

Abstract
Background
Hepatic non-parenchymal cells (NPCs), encompassing hepatic stellate cells (HSCs), macrophages and endothelial cells, synthesize new hepatocyte growth factor (HGF) during liver regeneration (LR), and also play an important function in matrix production at the end of regeneration.
Aims
The aim of this study was to determine whether ablating NPCs either during hepatocyte proliferation or during matrix resynthesis will have any effect on LR.
Methods
Rats were injected with either gliotoxin (which induces NPC apoptosis) or vehicle control at various stages during partial hepatectomy (PH). NPCs and hepatocytes were also treated in vitro with gliotoxin.
Results
Proliferating cells were abundant in control livers 24 hours after PH, while in gliotoxin-treated rats, mitosis was absent, apoptotic NPCs were apparent, and HGF was decreased. In vitro studies demonstrated a >50% decrease in cell viability in NPC cultures, while hepatocyte viability and proliferation were unaffected. Chronic elimination of NPCs over a period of 5 days after PH led to increased desmin-positive HSCs and fewer alpha-smooth muscle actin-expressing HSCs. Finally, there was continued proliferation of hepatocytes and decreased collagen I and TGF-β when HSCs, the matrix-producing NPCs, were ablated during later stages of LR.
Conclusions
Ablation of NPCs at early time points after PH interferes with liver regeneration, while their ablation at late stages causes impairment in the termination of LR, demonstrating a time-dependent regulatory role of NPCs in the regenerative process.
Keywords: liver regeneration, proliferation, apoptosis, stellate cells, desmin, hepatocyte growth factor, extracellular matrix
INTRODUCTION
Adult liver has the unique capacity to regenerate after insult and loss of liver mass, and hence is a useful model to study organ regeneration and controlled growth. The most common method of inducing regeneration experimentally is surgical removal of three of five lobes from the rodent liver, commonly referred to as a 2/3 or partial hepatectomy (PH) (1). The remaining two lobes grow in size until the liver mass is completely restored (2).
Matrix synthesis and remodeling is highly relevant in liver regeneration, and involves release of matrix-bound growth factors, including hepatocyte growth factor (HGF), by urokinase and matrix metalloproteinases. HGF is indispensible for liver regeneration because it is the main mitogenic stimulus driving hepatocyte proliferation (3). Pre-existing stores of HGF are rapidly consumed within the first 3 hours (4), and replenished by non-parenchymal cells (NPCs) such as hepatic stellate cells (HSCs) and endothelial cells, which synthesize new HGF (5, 6). Coinciding with the peak of HGF consumption, levels of HGF mRNA increase dramatically in liver and lungs, peaking at 12 hours (7). Thus, the kinetics of HGF consumption and de novo synthesis are crucial in the initiation and continuation of liver regeneration.
Although many of the processes involved in initiation of liver regeneration are well-documented, the mechanisms underlying its termination are not fully elucidated. The presence of extracellular matrix (ECM) is known to maintain hepatocytes in a quiescent, differentiated state (8). Further, re-synthesis and deposition of ECM after PH is tightly regulated (9, 10). The onset of ECM production occurs after the major wave of hepatocyte DNA synthesis (11). Because they play an important function in matrix production, activated HSCs are major mediators of liver fibrosis and cirrhosis (12). Indeed, it has been postulated that because of their role in collagen deposition, HSCs are also the source of remodeled ECM during liver regeneration (2, 13).
Gliotoxin, a fungal metabolite, causes potent immunosuppression through NF-κB inhibition to induce leukocyte apoptosis (14). Recently, gliotoxin has also been shown to induce apoptosis by a mitochondrial-dependent pathway (15–20). Administration of gliotoxin to rats treated with carbon tetrachloride, thioacetamide, or after bile duct ligation significantly reduced or resolved fibrosis via HSC apoptosis (21–23). However, in addition to stellate cells, gliotoxin is also known to affect the viability of Kupffer cells and endothelial cells, and hence non-selectively targets the non-parenchymal cell (NPC) population of the liver (17, 19).
We administered gliotoxin at different times after PH to address the role and possible mechanism by which NPCs may be regulating liver regeneration. We show that while early ablation of NPCs adversely affects liver regeneration, their loss at later stages interferes with termination of hepatic regeneration and prolongs hepatocyte proliferation, due to ablation of the HSC population.
MATERIALS AND METHODS
Animals, surgery, and gliotoxin treatments
8-week old male Fischer 344 rats were anesthetized with Isoflurane (Baxter, IL) and PH was performed (24). For the early-stage and chronic treatments, animals were injected intraperitoneally (i.p.) with either dimethylsulfoxide (DMSO; vehicle control) or gliotoxin (3mg/kg, unless otherwise indicated) one day prior to, as well as immediately after PH. This dose of gliotoxin has been shown to induce NPC apoptosis in vivo (19, 20). For the late-stage treatments, animals after PH were injected with either DMSO or 3mg/kg gliotoxin at days 4, 5, and 6. Rats were harvested after PH at one day (early-stage), 5 days (chronic), or 5, 6, and 7 days (late-stage), (n ≥3/condition/per time-point). Schematics are shown in figures. All animals received humane care and studies were approved by the University of Pittsburgh’s Institutional Animal Care and Use Committee in accordance with National Institutes of Health guidelines.
Primary rat hepatocyte and NPC cultures
A single-cell suspension of hepatocytes and NPCs was obtained from 3-month old rat livers (n=3) using a modified calcium two-step collagenase perfusion technique (25). Hepatocytes were separated from non-parenchymal cells (NPCs) by low-speed centrifugation (50g, 5 minutes, 4°C). Supernatant was centrifuged at 500g for 10 minutes at 4°C to isolate NPCs. Hepatocytes were seeded at a density of 200,000 live cells/ml onto collagen-coated plates and maintained in hepatocyte growth medium containing the growth factors HGF and epidermal growth factor (EGF) (25). NPCs were resuspended in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal bovine serum (FBS) and seeded onto collagen-coated plates. Media was changed 24 hours later and cells were maintained in DMEM with 10% FBS to select for HSC attachment and proliferation. Alternatively, hepatocytes were mixed with the NPC fraction and cultured together in DMEM + 10% FBS. All plates were incubated at 37°C in 5% CO2 until harvested for proliferation and viability assays as described below.
