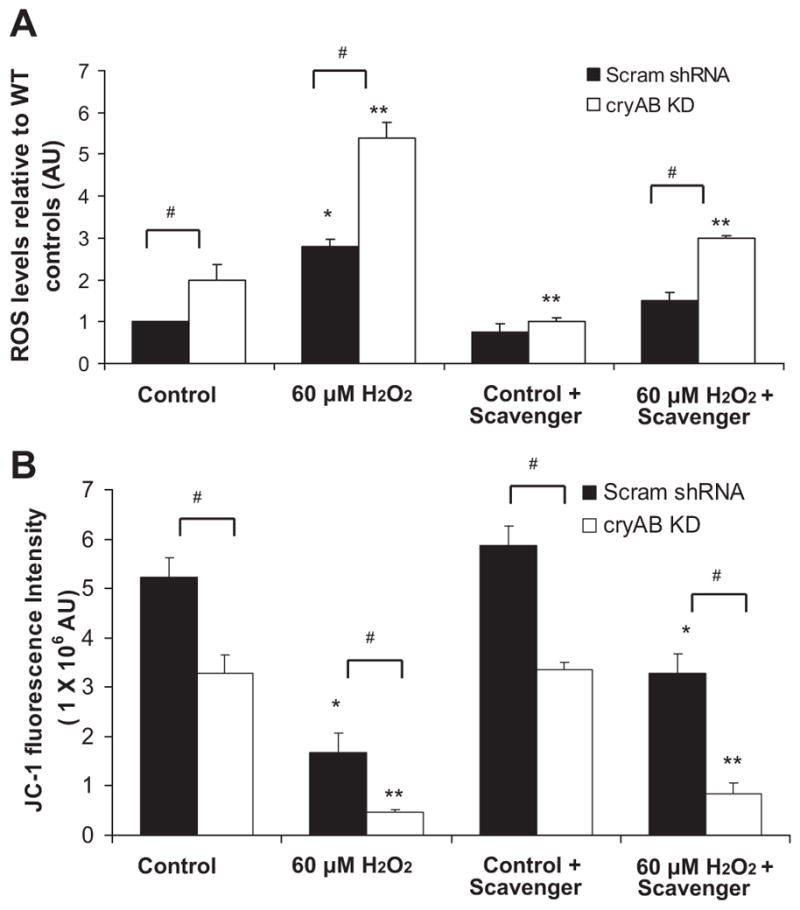
Effect of cryAB silencing on mitochondrial function. A: quantification of ROS levels in Scram shRNA and cryAB KD CMs at baseline and after exposure to H2O2 (*P < 0.05 vs. Scram shRNA controls and **P < 0.05 vs. cryAB KD controls). ROS were also assessed after treatment of cells with the ROS scavengers tiron and sodium pyruvate. #P < 0.05, differences between groups (cryAB KD vs. Scram shRNA). Values are reported as means ± SE; n = 6/treatment. B: quantification of JC-1 red fluorescence using a plate reader. JC-1 fluorescence was assessed after exposure to 60 μM H2O2 in Scram shRNA CMs (*P < 0.05) and cryAB KD CMs (**P < 0.05) compared with control conditions.
To determine the effect of reduced expression of cryAB and ROS production on mitochondrial function, we assessed the dissipation of mitochondrial membrane potential, an indicator of early apoptosis, using the mitochondrial dye JC-1. Increased dissipation of the mitochondrial membrane potential in cryAB KD CMs was observed compared with Scramb shRNA/control CMs at baseline. This effect was illustrated by the significant loss of JC-1 red fluorescence in cryAB KD CMs to 63 ± 8% of the levels that were observed in Scram shRNA/control CMs at baseline (P < 0.05). In the presence of ROS scavengers under control conditions, JC-1 fluorescence was increased by 12 ± 7% in control CMs relative to initial levels at rest (P < 0.05) and by 2 ± 0.5% in KD CMs (P < 0.05) relative to their initial levels at rest. These results suggest that both control and KD CMs were exposed to oxidative stress under cell culturing conditions but that dissipation of membrane potential only occurred in KD CMs in the absence or presence of ROS scavengers. The increased ROS observed in KD CMs may occur alongside mitochondrial structure alterations that cannot be salvaged by the removal of ROS alone, pointing to an involvement of cryAB at the mitochondrial level. After exposure to 60 μM H2O2, JC-1 fluorescence decreased to 32 ± 2% in Scram shRNA CMs (P < 0.05) and to only 9 ± 1% (P < 0.05) in cryAB KD CMs (Fig. 4B). In the presence of H2O2 together with ROS scavengers, JC-1 fluorescence was 63 ± 3% of its original value in control CMs. In KD CMs, JC-1 fluorescence was 26 ± 3% of its original value (P < 0.05). Together, these results suggest that the increased ROS levels in cryAB KD CMs contributed to the loss of mitochondrial membrane potential, which cannot be overcome with free radical scavengers (Fig. 4B).
Since we observed mitochondrial dysfunction in cryAB KD CMs, we investigated whether mitochondrial ultrastructure was disturbed by cryAB silencing. Electron microscopy images showed that in CMs transduced with Scram shRNA, the mitochondria were intact, with dense, well-ordered cristae (Fig. 5, A and B). However, in cryAB KD CMs, approximately half of the observed mitochondria exhibited altered morphology and loss of density and organization of cristae, suggestive of mitochondrial swelling and rupture of the inner membrane (Fig. 5, C and D). These results are consistent with our earlier results of mitochondrial dysfunction and increased ROS production (Fig. 4, A and B).