Cell growth and viability assays
[3H]thymidine incorporation assay on hepatocytes treated with 0.15μM gliotoxin was performed as described previously to measure cell proliferation (26).
Cell viability on primary rat hepatocytes, primary rat HSCs, human microvascular endothelial cells (HMVEC; ATCC, Manassas, VA), or HSC-T6 cells (an immortalized rat liver stellate cell line (27)) treated with 1.5 μM gliotoxin for 4 hours (21) was measured by (3,4.5-dimethylthiazol-2-yl)2,5-diphenyltetrazolium bromide (MTT) assay (28).
Protein extraction and Western blotting
Whole-cell lysates were prepared for Western blot (WB) as described previously (29). Primary antibodies used were against: desmin (1:500), TGF-β (1:200), glyceraldehyde 3-phosphate dehydrogenase (GAPDH; 1:500), collagen VI (1:100), SE-1/CD32-B (1:200), and glypican-3 (1:200) (all from Santa Cruz Biotechnology, Santa Cruz, CA), α-smooth muscle actin (α-SMA; 1:500) (Sigma Aldrich, St. Louis, MO), HGF (1:500), collagen I (1:500), and collagen IV (1:500) (Abcam, Boston, MA), glial fibrillary acidic protein (GFAP; 1:250) (Dako, Carpinteria, CA), E-cadherin (1:1000) (BD Biosciences, San Jose, CA), collagen III (1:200) and ED-1/CD68 (1:100) (AbD Serotec, Raleigh, NC), Met (1:1000) and phosphorylated-Met (Tyr1234/1235; 1:1000) (Cell Signaling, Danvers, MA), and β-actin (1:5000) (Chemicon, Temecula, CA). Horseradish-peroxidase conjugated secondary antibodies (Chemicon) were used at a concentration of 1:25,000 to 1:40,000.
Immunohistochemistry
Immunohistochemistry (IHC) was performed as described elsewhere (29). The primary antibodies used were: Ki67 and cyclin D1 (1:200 and 1:50; Fisher Scientific, Pittsburgh, PA), desmin and NF-κB p65 (1:50 and 1:10; Santa Cruz), α-SMA (1:50; Dako), and collagen I (1:100; Abcam). Biotinylated secondaryantibodies (Jackson ImmunoResearch Laboratories, Inc., West Grove, PA) were used at a 1:500 dilution. Apoptosis was determined using terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining (ApopTag Peroxidase In Situ Apoptosis Detection Kit, S7100, Chemicon International, Temecula, CA).
mRNA and real-time PCR
mRNA was isolated and purified from frozen livers and cDNA generated (26). Expression levels of HGF were determined by real-time PCR using SYBR green and the following primers:
Forward: 5′ – CATTGGTAAAGGAGGCAGCTATAAA – 3′
Reverse: 5′ – GGATTTCGACAGTAGTTTTCCTGTAGG – 3′
Expression levels of collagen 1a1 were determined using primers from SuperArray Bioscience (Valencia, CA, cat. no. PPR42922A) and SYBR green (26). HGF and collagen I were normalized relative to expression of cyclophilin in each sample. Gene expression was calculated by using the 2(−ΔΔCt) method, which was derived from average Ct and expressed as fold change.
RESULTS
Gliotoxin treatment during early liver regeneration leads to apoptosis of NPCs and absence of hepatocyte proliferation
In order to study the effects of eliminating NPCs during early liver regeneration, rats were injected i.p. with either 3mg/kg gliotoxin or DMSO one day before PH, as well as simultaneous with PH, as outlined in Methods (Figure 1A). This dosage has been previously reported in the literature to induce apoptosis of activated HSCs and other NPCs in vivo (18–20). However, since the exact kinetics of gliotoxin metabolism and NPC activation after PH are unknown, we treated rats prior to as well as at the time of PH to ensure immediate efficacy of the drug. Livers from all animals were harvested for IHC, protein, and RNA when the gliotoxin-treated rats began showing signs of morbidity (one day after PH). TUNEL IHC shows significant apoptosis in gliotoxin-treated livers, which was restricted to NPCs located in the perisinusoidal spaces (Figure 1C). Hematoxylin and eosin (H&E) and Ki67 IHC show the presence of many mitotic figures and proliferating cells in the control 24 hours after PH (Figure 1B), while in the gliotoxin-treated animals, mitosis was almost completely absent (Figure 1C).
Figure 1. Gliotoxin treatment during early liver regeneration induces apoptosis of HSCs and inhibits proliferation.

(A) Schematic of the early-stage gliotoxin study, in which rats were treated with DMSO or 3mg/kg gliotoxin, followed by a second dose 24 hours later at the time of PH. Livers were harvested 24 hours following treatment. (B) H&E staining in control and gliotoxin-treated livers (arrows point to mitotic figures; 200X). (C) Ki67 and TUNEL staining in control and gliotoxin-treated groups.
To determine the molecular mechanisms by which gliotoxin blocks proliferation, we examined cyclin D1 expression, which controls G1/S transition in hepatocytes, by IHC. Figure 2A shows that while around 90% of hepatocytes express nuclear cyclin D1 18 hours after PH, fewer than 10% of hepatocytes in gliotoxin-treated livers are positive. WB confirms that glial fibrillary acidic protein (GFAP), a common marker of quiescent stellate cells which increases during the acute response to liver injury in rats (30), decreases 24 hours after PH in the gliotoxin-treated livers as compared to DMSO, verifying depletion of HSCs (Figure 2B). A modest decrease in macrophage/Kupffer cell (KC) population (represented by ED-1 Western blotting) and a greater decrease in SE-1 expressing endothelial cells, which are the cells known to express HGF during liver regeneration, was evident in the gliotoxin-treated animals (Figure 2B).
Figure 2. Several signaling pathways essential to early liver regeneration are inhibited after gliotoxin treatment.

(A) Cyclin D1 IHC in control and gliotoxin-treated livers (200X). (B) Expression of HGF mRNA in control and gliotoxin-treated livers 1 day after PH, as measured by real-time PCR. (C) WB analysis utilizing whole cell lysates from T0 and PH control and gliotoxin-treated livers. (D) IHC for the NF-κB subunit p65 in control and gliotoxin-treated livers (200X).
To determine the effect of loss of NPC populations on liver regeneration, we next examined several signaling pathways implicated in the initiation of regeneration. WB showed a notable decrease in HGF protein expression after gliotoxin treatment (Figure 2B), which was further corroborated by a 50% reduction in HGF mRNA expression (Figure 2C). Signaling through the HGF receptor Met was also inhibited after gliotoxin, as shown by decreases in total and tyrosine-phosphorylated Met (Figure 2B). Finally, IHC shows that NF-κB, which is a target of both TNF-and HGF signaling (31, 32), is active in the control livers, with diffuse and abundant cytoplasmic and nuclear staining evident; this activation, however, is absent in gliotoxin-treated livers (Figure 2D). Thus, administration of gliotoxin before and shortly after PH increases NPC apoptosis that blocks cell cycle progression and proliferation of hepatocytes. This observation reaffirms the role of HSCs, ECs, and KCs in driving liver regeneration by producing HGF and additional cytokines.
Gliotoxin treatment in vitro negatively affects viability and survival of NPCs but not hepatocytes
To verify that gliotoxin primarily affects NPCs and not hepatocytes, primary rat hepatocytes, primary rat HSCs, mixed cultures of rat hepatocytes and NPCs, human microvascular endothelial cells, or HSC-T6 cells were treated for 4 hours with 1.5 μM gliotoxin and harvested for assessment of cell viability by MTT assay. As shown in Figure 3A, gliotoxin treatment does not affect hepatocyte viability; however, it causes a >50% decrease in viability of all NPC types. TUNEL staining demonstrated massive apoptosis in primary rat HSCs, which was absent in hepatocytes (Figure 3C). Viability and apoptosis of mixed cultures containing both primary hepatocytes and NPCs is intermediate to that of cultures containing either cell type alone. Furthermore, proliferation was unaffected in hepatocytes exposed to gliotoxin for 4 days in culture, as measured by thymidine incorporation assay (Figure 3B). To demonstrate integrity of cell cycling, this assay was performed both with and without growth factor stimulation (HGF and EGF; data from basal media not shown). These observations substantiate that the lack of proliferation in hepatocytes after PH/gliotoxin is secondary to NPC loss.
Figure 3. Primary hepatocytes are resistant to gliotoxin toxicity in vitro.

(A) MTT assay on primary rat hepatocytes, primary rat HSCs, mixed cultures of rat hepatocytes and NPCs, human microvascular endothelial cells, and HSC-T6 cells treated with gliotoxin for 4 hours. (B) Thymidine incorporation assay on primary rat hepatocytes treated with a low dose of gliotoxin for 4 consecutive days in culture. (C) TUNEL staining on primary rat hepatocytes, HSCs, and mixed cultures (hepatocytes and NPCs) treated with gliotoxin for 4 hours.
Chronic gliotoxin treatment induces an alteration in HSC phenotype concomitant with a change in ECM composition
At days 3–5, only mild hepatocyte proliferation is ongoing and there is extensive new matrix synthesis (2, 9, 10). To address ongoing changes in matrix, we administered gliotoxin over a period of 7 days, including 5 consecutive days after PH (Figure 4A), to eliminate NPCs chronically. The dose of gliotoxin was reduced to 1.2mg/kg to adjust for decrease in liver weight after PH. Chronic gliotoxin administration increased TUNEL-positive apoptotic nuclei primarily in non-parenchymal cells (Figure 4D). Intriguingly, this treatment results in a higher expression of desmin in gliotoxin-treated rats as compared to control rats, which had more α-smooth muscle actin (α-SMA) (Figure 4C, D). Concomitantly, gliotoxin treatment led to decrease in the mature processed form of collagen I, an essential component of de novo matrix synthesis (9), as assessed by IHC, mRNA, and protein expression (Figure 4B, C, D). Simultaneous increases in collagen IV, as well as glypican-3, a hepatocyte protein residing in the extracellular matrix space (33), and E-cadherin, an epithelial marker (34)) (Figure 4C) in gliotoxin-treated livers suggested a change in ECM composition that is not stimulated by transdifferentiation of hepatocytes to a mesenchymal phenotype.
Figure 4. Chronic gliotoxin treatment induces a change in HSC phenotype and ECM composition.

(A) Schematic of the chronic gliotoxin study, in which rats were treated with DMSO or 3mg/kg gliotoxin, followed by a second dose of either DMSO or 1.2 mg/kg gliotoxin 24 hours later simultaneous with PH, and then 4 consecutive doses of either DMSO or 1.2 mg/kg gliotoxin for 5 consecutive days. Rats were harvested 5 days following PH. (B) Expression of collagen I mRNA in control and gliotoxin-treated livers 5 days after PH, as measured by real-time PCR. (C) WB analysis of whole cell lysates from control and gliotoxin-treated livers. (D) Ki67, TUNEL, desmin, α-SMA, and collagen I IHC on control and gliotoxin-treated livers (200X).