Fig. 5.
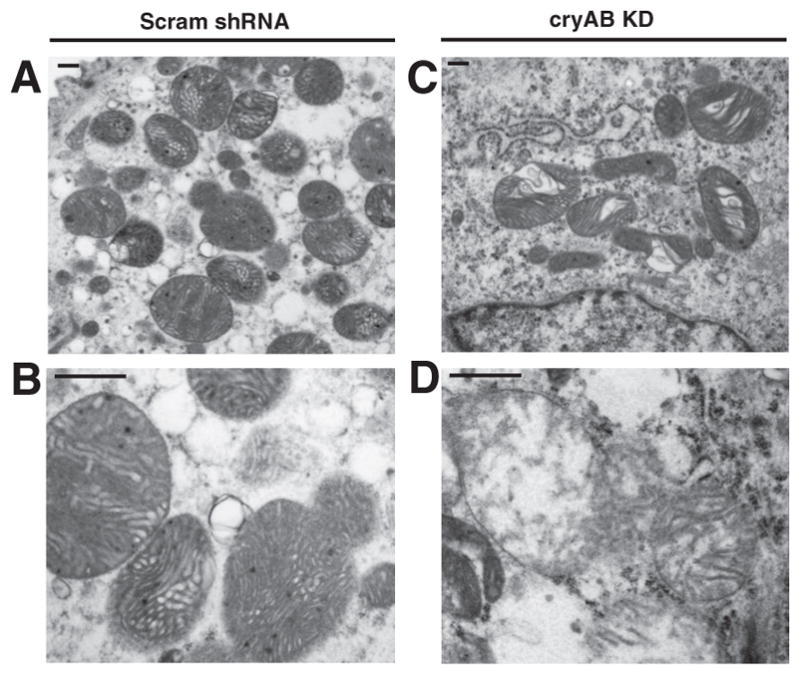
Effect of cryAB silencing on mitochondrial ultrastructure in mouse neonatal CMs. A and B: transmission electron microscopy images of Scram shRNA (A) and cryAB KD (B) CMs examining mitochondria. C and D are higher magnifications of mitochondria in CMs transduced with Scram shRNA or cryAB-targeting shRNAs, respectively. Scale bars = 500 nm.
Identification of cryAB interactions with proteins involved in apoptosis after exposure to H2O2
Our results suggest an involvement of cryAB in preventing ROS-induced apoptosis at the level of the mitochondria. Furthermore, in response to oxidative stress, cryAB and PcryAB have been observed to associate with the mitochondria (19); thus, an interaction with mitochondrial proteins (such as VDAC and TOM 20) may stabilize the mitochondria and, ultimately, prevent mitochondria-induced apoptosis. We hypothesized that cryAB also potentially regulates the apoptotic cascade downstream of the mitochondria by interacting with cytochrome c, caspase 9, caspase 3, and caspase 12. To mimic and exacerbate the oxidative stress observed after cryAB KD, we exposed adult WT CMs to 100 μM H2O2 (40). Colocalization of cryAB with these proteins was shown by qualitative fluorescence images of adult CMs under control conditions and after exposure to 100 μM H2O2 imaged for cryAB, in the green channel at 488 nm, and TOM 20, VDAC, cytochrome c, or caspase 3, imaged in the red channel at 633 nm (Fig. 6, A and B). The right images in Fig. 6, A and B, show the overlap of cryAB with TOM 20, VDAC, cytochrome c, and caspase 3, suggestive of a potential interaction of cryAB with these proteins. Colocalization statistics and Pearson and Manders’ coefficients (with M1 being indicative of the fraction of cryAB green staining overlap with red staining of TOM 20, VDAC, cytochrome c or caspase 3 and M2 being indicative of the fraction of TOM 20, VDAC, cytochrome c, or caspase 3 red staining overlap with cryAB green staining) were determined for the two-dimensional images using ImageJ JACoP (Table 1). This analysis indicated a significant degree of colocalization between cryAB and TOM 20, between cryAB andVDAC, between cryAB and cytochrome c, and between cryAB and caspase 3. Colocalization was significantly increased after treatment with 100 μM H2O2 for cryAB and TOM 20, cryAB and VDAC, cryAB and cytochrome c, and cryAB and caspase 3. In addition, we performed a comprehensive 3-D analysis of ~100-μm-thick z-stacks to assess colocalization of cryAB with TOM 20, VDAC, cytochrome c, and caspase 3 under control conditions (Fig. 6C) and after 100 μM H2O2 (Fig. 6D). This analysis indicated a significant degree of colocalization between cryAB and TOM 20, cryAB and VDAC, cryAB and cytochrome c, and cryAB and caspase 3. The extent of colocalization was enhanced after treatment with 100 μM H2O2 for all proteins (Fig. 6D).
Fig. 6.