Inverse relationship of desmin and α-SMA expression over the course of PH
In order to determine whether the phenotypic alteration in HSC marker expression after gliotoxin treatment may be of biological significance, we cultured primary rat HSCs and harvested at two different time points: D2, during which HSCs are still relatively quiescent, and D10, when HSCs have become activated in culture. Figure 5A shows that activated HSCs, which are characterized by expression of α-SMA, do not express HGF, while HSCs that are not yet expressing α-SMA are capable of producing HGF. We next examined expression of HSC markers after regular PH (independent of any gliotoxin administration) to determine the kinetics of phenotypic changes in the expression of HSC markers during liver regeneration. α-SMA, which is expressed at low levels in the first few hours after PH, is strongly expressed at D2 and D5 (Figure 5B), which corresponds with peak TGF-β mRNA expression and beginning of matrix re-deposition (11). Conversely, desmin is expressed early in liver regeneration but decreases over time, except at days 3 and 7, when α-SMA is decreased (Figure 5B). Thus, desmin and α-SMA expression show an inverse relationship after PH, which suggests the HSC population is able to assume different functions during liver regeneration.
Figure 5. Expression of desmin is inversely correlated with α-SMA expression over the course of PH.
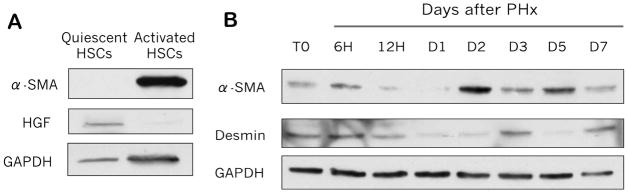
(A) WB analysis of α-SMA and HGF expression in cultured quiescent and activated HSCs. (B) WB of α-SMA and desmin expression at various time points after PH in rat livers.
Treatment with gliotoxin at the time of matrix re-deposition results in increased hepatocyte proliferation
As HSCs can assume an altered phenotype in response to chronic gliotoxin treatment, and as HSCs are the only cells implicated in ECM synthesis (not EC or KC), the impact of gliotoxin-mediated HSC loss was assessed at the time of matrix deposition. To determine the impact of HSC ablation during termination of liver regeneration, we administered gliotoxin or DMSO at 4, 5, and 6 days after PH and harvested at D5, 6, and 7 (Fig. 6A). After an initial decrease, continuous administration of gliotoxin led to a statistically significant increase in the number of Ki-67-positive hepatocytes indicating continued proliferation at the later stages of liver regeneration (Figure 6B, C). Liver weight to body weight (lw/bw) ratios were decreased in gliotoxin-treated rats at days 5 and 6 after PH; however, this trend was reversed at D7, when gliotoxin-treated animals began to show a marginal (but not significant) increase in lw/bw compared to controls (Figure 6D). We next investigated the mechanisms that may be driving sustained hepatocyte proliferation after PH/gliotoxin. Table 1 shows that the expression of several matrix-related collagens and other HSC-related markers are altered in gliotoxin-treated rats at D7 after PH, as assessed by gene array. WB also demonstrated a decrease in the mature form of collagen I at D7, a decrease in TGF-β, a known mito-inhibitor which also stimulates collagen synthesis (11), and increased expression of HGF at Day 7 after PH (Figure 6E). Interestingly, we did not observe a compensatory increase in collagen IV expression, as we did with chronic gliotoxin treatment (not shown). WB also confirmed that gliotoxin induced an increase in desmin-positive HSCs, only a mild decrease in KCs, and had no effect on the EC population at D7 (data not shown). Finally, TUNEL staining showed that while control livers at D7 after PH showed occasional apoptotic hepatocytes per field, apoptosis in gliotoxin-treated livers primarily occurred in activated HSCs (Figure 6F). Thus, administration of gliotoxin during the late stages of liver regeneration causes a phenotypic change in HSC marker expression, either by selection or by induced phenotypic change. This is associated with changes in ECM composition and a resultant increase in proliferating hepatocytes, suggesting impaired termination of regeneration.
Figure 6. Treatment with gliotoxin during the later stages of liver regeneration results in increased proliferation and decreased matrix re-deposition.

(A) Schematic of the late-stage gliotoxin study, in which rats were subjected to PH followed by treatment with DMSO or 3mg/kg gliotoxin on days 4, 5, and 6 after PH. Livers were harvested 5, 6, or 7 days following PH. (B) Quantification of the Ki67 staining in (C). (C) Representative images of Ki67 IHC on livers harvested at D5, D6, and D7 after PH in gliotoxin and control groups (100X). (D) Graph of liver weight to body weight ratios after late DMSO/gliotoxin treatment. (E) WB analysis of whole cell lysates from control and gliotoxin-treated livers harvested at D7. (F) TUNEL staining on livers from the late-stage experiment (200X).
Table 1.
Changes in expression of collagens and other stellate-cell related markers in gliotoxin-treated rats at D7 after partial hepatectomy
| Gene | DMSO D7 PHx | Gliotoxin D7 PHx | Fold change |
|---|---|---|---|
|
| |||
| Smooth muscle alpha-actin | 686.5 | 441.1 | −1.56 |
| Procollagen, type I, alpha 2 | 551.3 | 359.3 | −1.53 |
| Procollagen, type I, alpha 1 | 377.5 | 247.8 | −1.52 |
| Collagen alpha1 type I | 575.2 | 425.1 | −1.35 |
| Procollagen, type I, alpha 1 | 23011.6 | 16586.5 | −1.39 |
| Procollagen type XII alpha 1 | 216.3 | 162.8 | −1.33 |
| Procollagen, type I, alpha 2 | 415.8 | 335.4 | −1.24 |
| Collagen type V, alpha 2 | 795.3 | 645.8 | −1.23 |
| Procollagen, type I, alpha 2 | 643.7 | 617.6 | −1.04 |
| Desmin | 191 | 219.2 | 1.15 |
| Collagen type X alpha 1 | 89.9 | 113.3 | 1.26 |
| Type II collagen | 334.7 | 436.6 | 1.30 |
| HGF | 365.9 | 511.2 | 1.40 |
DISCUSSION
Liver regeneration is a complex process involving cooperation between different signaling pathways and cell types. Although hepatocytes themselves are fully capable of repopulating the liver after PH, the involvement of other cell types, such as HSCs, ECs, and KCs, is also required for proper restoration of liver mass. However, very little is known about the role, function, and activation state of these non-parenchymal cells during liver regeneration. Foxf1+/− mice exhibit defective HSC activation and abnormal liver repair after carbon tetrachloride injury (35).
Conversely, mice resistant to collagen I degradation show persistent activation of HSCs and fail to regenerate properly after CCl4 injury (36). Similarly, ablation or inhibition of EC proliferation inhibited liver regeneration (37, 38). Conversely, the data on liver regeneration in KC-depleted animals is contradictory, with some reports indicating that absence of KCs enhances liver regeneration, while others demonstrated an inhibitory effect (39, 40). Although these studies highlight the importance of a tightly regulated and properly functioning NPC compartment, a more direct method is needed to address the role of the entire NPC population during liver regeneration. To our knowledge, ours is the first study to elucidate the importance of NPCs in both the initiation and termination of liver regeneration through elimination of these cell types.
Treatment with gliotoxin at the time of peak hepatocyte DNA synthesis (41) led to apparent apoptosis in NPCs and a dramatic suppression of hepatocyte proliferation. Concomitantly, cyclin D1, which is expressed as early as 6 hours after PH (42), is decreased, suggesting a block in upstream signaling, most likely due to decreased HGF production. Consistent with previous findings showing Met induction by HGF (43, 44), we found decreased total Met protein in the absence of HGF. Additionally, NF-κB, which controls transcription of cyclin-D1 and is activated very early during liver regeneration by HGF as well as by macrophage-derived TNF-α, is induced in controls at 3H after PH but not after gliotoxin treatment (45, 46). Therefore, a previous study implicating lack of NF-κB activation and DNA synthesis after pre-treatment with gliotoxin before PH (47) could possibly be attributed to absence of HGF and Met derived mitogenic signals necessary for proliferation. Previous studies have shown that HGF/Met signaling elimination cannot be compensated by alternative signaling from EGFR (48).
Likewise, we cannot rule out the possibility that the absence of other macrophage-derived cytokines important for liver regeneration, such as Il-6 and IL-1β, contribute to the phenotype seen after gliotoxin treatment. Thus, we believe that NPC loss blocks hepatocyte proliferation, either indirectly through loss of growth factors and/or cytokines, such as HGF, that would normally be provided by NPCs during early liver regeneration, or through loss of physical interactions between hepatocytes and NPCs. Indeed, HSC depletion with gliotoxin led to a dramatic decrease in hepatocyte proliferation and oval cell activation during the regenerative phase after acetaminophen-induced injury (49).
Data on gliotoxin toxicity in hepatocytes is somewhat controversial, with some groups claiming it causes apoptosis (17, 47), and others showing hepatocytes being overall resistant and prone to damage only at high concentrations (15, 19, 21, 49). In our studies, we show that in vitro, hepatocytes are resistant to gliotoxin toxicity at concentration that causes NPC apoptosis and decreased viability. Direct cytotoxic effects of gliotoxin on hepatocytes in vivo are also unlikely since histology shows highly selective NPC apoptosis. Thus, from our short-term study we conclude that the ablation of NPCs by gliotoxin leads to absence of proliferative signals necessary for liver regeneration.
The presence of increased numbers of desmin-positive cells after PH and chronic gliotoxin treatment is intriguing. We found that the net increase in desmin expression corresponded with decreased α-SMA expression. It is possible that loss of α-SMA reflects the expected depletion of activated HSCs, while the increase in desmin could be explained by replenishment of quiescent HSCs. Thus, newly appearing HSC may proceed into a desmin-expressing inactive phenotype prior to expressing α-SMA and undergoing activation. An alternative explanation is that HSCs are capable of switching their phenotype, which may thus be a mechanism by which HSCs evade gliotoxin-induced apoptosis. In the case of chronic treatment, then, it is unclear whether gliotoxin truly ablates activated HSCs, or whether it merely inhibits HSC activation. Also unknown is whether the function of desmin-positive cells differs from that of α-SMA-positive cells after PH. In light of the results in Figure 5, it is tempting to speculate that different sub-populations of HSCs may be distinguishable during liver regeneration, with desmin indicating quiescent HSC producing HGF, and α-SMA identifying activated HSC not involved in HGF production. Further investigation will be necessary to determine the significance of these findings.
The long-term effect of HSC depletion on matrix synthesis after PH has not been reported, although depletion of activated HSCs with gliotoxin has shown to decrease engraftment of transplanted hepatocytes, perhaps through a matrix-dependent mechanism (50). Previously, our laboratory has shown that loss of Integrin Linked Kinase (ILK) involved in hepatocyte/ECM signaling causes hepatomegaly before and after PH (51, 52), suggesting that the presence of a functioning ECM-to-hepatocyte signal is critical for proper termination of liver regeneration. We observed a decrease in collagen I, an essential component of the matrix which is normally re-synthesized towards the end of the regenerative process (9), in rats treated with gliotoxin during the late stages of regeneration, simultaneous with sustained hepatocyte proliferation. It is conceivable that lack of HSCs during late liver regeneration may result in continued proliferation either through a direct mechanism, such as loss of collagen, or indirectly, through loss of the termination signal TGF-β, as HSCs are a major source of active TGF-β during liver regeneration and injury (53). Furthermore, the increase in HGF seen concomitantly could be secondary to lack of termination signals, which would allow the liver to continue providing signals to proliferate. Indeed, TGF-β has been shown to negatively regulate HGF expression (54), and others have shown that HSCs are negative regulators of hepatocyte proliferation in liver regeneration through activation of TGF-β (55).