CryAB colocalization with proteins in the apoptotic cascade. A and B: two-dimensional immunofluorescence of adult mouse CMs under control conditions (A) and CMs exposed to 100 μM H2O2 (B) using antibodies against cryAB (left) as well as translocase to outer mitochondrial membrane 20 kDa (TOM 20), VDAC, cytochrome c (cyto C), and caspase 3 (middle). The right images are indicative of colocalization. Scale bars = 4 μm. C and D: three-dimensional analysis of colocalization of cryAB with TOM 20, VDAC, cyto C, and caspase 3 in adult mouse CMs under control conditions (C) and after exposure to 100 μM H2O2 (D). Scale bar = 5 μm. In all images, cryAB is shown in green and the numbers indicate the amount of colocalization as calculated using Imaris.
Table 1.
Two-dimensional immunofluorescence colocalization analysis of adult CMs
| Pearson Coefficient | Manders’ Coefficient
|
||
|---|---|---|---|
| M1 | M2 | ||
| Control | |||
| cryAB + TOM 20 | 0.46 ± 0.07 | 0.54 ± 0.07 | 0.69 ± 0.09 |
| cryAB + VDAC | 0.66 ± 0.03 | 0.77 ± 0.03 | 0.65 ± 0.03 |
| cryAB + cyto C | 0.42 ± 0.12 | 0.56 ± 0.08 | 0.72 ± 0.09 |
| cryAB + caspase 3 | 0.55 ± 0.06 | 0.65 ± 0.09 | 0.59 ± 0.08 |
| After 100 μM H2O2 | |||
| cryAB + TOM 20 | 0.70 ± 0.06* | 0.74 ± 0.04* | 0.93 ± 0.04* |
| cryAB + VDAC | 0.79 ± 0.03* | 0.89 ± 0.02* | 0.72 ± 0.03* |
| cryAB + cyto C | 0.87 ± 0.04* | 0.80 ± 0.05* | 0.80 ± 0.05 |
| cryAB + caspase 3 | 0.78 ± 0.05* | 0.85 ± 0.05* | 0.77 ± 0.06* |
Values are shown as means ± SE; n = 5. Colocalization coefficients of two-dimensional immunofluorescence of adult mouse cardiomyocytes (CMs) were determined under control conditions and after exposure to 100 μM H2O2 using antibodies against α-crystallin B (cryAB), translocase to outer mitochondrial membrane 20 kDa (TOM 20), voltage-dependent anion channel (VDAC), cytochrome c (cyto C), and caspase 3. Pearson coefficient determined the colocalization of cryAB staining (in green) with TOM 20, VDAC, cyto C, and caspase 3 staining (in red). Manders’ overlap coefficients are based on Pearson’s correlation coefficient, with M1 indicative of the proportion of the green signal coincident with the red signal, or cryAB staining (in green) colocalized with TOM 20, VDAC, cyto C, and caspase 3 staining (in red) and with M2 indicative of the red signal coincident with green signal, or TOM 20, VDAC, cyto C, and caspase 3 staining (in red) overlapped with cryAB staining (in green). Numbers were calculated using ImageJ JACoP (4).
Significantly different vs. control (P < 0.05).
After exposure to 100 μM H2O2, immunoblot analysis of cellular lysates showed significant increases in the levels of cryAB (82.7 ± 2.9 AU in H2O2 vs. 24.2 ± 7.7 AU in controls, P < 0.05) and PcryAB (17.9 ± 1.1 AU in H2O2 vs. 5.1 ± 1.5 AU in controls, P < 0.05; Fig. 7A). Levels of cleaved caspase 3 (9.7 ± 0.8 AU in H2O2 vs. 2.6 ± 0.3 AU in controls, P < 0.05) and cleaved caspase 12 (56.4 ± 4.3 AU in H2O2 vs. 13.3 ± 0.9 AU in controls, P < 0.05) showed significantly increased expression levels. However, expression levels of TOM 20 (39.7 ± 6.7 AU in H2O2 vs. 36.9 ± 5.0 AU in controls) and VDAC (36.2 ± 0.5 AU in H2O2 vs. 35.0 ± 4.6 AU in controls) were not significantly different from controls.
Fig. 7.

CryAB protein interactions with proteins in the apoptotic cascade. A: immunoblots showing cryAB, PcryAB, TOM 20, VDAC, and caspase 3, 9, and 12 levels along with actin standards in adult mouse CMs under control conditions and following exposure to 100 μM H2O2. B: immunoblots showing the interactions of cryAB and PcryAB with TOM 20, VDAC, caspase 3, caspase 9, and caspase 12. CryAB and PcryAB were immunoprecipitated (IP) from adult cardiac tissue (100 μM H2O2 for 1 h) using protein A/G-agarose beads, and the eluate was run on an SDS-PAGE gel, probing for interactors. Gels were also probed for cryAB and PcryAB as controls. IgG beads alone were used as negative controls, as shown in the -ve control lane. Protein (20 μg) was loaded, and equal loading of cardiac lysate is shown as a positive control. C: immunoblots showing uncleaved and cleaved caspase 12 and actin levels in CMs transduced with either Scram shRNA or cryAB KD under control conditions and after exposure to 60 μM H2O2. D: densitometry quantification of levels of cleaved (active) caspase 12 in Scram shRNA and cryAB KD CMs at baseline and after exposure to H2O2 (*P < 0.05 vs. Scram shRNA controls and **P < 0.05 vs. cryAB KD controls). #P < 0.05, differences between groups (cryAB KD vs. Scram shRNA).
Next, we immunoprecipitated cryAB and PcryAB under control conditions from adult mouse cardiac tissue. CryAB and PcryAB both formed complexes with TOM 20, VDAC, un-cleaved and cleaved caspase 3, caspase 9, and caspase 12 (Fig. 7B). We performed similar experiments using cardiac tissue exposed to 100 μM H2O2 and observed that cryAB and PcryAB appeared to precipitate greater amounts of TOM 20, VDAC, uncleaved and cleaved caspase 3, and caspase 12 in the eluate from CMs exposed to 100 μM H2O2 compared with control conditions (results not shown), suggesting a potential increase in the level of interaction after exposure to oxidative stress.
We made numerous attempts to determine interactions of either cryAB or PcryAB with Bcl-XS and Bax (26) but were unable to detect signals under these conditions. Neither cryAB nor PcryAB were found to precipitate with cytochrome c, suggesting that their colocalization (Fig. 6) was not indicative of an interaction between these proteins.
Since cryAB and PcryAB appeared to precipitate caspase 12 under control conditions (Fig. 7B), we hypothesized that this potential interaction led to decreased caspase 12 activation. Cleaved caspase 12 levels (30) were assessed in control CMs and KD CMs in the absence or presence of H2O2. Immunoblot analysis and quantification showed a significant sixfold increase in levels of cleaved caspase 12 in cryAB KD CMs compared with Scram shRNA control CMs at baseline (P < 0.05; Fig. 7, C and D). After H2O2, cleaved caspase 12 showed a 2.1-fold increase in Scram shRNA CMs and an ~10-fold increase in cryAB KD CMs, suggestive of an exacerbation of caspase 12 activation in cryAB KD CMs after exposure to H2O2.
DISCUSSION
This study provides evidence of a cytosolic to mitochondrial translocation of cryAB and PcryAB in adult mouse CMs in response to H2O2-induced oxidative stress. Upregulation of total cryAB levels after H2O2 exposure and the significant reduction in viability in mouse neonatal cryAB KD CMs suggest that cryAB is protective against apoptosis. Immuno-precipitation assays indicated that cryAB intervenes at multiple points in the intrinsic apoptotic cascade by interacting with TOM 20, VDAC, caspase 3, and caspase 12 [activated by ER stress (30)] and that these interactions may be part of the protective mechanism of cryAB in CMs.
One of the conclusions of this study is that the protective mechanism of cryAB in ROS-induced cell death may involve its stabilization of the mitochondria. Our results extend earlier findings regarding cryAB localization (28). PcryAB was associated with the mitochondria even under control conditions (Fig. 1), and this association increased significantly under conditions of oxidative stress (Fig. 3). It is possible that through this association, PcryAB binds and stabilizes or modulates the activity of MPTP proteins, preventing pore opening and ensuing apoptosis. Furthermore, our results indicate that the interaction of cryAB or PcryAB with VDAC may be important for the protective mechanism, as it was significantly increased under stress conditions (Fig. 7 and results not shown). VDAC is a protein located in the outer mitochondrial membrane and mediates the transport of anions, cations, ATP, Ca2+, and many metabolites between the mitochondria and the cytosol (36). VDAC has also been recognized as having a key role in mitochondria-mediated apoptosis by interacting with both anti- and proapoptotic proteins to regulate cytochrome c release and thus mitochondria-mediated apoptosis (36). By interacting with VDAC, cryAB and/or PcryAB may stabilize the mitochondrial membrane, thus blocking the first step in the mitochondrial pathway of apoptosis. In agreement with this, a previous study (1) has shown that VDAC directly interacts with Bcl-xL in HEK-293 cells and that this interaction results in decreased channel conductivity and decreased staurosporine-induced apoptosis. The interaction of cryAB and PcryAB with TOM 20 can also act as a stabilizing force. TOM 20 is a member of the outer mitochondrial membrane protein translocase involved in transporting preproteins into the mitochondria (13). A previous study (6) has shown that TOM 20 is prone to unfolding in response to I/R in the myocardium. By maintaining the structure of TOM 20, HSPs (including Hsp70) have been suggested to contribute to protection of the myocardium in ischemic preconditioning (6). Furthermore, the unfolding of proteins when transported through TOM 20 may act as a stimulus to attract cryAB and PcryAB to the mitochondria. We have shown by electron microscopy that PcryAB was present in the mitochondria; thus, an interaction with proteins found in the outer and inner mitochondrial membrane would be feasible. Furthermore, a previous study (8) has shown that cytosolic HSPs can localize to the interior of the mitochondria, for instance, Hsp25 binds to and protects mitochondria cytochrome complex I from oxidative stress in PC12 cells. Therefore, the interaction of cryAB with proteins in the inner mitochondrial membrane may be a central mechanism by which cryAB prevents apoptosis. Interestingly, cryAB has been shown to interact with the proapoptotic proteins Bax and Bcl-XS, preventing their translocation from the cytosol into mitochondria, thus maintaining mitochondrial integrity and leading to decreased apoptosis (26).