Since fulminant hepatic failure (FHF) is a major clinical problem, animal models that mimic this condition would be useful to test novel therapeutic strategies that activate pro-proliferative pathways, which would augment liver regeneration after insult or injury. Additionally, acute inhibition of liver regeneration in rats or mice may help identify biomarkers of FHF onset, allowing more accurate prediction of successful transplantation outcomes in humans. As our data has shown, eliminating NPCs at the time of PH leads to a failure in hepatocyte regeneration and thus may provide a useful model to study FHF. Also, understanding the interactions between hepatocytes and ECM during regeneration is essential in creating therapies to limit growth after adequate regeneration (56). Our data demonstrate the role of HSCs in matrix remodeling during late states of liver regeneration, which suggests that modulation of HSC function may be a beneficial strategy to blunt excess hepatocyte growth in certain pathologies such as hepatomegaly.
Acknowledgments
Financial Support: This study was funded by NIH grants 5R01CA035373-27 and 5R01CA103958-05 to GKM and a Pathology Postdoctoral Research Training Grant (PPRTP) from the University of Pittsburgh to KNB.
We thank Dr. Satdarshan Pal Singh Monga for invaluable insight and advice on this project.
Abbreviations
- PH
Partial hepatectomy
- LR
liver regeneration
- H&E
hematoxylin and eosin
- NPC
non-parenchymal cell
- HSC
hepatic stellate cell
- HGF
hepatocyte growth factor
- α-SMA
alpha smooth muscle actin
- GFAP
glial fibrillary acidic protein
- ECM
extracellular matrix
- TGF-β
transforming growth factor β
- IHC
immunohistochemistry
- DMSO
dimethylsulfoxide
- TUNEL
terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- p-Met
tyrosine-phosphorylated Met
- i.p
intraperitoneal
- BW
body weight
- LW
liver weight
- WB
Western blot
- FHF
fulminant hepatic failure
Footnotes
Disclosure: None of the authors have any potential conflicts of interest pertaining to the current manuscript.
References
- 1.Higgins GM, Anderson RM. Experimental pathology of the liver, 1:Restoration of the liver of the white rat following partial surgical removal. Arch Pathol. 1931;12:186–202. [Google Scholar]
- 2.Michalopoulos GK. Liver regeneration. J Cell Physiol. 2007;213(2):286–300. doi: 10.1002/jcp.21172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nakamura T, Nishizawa T, Hagiya M, et al. Molecular cloning and expression of human hepatocyte growth factor. Nature. 1989;342(6248):440–3. doi: 10.1038/342440a0. [DOI] [PubMed] [Google Scholar]
- 4.Pediaditakis P, Lopez-Talavera JC, Petersen B, Monga SP, Michalopoulos GK. The processing and utilization of hepatocyte growth factor/scatter factor following partial hepatectomy in the rat. Hepatology. 2001;34(4 Pt 1):688–93. doi: 10.1053/jhep.2001.27811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schirmacher P, Geerts A, Jung W, Pietrangelo A, Rogler CE, Dienes HP. The role of Ito cells in the biosynthesis of HGF-SF in the liver. EXS. 1993;65:285–99. [PubMed] [Google Scholar]
- 6.Lecouter J, Moritz DR, Li B, et al. Angiogenesis-independent endothelial protection of liver: role of VEGFR-1. Science. 2003;299(5608):890–3. doi: 10.1126/science.1079562. [DOI] [PubMed] [Google Scholar]
- 7.Zarnegar R, Defrances MC, Kost DP, Lindroos P, Michalopoulos GK. Expression of hepatocyte growth factor mRNA in regenerating rat liver after partial hepatectomy. Biochem Biophys Res Commun. 1991;177(1):559–65. doi: 10.1016/0006-291x(91)92020-k. [DOI] [PubMed] [Google Scholar]
- 8.Rana B, Mischoulon D, Xie Y, Bucher NL, Farmer SR. Cell-extracellular matrix interactions can regulate the switch between growth and differentiation in rat hepatocytes: reciprocal expression of C/EBP alpha and immediate-early growth response transcription factors. Mol Cell Biol. 1994;14(9):5858–69. doi: 10.1128/mcb.14.9.5858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rudolph KL, Trautwein C, Kubicka S, et al. Differential regulation of extracellular matrix synthesis during liver regeneration after partial hepatectomy in rats. Hepatology. 1999;30(5):1159–66. doi: 10.1002/hep.510300502. [DOI] [PubMed] [Google Scholar]
- 10.Gallai M, Sebestyen A, Nagy P, Kovalszky I, Onody T, Thorgeirsson SS. Proteoglycan gene expression in rat liver after partial hepatectomy. Biochem Biophys Res Commun. 1996;228(3):690–4. doi: 10.1006/bbrc.1996.1718. [DOI] [PubMed] [Google Scholar]
- 11.Nakatsukasa H, Evarts RP, Hsia CC, Thorgeirsson SS. Transforming growth factor-beta 1 and type I procollagen transcripts during regeneration and early fibrosis of rat liver. Lab Invest. 1990;63(2):171–80. [PubMed] [Google Scholar]
- 12.Friedman SL. Mechanisms of hepatic fibrogenesis. Gastroenterology. 2008;134(6):1655–69. doi: 10.1053/j.gastro.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sato M, Suzuki S, Senoo H. Hepatic stellate cells: unique characteristics in cell biology and phenotype. Cell Struct Funct. 2003;28(2):105–12. doi: 10.1247/csf.28.105. [DOI] [PubMed] [Google Scholar]
- 14.Pahl HL, Krauss B, Schulze-Osthoff K, et al. The immunosuppressive fungal metabolite gliotoxin specifically inhibits transcription factor NF-kappaB. J Exp Med. 1996;183(4):1829–40. doi: 10.1084/jem.183.4.1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Orr JG, Leel V, Cameron GA, et al. Mechanism of action of the antifibrogenic compound gliotoxin in rat liver cells. Hepatology. 2004;40(1):232–42. doi: 10.1002/hep.20254. [DOI] [PubMed] [Google Scholar]
- 16.Kweon YO, Paik YH, Schnabl B, Qian T, Lemasters JJ, Brenner DA. Gliotoxin-mediated apoptosis of activated human hepatic stellate cells. J Hepatol. 2003;39(1):38–46. doi: 10.1016/s0168-8278(03)00178-8. [DOI] [PubMed] [Google Scholar]
- 17.Anselmi K, Stolz DB, Nalesnik M, Watkins SC, Kamath R, Gandhi CR. Gliotoxin causes apoptosis and necrosis of rat Kupffer cells in vitro and in vivo in the absence of oxidative stress: exacerbation by caspase and serine protease inhibition. J Hepatol. 2007;47(1):103–13. doi: 10.1016/j.jhep.2007.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Elsharkawya M, Wright MC, Hay RT, et al. Persistent activation of nuclear factor-kappaB in cultured rat hepatic stellate cells involves the induction of potentially novel Rel-like factors and prolonged changes in the expression of IkappaB family proteins. Hepatology. 1999;30(3):761–9. doi: 10.1002/hep.510300327. [DOI] [PubMed] [Google Scholar]
- 19.Hagens WI, Olinga P, Meijer DK, Groothuis GM, Beljaars L, Poelstra K. Gliotoxin non-selectively induces apoptosis in fibrotic and normal livers. Liver Int. 2006;26(2):232–9. doi: 10.1111/j.1478-3231.2005.01212.x. [DOI] [PubMed] [Google Scholar]
- 20.Elsharkawya M, Oakley F, Mann DA. The role and regulation of hepatic stellate cell apoptosis in reversal of liver fibrosis. Apoptosis. 2005;10(5):927–39. doi: 10.1007/s10495-005-1055-4. [DOI] [PubMed] [Google Scholar]
- 21.Wright MC, Issa R, Smart DE, et al. Gliotoxin stimulates the apoptosis of human and rat hepatic stellate cells and enhances the resolution of liver fibrosis in rats. Gastroenterology. 2001;121(3):685–98. doi: 10.1053/gast.2001.27188. [DOI] [PubMed] [Google Scholar]
- 22.Dekel R, Zvibel I, Brill S, Brazovsky E, Halpern Z, Oren R. Gliotoxin ameliorates development of fibrosis and cirrhosis in a thioacetamide rat model. Dig Dis Sci. 2003;48(8):1642–7. doi: 10.1023/a:1024792529601. [DOI] [PubMed] [Google Scholar]
- 23.Hagens WI, Beljaars L, Mann DA, et al. Cellular targeting of the apoptosis-inducing compound gliotoxin to fibrotic rat livers. J Pharmacol Exp Ther. 2008;324(3):902–10. doi: 10.1124/jpet.107.132290. [DOI] [PubMed] [Google Scholar]
- 24.Stolz DB, Mars WM, Petersen BE, Kim TH, Michalopoulos GK. Growth factor signal transduction immediately after two-thirds partial hepatectomy in the rat. Cancer Res. 1999;59(16):3954–60. [PubMed] [Google Scholar]
- 25.Block GD, Locker J, Bowen WC, et al. Population expansion, clonal growth, and specific differentiation patterns in primary cultures of hepatocytes induced by HGF/SF, EGF and TGF alpha in a chemically defined (HGM) medium. J Cell Biol. 1996;132(6):1133–49. doi: 10.1083/jcb.132.6.1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bhave VS, Paranjpe S, Bowen WC, et al. Genes inducing iPS phenotype play a role in hepatocyte survival and proliferation in vitro and liver regeneration in vivo. Hepatology. 2011;54(4):1360–70. doi: 10.1002/hep.24507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vogel S, Piantedosi R, Frank J, et al. An immortalized rat liver stellate cell line (HSC-T6): a new cell model for the study of retinoid metabolism in vitro. J Lipid Res. 2000;41(6):882–93. [PubMed] [Google Scholar]
- 28.Zeng G, Apte U, Cieply B, Singh S, Monga SP. siRNA-mediated beta-catenin knockdown in human hepatoma cells results in decreased growth and survival. Neoplasia. 2007;9(11):951–9. doi: 10.1593/neo.07469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nejak-Bowen K, Kikuchi A, Monga SP. Beta-catenin-NF-kappaB interactions in murine hepatocytes: A complex to die for. Hepatology. 2012 doi: 10.1002/hep.26042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Carotti S, Morini S, Corradini SG, et al. Glial fibrillary acidic protein as an early marker of hepatic stellate cell activation in chronic and posttransplant recurrent hepatitis C. Liver Transpl. 2008;14(6):806–14. doi: 10.1002/lt.21436. [DOI] [PubMed] [Google Scholar]
- 31.Taub R. Liver regeneration 4: transcriptional control of liver regeneration. Faseb J. 1996;10(4):413–27. [PubMed] [Google Scholar]
- 32.Muller M, Morotti A, Ponzetto C. Activation of NF-kappaB is essential for hepatocyte growth factor-mediated proliferation and tubulogenesis. Mol Cell Biol. 2002;22(4):1060–72. doi: 10.1128/MCB.22.4.1060-1072.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Song HH, Filmus J. The role of glypicans in mammalian development. Biochim Biophys Acta. 2002;1573(3):241–6. doi: 10.1016/s0304-4165(02)00390-2. [DOI] [PubMed] [Google Scholar]
- 34.Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119(6):1420–8. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kalinichenko VV, Bhattacharyya D, Zhou Y, et al. Foxf1 +/− mice exhibit defective stellate cell activation and abnormal liver regeneration following CCl4 injury. Hepatology. 2003;37(1):107–17. doi: 10.1053/jhep.2003.50005. [DOI] [PubMed] [Google Scholar]
- 36.Issa R, Zhou X, Trim N, et al. Mutation in collagen-1 that confers resistance to the action of collagenase results in failure of recovery from CCl4-induced liver fibrosis, persistence of activated hepatic stellate cells, and diminished hepatocyte regeneration. FASEB J. 2003;17(1):47–9. doi: 10.1096/fj.02-0494fje. [DOI] [PubMed] [Google Scholar]