A second conclusion that can be drawn from the present study is that while cryAB KD does not seem to have a significant effect on CM viability after H2O2, it does, however, interact with oxidative stress-mediated apoptosis. This effect can be explained by the fact that viability measures are not sensitive enough to detect interactions between cryAB KD and H2O2 exposure, as they take into account various types of cell death (autophagy, necrosis, and apoptosis), not apoptosis alone. Our results suggest that cryAB is protective against mitochondrial oxidative stress-induced apoptosis in CMs at baseline and after extrinsically exacerbated oxidative stress. Much like other HSPs that confer a protective effect against a wide range of cellular stresses, cryAB is induced by cellular stresses (9), such as oxidative stress, and is involved in protection against such stress. We found that at baseline, cryAB KD CMs were already subject to oxidative stress by increased ROS production, which decreased their viability. Previously, it has been shown that in heat shock factor 1 knockout mice, which have reduced expression of Hsp70, Hsp25, and cryAB, there is increased mitochondrial oxidative damage (44), and, here, we show that silencing of cryAB alone induced increased oxidative damage at the mitochondrial level as well as loss of mitochondrial membrane potential. The mitochondria are a major source of ROS and oxidative stress, and they trigger ROS-induced activation of the intrinsic apoptotic pathway by releasing cytochrome c (12). Hence, KD of cryAB could contribute to increased mitochondria-induced apoptosis by ROS-induced ROS production (45). Increased ROS production as a result of alterations to the mitochondrial membrane potential and electron transport chain would trigger a feedforward, subsequent increased level of ROS production, leading to apoptosis. The electron microscopy images in the present study support this speculation, as they showed significantly altered mitochondrial ultrastructure in cryAB-silenced cardiomyocytes, suggestive of mitochondrial permeabilization and rupture (Fig. 5), which would lead to apoptosis induction. Furthermore, given the ability of anti-oxidants to protect against apoptosis in I/R injury by blocking the increased expression of p53, Bax, and caspase 3 and by inhibiting caspase 3 activation (31), it may be that the oxidative stress imposed by cryAB KD functions by the opposite mechanism, increasing the expression of proapoptotic proteins and stimulating apoptosis.
Downstream of the interactions with mitochondrial proteins, cryAB may also protect from apoptosis induced by oxidative stress by interacting with caspase 3 and caspase 12, further suggesting an involvement of cryAB in protection against not only the intrinsic pathway but also ER stress-induced apoptosis (41). In the present study and those of others (20, 21), cytosolic cryAB appears to interact with uncleaved caspase 3 to prevent its cleavage, and therefore its activation, thus preventing progression along the apoptotic cascade. In the absence of cryAB, caspase 3 activity increased, and it resulted in the higher levels of apoptosis observed in cryAB KD CMs. The apparent increased interactions of cryAB with cleaved caspase 3 observed in the present study have not been reported previously, to our knowledge. This may be indicative of the potential ability of cryAB to bind and inhibit active caspase 3, even after its cleavage, thus potentially inhibiting its proteolytic activity and cleavage of substrates in the cell and providing an additional point for intervention in preventing apoptosis in CMs. CryAB has been shown to bind caspase 12. Caspase 12 is activated by ER stress (30), suggesting that cytosolic cryAB is also potentially involved in preventing ER stress-induced apoptosis. ER stress also contributes to apoptosis, by leading to the release of Ca2+ from the ER, causing depletion of ER Ca2+ and activating Ca2+ -dependent endonucleases involved in DNA fragmentation (14). Furthermore, Ca2+ is also believed to modulate cytochrome c release directly by regulating the MPTP, leading to apoptosis (15). As a molecular chaperone, cryAB would be induced in response to ER stress, suggesting that in cryAB-silenced cells, ER stress would lead to rampant apoptosis. This comes in agreement with our previous findings showing an upregulation of cryAB in response to ER stress as well as decreased viability of cMs in the absence of cryAB under conditions of augmented ER stress (5). This study did not differentiate between apoptosis by the intrinsic pathway and apoptosis induced by ER stress, but determining the contribution of ER stress to apoptosis in the absence of cryAB would be of interest.
Regulating the levels of apoptosis is an attractive target for ameliorating any ischemic injury in the heart. Our results contribute to delineating the involvement of cryAB in the intrinsic and ER stress pathway of apoptosis activated by ROS-induced oxidative damage. ROS are known to play a major role in the pathogenesis of myocardial dysfunction in a variety of conditions, including I/R injury (16), with H2O2 playing a significant role in oxidative stress injury (39). Therefore, determining where in the apoptotic cascade cryAB intervenes to prevent H2O2 injury can have therapeutic implications for cardiac disease. For instance, it has been shown that administration of exogenous cryAB significantly improves murine cardiac function after I/R injury and that it decreases caspase 3 activity and apoptosis in hypoxic human endothelial cells but not in mouse atrial CMs (40). Consistent with the work by Velotta et al. (40), who investigated cryAB in human endothelial cells, our study in ventricular CMs showed that cryAB intervenes and modulates apoptosis by interacting with VDAC, TOM 20, caspase 3, and caspase 12 in ventricular CMs. This suggests that therapeutic administration of cryAB could target and inhibit the intrinsic and ER stress pathways of apoptosis in ventricular CMs, thus potentially contributing to improved cardiac function. Further studies focusing on the extrinsic pathway of apoptosis and alternate death pathways, such as autophagy and necrosis, would therefore prove quite insightful.
Acknowledgments
The authors thank Dr. Andrea Jurisicova and Dr. Rudiger von Harsorf for valuable discussions and insight.