- 37.Greenea K, Wiener S, Puder M, et al. Endothelial-directed hepatic regeneration after partial hepatectomy. Ann Surg. 2003;237(4):530–5. doi: 10.1097/01.SLA.0000059986.96051.EA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang L, Wang X, Xie G, Hill CK, Deleve LD. Liver sinusoidal endothelial cell progenitor cells promote liver regeneration in rats. J Clin Invest. 2012;122(4):1567–73. doi: 10.1172/JCI58789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Abshagen K, Eipel C, Kalff JC, Menger MD, Vollmar B. Loss of NF-kappaB activation in Kupffer cell-depleted mice impairs liver regeneration after partial hepatectomy. Am J Physiol Gastrointest Liver Physiol. 2007;292(6):G1570–7. doi: 10.1152/ajpgi.00399.2006. [DOI] [PubMed] [Google Scholar]
- 40.Boulton RA, Alison MR, Golding M, Selden C, Hodgson HJ. Augmentation of the early phase of liver regeneration after 70% partial hepatectomy in rats following selective Kupffer cell depletion. J Hepatol. 1998;29(2):271–80. doi: 10.1016/s0168-8278(98)80013-5. [DOI] [PubMed] [Google Scholar]
- 41.Michalopoulos GK, Defrances MC. Liver regeneration. Science. 1997;276(5309):60–6. doi: 10.1126/science.276.5309.60. [DOI] [PubMed] [Google Scholar]
- 42.Albrecht JH, Hu MY, Cerra FB. Distinct patterns of cyclin D1 regulation in models of liver regeneration and human liver. Biochem Biophys Res Commun. 1995;209(2):648–55. doi: 10.1006/bbrc.1995.1548. [DOI] [PubMed] [Google Scholar]
- 43.Abounader R, Ranganathan S, Kim BY, Nichols C, Laterra J. Signaling pathways in the induction of c-met receptor expression by its ligand scatter factor/hepatocyte growth factor in human glioblastoma. J Neurochem. 2001;76(5):1497–508. doi: 10.1046/j.1471-4159.2001.00158.x. [DOI] [PubMed] [Google Scholar]
- 44.Seol DW, Chen Q, Zarnegar R. Transcriptional activation of the hepatocyte growth factor receptor (c-met) gene by its ligand (hepatocyte growth factor) is mediated through AP-1. Oncogene. 2000;19(9):1132–7. doi: 10.1038/sj.onc.1203404. [DOI] [PubMed] [Google Scholar]
- 45.Fitzgerald MJ, Webber EM, Donovan JR, Fausto N. Rapid DNA binding by nuclear factor kappa B in hepatocytes at the start of liver regeneration. Cell Growth Differ. 1995;6(4):417–27. [PubMed] [Google Scholar]
- 46.Guttridge DC, Albanese C, Reuther JY, Pestell RG, Baldwina S., Jr NF-kappaB controls cell growth and differentiation through transcriptional regulation of cyclin D1. Mol Cell Biol. 1999;19(8):5785–99. doi: 10.1128/mcb.19.8.5785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Plumpe J, Malek NP, Bock CT, Rakemann T, Manns MP, Trautwein C. NF-kappaB determines between apoptosis and proliferation in hepatocytes during liver regeneration. Am J Physiol Gastrointest Liver Physiol. 2000;278(1):G173–83. doi: 10.1152/ajpgi.2000.278.1.G173. [DOI] [PubMed] [Google Scholar]
- 48.Paranjpe S, Bowen WC, Bella W, Nejak-Bowen K, Luo JH, Michalopoulos GK. Cell cycle effects resulting from inhibition of hepatocyte growth factor and its receptor c-Met in regenerating rat livers by RNA interference. Hepatology. 2007;45(6):1471–7. doi: 10.1002/hep.21570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shen K, Chang W, Gao X, et al. Depletion of activated hepatic stellate cell correlates with severe liver damage and abnormal liver regeneration in acetaminophen-induced liver injury. Acta Biochim Biophys Sin (Shanghai) 43(4):307–15. doi: 10.1093/abbs/gmr005. [DOI] [PubMed] [Google Scholar]
- 50.Benten D, Kumaran V, Joseph B, et al. Hepatocyte transplantation activates hepatic stellate cells with beneficial modulation of cell engraftment in the rat. Hepatology. 2005;42(5):1072–81. doi: 10.1002/hep.20889. [DOI] [PubMed] [Google Scholar]
- 51.Apte U, Gkretsi V, Bowen WC, et al. Enhanced liver regeneration following changes induced by hepatocyte-specific genetic ablation of integrin-linked kinase. Hepatology. 2009;50(3):844–51. doi: 10.1002/hep.23059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liu B, Paranjpe S, Bowen WC, et al. Investigation of the role of glypican 3 in liver regeneration and hepatocyte proliferation. Am J Pathol. 2009;175(2):717–24. doi: 10.2353/ajpath.2009.081129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bissell DM, Wang SS, Jarnagin WR, Roll FJ. Cell-specific expression of transforming growth factor-beta in rat liver. Evidence for autocrine regulation of hepatocyte proliferation. J Clin Invest. 1995;96(1):447–55. doi: 10.1172/JCI118055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sakurai H, Nigam SK. Transforming growth factor-beta selectively inhibits branching morphogenesis but not tubulogenesis. Am J Physiol. 1997;272(1 Pt 2):F139–46. doi: 10.1152/ajprenal.1997.272.1.F139. [DOI] [PubMed] [Google Scholar]
- 55.Ebrahimkhani MR, Oakley F, Murphy LB, et al. Stimulating healthy tissue regeneration by targeting the 5-HT(2)B receptor in chronic liver disease. Nat Med. 2011;17(12):1668–73. doi: 10.1038/nm.2490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Karp SJ. Clinical implications of advances in the basic science of liver repair and regeneration. Am J Transplant. 2009;9(9):1973–80. doi: 10.1111/j.1600-6143.2009.02731.x. [DOI] [PubMed] [Google Scholar]