GRANTS
This work was funded by an Ontario Graduate Scholarship (to R. Chis), Heart and Stroke Foundation of Ontario Grant T-6281 (to A. O. Gramolini), an Early Research Award from the Ontario Ministry of Research and Innovation (to A. O. Gramolini), an unrestricted grant from Boehringer Ingelheim Canada (to A. O. Gramolini), and a Heart and Stroke/Lewar Centre Fellowship (to N. Bousette). A. O. Gramolini was a New Investigator of Heart and Stroke Foundation Canada and holds a Canada Research Chair in Cardiovascular Proteomics and Molecular Therapeutics.
Footnotes
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: R.C., P.S., N.B., T.M., A.W., and A.O.G. conception and design of research; R.C., P.S., N.B., T.M., and A.W. performed experiments; R.C., P.S., N.B., T.M., and A.O.G. analyzed data; R.C., P.S., N.B., T.M., A.W., P.H.B., and A.O.G. interpreted results of experiments; R.C., P.S., N.B., and A.O.G. prepared figures; R.C., P.S., and A.O.G. drafted manuscript; R.C., P.S., N.B., T.M., A.W., P.H.B., and A.O.G. edited and revised manuscript; R.C., P.S., N.B., T.M., A.W., P.H.B., and A.O.G. approved final version of manuscript.
References
- 1.Arbel N, Ben-Hail D, Shoshan-Barmatz V. Mediation of the anti-apoptotic activity of BCL-xL upon interaction with VDAC1. J Biol Chem. 2012;31:227–285. doi: 10.1074/jbc.M112.345918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aschkenasy G, Bromberg Z, Raj N, Deutschman CS, Weiss YG. Enhanced Hsp70 expression protects against acute lung injury by modulating apoptotic pathways. PLos One. 2011;6:e26956. doi: 10.1371/journal.pone.0026956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bennardini F, Wrzosek A, Chiesi M. Alpha B-crystallin in cardiac tissue. Association with actin and desmin filaments. Circ Res. 1992;71:288–294. doi: 10.1161/01.res.71.2.288. [DOI] [PubMed] [Google Scholar]
- 4.Bolte S, Cordelieres FP. A guided tour into subcellular colocalization analysis in light microscopy. J Microsc. 2006;224:213–232. doi: 10.1111/j.1365-2818.2006.01706.x. [DOI] [PubMed] [Google Scholar]
- 5.Bousette N, Chugh S, Fong V, Isserlin R, Kim KH, Volchuk A, Backx PH, Liu P, Kislinger T, MacLennan DH, Emili A, Gramolini AO. Constitutively active calcineurin induces cardiac endoplasmic reticulum stress and protects against apoptosis that is mediated by α-crystallin-B. Proc Natl Acad Sci USA. 2010;107:18481–18486. doi: 10.1073/pnas.1013555107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bowers M, Ardehali H. TOM20 and the Heartbreakers: evidence for the role of mitochondrial transport proteins in cardioprotection. J Mol Cell Cardiol. 2006;41:406–409. doi: 10.1016/j.yjmcc.2006.06.073. [DOI] [PubMed] [Google Scholar]
- 7.Coutinho ARS, Assumpção MEO, Bordignon V. Presence of cleaved caspase 3 in swine embryos of different developmental capacities produced by parthenogenetic activation. Mol Reproduct Dev. 2011;78:673–683. doi: 10.1002/mrd.21368. [DOI] [PubMed] [Google Scholar]
- 8.Downs CA, Jones LR, Heckathorn SA. Evidence for a novel set of small heat-shock proteins that associates with the mitochondria of murine PC12 cells and protects NADH:ubiquinone oxidoreductase from heat and oxidative stress. Arch Biochem Biophys. 1999;365:344–350. doi: 10.1006/abbi.1999.1177. [DOI] [PubMed] [Google Scholar]
- 9.Gramolini AO, Kislinger T, Alikhani-Koopaei R, Fong V, Thompson NJ, Isserlin R, Sharma P, Oudit GY, Trivieri MG, Fagan A, Kannan A, Higgins DG, Huedig H, Hess G, Arab S, Seidman JG, Seidman CE, Frey B, Perry M, Backx PH, Liu PP, MacLennan DH, Emili A. Comparative proteomics profiling of a phospholamban mutant mouse model of dilated cardiomyopathy reveals progressive intracellular stress responses. Mol Cell Proteomics. 2008;7:519–533. doi: 10.1074/mcp.M700245-MCP200. [DOI] [PubMed] [Google Scholar]
- 10.Gravance CG, Garner DL, Baumber J, Ball BA. Assessment of equine sperm mitochondrial function using JC-1. Theriogenology. 2000;53:1691–1703. doi: 10.1016/s0093-691x(00)00308-3. [DOI] [PubMed] [Google Scholar]
- 11.Green DR, Reed JC. Mitochondria and apoptosis. Science. 1998;281:1309–1312. doi: 10.1126/science.281.5381.1309. [DOI] [PubMed] [Google Scholar]
- 12.Green DR, Kroemer G. The pathophysiology of mitochondrial cell death. Science. 2004;305:626–629. doi: 10.1126/science.1099320. [DOI] [PubMed] [Google Scholar]
- 13.Grey JY, Connor MK, Gordon JW, Yano M, Mori M, Hood DA. Tom20-mediated mitochondrial protein import in muscle cells during differentiation. Am J Physiol Cell Physiol. 2000;279:C1393–C1400. doi: 10.1152/ajpcell.2000.279.5.C1393. [DOI] [PubMed] [Google Scholar]
- 14.Hajnóczky G, Csordás G, Madesh M, Pacher P. Control of apoptosis by IP3 and ryanodine receptor driven calcium signals. Cell Calcium. 2000;28:349–363. doi: 10.1054/ceca.2000.0169. [DOI] [PubMed] [Google Scholar]
- 15.Halestrap AP, Clarke SJ, Javadov SA. Mitochondrial permeability transition pore opening during myocardial reperfusion-a target for cardio-protection. Cardiovasc Res. 2004;61:372–385. doi: 10.1016/S0008-6363(03)00533-9. [DOI] [PubMed] [Google Scholar]
- 16.Hess ML, Manson NH, Okabe E. Involvement of free radicals in the pathophysiology of ischemic heart disease. Can J Physiol Pharmacol. 1982;60:1382–1389. doi: 10.1139/y82-206. [DOI] [PubMed] [Google Scholar]
- 17.Hoover HE, Thuerauf DJ, Martindale JJ, Glembotski CC. αB-crystallin gene induction and phosphorylation by MKK6-activated p38. J Biol Chem. 2000;275:23825–23833. doi: 10.1074/jbc.M003864200. [DOI] [PubMed] [Google Scholar]
- 18.Ito H, Okamoto K, Nakayama H, Isobe T, Kato K. Phosphorylation of αB-crystallin in response to various types of stress. J Biol Chem. 1997;272:29934–29941. doi: 10.1074/jbc.272.47.29934. [DOI] [PubMed] [Google Scholar]
- 19.Jin JK, Whittaker R, Glassy MS, Barlow SB, Gottlieb RA, Glembotski CC. Localization of phosphorylated αB-crystallin to heart mitochondria during ischemia-reperfusion. Am J Physiol Heart Circ Physiol. 2008;294:H337–H344. doi: 10.1152/ajpheart.00881.2007. [DOI] [PubMed] [Google Scholar]
- 20.Kamradt MC, Chen F, Cryns VL. The small heat shock protein αB-crystallin negatively regulates cytochrome c- and caspase-8-dependent activation of caspase-3 by inhibiting its autoproteolytic maturation. J Biol Chem. 2001;276:16059–16063. doi: 10.1074/jbc.C100107200. [DOI] [PubMed] [Google Scholar]
- 21.Kamradt MC, Chen F, Sam S, Cryns VL. The small heat shock protein αB-crystallin negatively regulates apoptosis during myogenic differentiation by inhibiting caspase-3 activation. J Biol Chem. 2002;277:38731–38736. doi: 10.1074/jbc.M201770200. [DOI] [PubMed] [Google Scholar]
- 22.Kraev A, Chumakov I, Carafoli E. The organization of the human gene NCX1 encoding the sodium-calcium exchanger. Genomics. 1996;37:105–112. doi: 10.1006/geno.1996.0526. [DOI] [PubMed] [Google Scholar]
- 23.Longoni SJ, Chiesi M. Cardiac alpha-crystallin. I. Isolation and identification. Mol Cell Biochem. 1990;99:113–120. [PubMed] [Google Scholar]
- 24.Maloyan A, Sanbe A, Osinska H, Westfall M, Robinson D, Imahashi Ki Murphy E, Robbins J. Mitochondrial dysfunction and apoptosis underlie the pathogenic process in α-B-crystallin desmin-related cardiomyopathy. Circulation. 2005;112:3451–3461. doi: 10.1161/CIRCULATIONAHA.105.572552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mao YW, Xiang H, Wang J, Korsmeyer S, Reddan J, Li DW. Human bcl-2 gene attenuates the ability of rabbit lens epithelial cells against H2O2-induced apoptosis through down-regulation of the αB-crystallin gene. J Biol Chem. 2001;276:43435–43445. doi: 10.1074/jbc.M102195200. [DOI] [PubMed] [Google Scholar]
- 26.Mao YW, Liu JP, Xiang H, Li DW. Human αA- and αB-crystallins bind to Bax and Bcl-Xs to sequester their translocation during staurosporine- induced apoptosis. Cell Death Differ. 2004;11:512–526. doi: 10.1038/sj.cdd.4401384. [DOI] [PubMed] [Google Scholar]
- 27.Martindale JJ, Wall JA, Martinez-Longoria DM, Aryal P, Rockman HA, Guo Y, Bolli R, Glembotski CC. Overexpression of mitogen-activated protein kinase kinase 6 in the heart improves functional recovery from ischemia in vitro and protects against myocardial infarction in vivo. J Biol Chem. 2005;280:669–676. doi: 10.1074/jbc.M406690200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Morrison LE, Hoover HE, Thuerauf DJ, Glembotski CC. Mimicking phosphorylation of αB-crystallin on serine-59 is necessary and sufficient to provide maximal protection of cardiac myocytes from apoptosis. Circ Res. 2003;92:203–211. doi: 10.1161/01.res.0000052989.83995.a5. [DOI] [PubMed] [Google Scholar]
- 29.Morrison LE, Whittaker RJ, Klepper RE, Wawrousek EF, Glembotski CC. Roles for αB-crystallin and HSPB2 in protecting the myocardium from ischemia-reperfusion-induced damage in a KO mouse model. Am J Physiol Heart Circ Physiol. 2004;286:H847–H855. doi: 10.1152/ajpheart.00715.2003. [DOI] [PubMed] [Google Scholar]
- 30.Nakagawa T, Zhu H, Morishima N, Li E, Xu J, Yankner BA, Yuan J. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-β. Nature. 2000;403:98–103. doi: 10.1038/47513. [DOI] [PubMed] [Google Scholar]
- 31.Oskarsson HJ, Coppey L, Weiss RM, Li WG. Antioxidants attenuate myocyte apoptosis in the remote non-infarcted myocardium following large myocardial infarction. Cardiovasc Res. 2000;45:679–687. doi: 10.1016/s0008-6363(99)00400-9. [DOI] [PubMed] [Google Scholar]
- 32.Ray PS, Martin JL, Swanson EA, Otani H, Dillman WH, Das DK. Transgene overexpression of αB crystallin confers simultaneous protection against cardiomyocyte apoptosis and necrosis during myocardial ischemia and reperfusion. FASEB J. 2001;15:393–402. doi: 10.1096/fj.00-0199com. [DOI] [PubMed] [Google Scholar]
- 33.Sharma P, Shathasivam T, Ignatchenko V, Kislinger T, Gramolini AO. Identification of an FHL1 protein complex containing ACTN1, ACTN4, and PDLIM1 using affinity purifications and MS-based protein-protein interaction analysis. Mol Biosys. 2011;7:1185–1196. doi: 10.1039/c0mb00235f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sharma PIV, Grace K, Ursprung C, Kislinger T, Gramolini AO. Endoplasmic reticulum protein targeting of phospholamban: a common role for an N-terminal diarginine motif in ER retention? PLos One. 2010;5:e11496. doi: 10.1371/journal.pone.0011496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shin JH, Jeong JY, Jin Y, Kim ID, Lee JK. p38β MAPK affords cytoprotection against oxidative stress-induced astrocyte apoptosis via induction of αB-crystallin and its anti-apoptotic function. Neurosci Lett. 2011;501:132–137. doi: 10.1016/j.neulet.2011.06.061. [DOI] [PubMed] [Google Scholar]
- 36.Shoshan-Barmatz V, De Pinto V, Zweckstetter M, Raviv Z, Keinan N, Arbel N. VDAC, a multi-functional mitochondrial protein regulating cell life and death. Mol Aspects Med. 2010;31:227–285. doi: 10.1016/j.mam.2010.03.002. [DOI] [PubMed] [Google Scholar]
- 37.Slezak J. Hydrogen peroxide changes in ischemic and reperfused heart. Am J Pathol. 1995;147:772–781. [PMC free article] [PubMed] [Google Scholar]
- 38.Taylor RP, Benjamin IJ. Small heat shock proteins: a new classification scheme in mammals. J Mol Cell Cardiol. 2005;38:433–444. doi: 10.1016/j.yjmcc.2004.12.014. [DOI] [PubMed] [Google Scholar]
- 39.Vandeplassche G, Hermans C, Thoné F, Borgers M. Mitochondrial hydrogen peroxide generation by NADH-oxidase activity following regional myocardial ischemia in the dog. J Mol Cell Cardiol. 1989;21:383–392. doi: 10.1016/0022-2828(89)90649-4. [DOI] [PubMed] [Google Scholar]
- 40.Velotta JB, Kimura N, Chang SH, Chung J, Itoh S, Rothbard J, Yang PC, Steinman L, Robbins RC, Fischbein MP. αB-crystallin improves murine cardiac function and attenuates apoptosis in human endothelial cells exposed to ischemia-reperfusion. Ann Thoracic Surg. 2011;91:1907–1913. doi: 10.1016/j.athoracsur.2011.02.072. [DOI] [PubMed] [Google Scholar]
- 41.von Harsdorf R, Li PF, Dietz R. Signaling pathways in reactive oxygen species-induced cardiomyocyte apoptosis. Circulation. 1999;99:2934–2941. doi: 10.1161/01.cir.99.22.2934. [DOI] [PubMed] [Google Scholar]
- 42.Wang X, Perez E, Liu R, Yan LJ, Mallet RT, Yang SH. Pyruvate protects mitochondria from oxidative stress in human neuroblastoma SK-N-SH cells. Brain Res. 2007;1132:1–9. doi: 10.1016/j.brainres.2006.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Whelan RS, Kaplinskiy V, Kitsis RN. Cell death in the pathogenesis of heart disease: mechanisms and significance. Ann Rev Physiol. 2010;72:19–44. doi: 10.1146/annurev.physiol.010908.163111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yan LJ, Christians ES, Liu L, Xiao X, Sohal RS, Benjamin IJ. Mouse heat shock transcription factor 1 deficiency alters cardiac redox homeostasis and increases mitochondrial oxidative damage. EMBO J. 2002;21:5164–5172. doi: 10.1093/emboj/cdf528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zorov DB, Filburn CR, Klotz LO, Zweier JL, Sollott SJ. Reactive oxygen species (ROS-induced) ROS release. J Exp Med. 2000;192:1001–1014. doi: 10.1084/jem.192.7.1001. [DOI] [PMC free article] [PubMed] [Google Scholar]