

Abstract
Reversibility of airway obstruction in response to β2-agonists is highly variable among asthmatics, which is partially attributed to genetic factors. In a genome-wide association study of acute bronchodilator response (BDR) to inhaled albuterol, 534,290 single nucleotide polymorphisms (SNPs) were tested in 403 white trios from the Childhood Asthma Management Program using five statistical models to determine the most robust genetic associations. The primary replication phase included 1397 polymorphisms in three asthma trials (pooled n=764). The second replication phase tested 13 SNPs in three additional asthma populations (n=241, n=215, and n=592). An intergenic SNP on chromosome 10, rs11252394, proximal to several excellent biological candidates, significantly replicated (p=1.98×10−7) in the primary replication trials. An intronic SNP (rs6988229) in the collagen (COL22A1) locus also provided strong replication signals (p=8.51×10−6). This study applied a robust approach for testing the genetic basis of BDR and identified novel loci associated with this drug response in asthmatics.
Keywords: pharmacogenetics, asthma, bronchodilator response, genome-wide association study, albuterol
Introduction
Asthma is a complex respiratory disease characterized by hyper-responsiveness of the bronchial muscles, chronic inflammation and reversible narrowing of the airways. It affects approximately 300 million individuals worldwide and its prevalence is expected to increase to 400 million by 2025.1 Asthma is the most common chronic illness in children,2,3 accounting for half a million hospitalizations a year in the United States. In 2007, asthma related health care costs in the US were estimated to be $56 billion, with the majority attributed to medications and hospitalizations.4 Taken together, asthma has a significant public health impact and steps towards its prevention or better management will decrease the overall disease burden.
β2-agonists are the most commonly used drugs for treating asthma.2 The therapeutic effects result from binding to the transmembrane β2-adrenergic receptor (β2-AR) located on airway smooth muscle cells to relieve bronchoconstriction. These are available as short-acting β2-agonists (SABA; e.g. albuterol) for rescuing acute asthma symptoms or as long-acting β2-agonists (LABA; e.g. salmeterol and formoterol) for controlling chronic asthma that is usually administered in combination with an inhaled corticosteroid. The reversibility of airway obstruction in response to these medications, known as bronchodilator response (BDR), may be measured as a change in lung function (forced expiratory volume in one second (FEV1)) or as fall in peak expiratory flow rate (PEFR), indicating a down-regulation of B2-agonist responsivity (tachyphylaxis) with prolonged drug use. Inter-individual variability in response to these drugs have been previously described and research suggests that genetic variants are major contributing factors.3 The identification of genetic loci associated with BDR to β2-agonists will help to facilitate personalized asthma treatment regimens.
Pharmacogenetic investigations of BDR have identified a number of genetic associations for this variable drug response. The majority had been candidate gene studies, which reported genetic associations to SNPs and/or haplotypes in the arginase 1 (ARG1) locus,5 the β2-adrenergic receptor (ADRB2) gene,6–9 the corticotropin-releasing hormone receptor (CRHR)-2 locus,10 and the adenylyl cyclase type 9 (AC9) gene.11 A recent genome-wide association study (GWAS) of BDR by our group identified a functional variant in the serine-rich 2-like (SPATS2L) gene, albeit the mechanism by which it regulates BDR remains unknown.12 In this manuscript, we expand on the previous literature by using a novel approach to identify genetic associations with BDR (defined by a change in lung function) whereby we apply five statistical models in a GWAS of this drug response phenotype to decrease the likelihood of false positive associations. Novel aspects of the current GWAS include use of genetic data from the parents of asthmatics in a family-based test, which is more robust against population stratification, as well as analysis of 11 BDR measures for each subject taken over a four year period in addition to BDR at randomization (taken upon entry into the clinical trial). Moreover, we considered both additive and recessive transmissions of the associated alleles. We then pooled the results from these multiple genome-wide analytical models to identify common genetic association signals to carry forward for replication analysis in additional asthma populations. This manuscript describes the findings of our innovative GWAS of BDR in asthmatic subjects.
Methods
Asthma Trial Populations
The asthma trial populations are summarized in Table 1 and details are available in the Supplemental Material. All patients or their legal guardians consented to each study protocol and ancillary genetic testing. All studies were approved by the respective Institutional Review Boards and/or Ethics Committees of the participating institutions.
Table 1.
Baseline Characteristics of Participants in the Asthma Populations used in this Analysis.
| CAMP | CAMP (Placebo) | AT | LOCCS | LODO | AT/LOCCS/LODO | CARE | ACRN | GACRS | |
|---|---|---|---|---|---|---|---|---|---|
| n=403 | n=171 | n=444 | n=165 | n=155 | n = 764 | n=215 | n=241 | n=592 | |
| Age, mean (sd) | 8.8 (2.1) | 8.7 (2.1) | 32.4 (13.6) | 34.4 (15.3) | 42.9 (14.7) | 34.9 (14.8) | 10.6 (2.9) | 31.7 (42.2) | 9.0 (1.8) |
| Range | 5.2–13.2 | 5.2 – 13.2 | 12.0 – 80 | 7 – 71 | 15 – 76 | 7 – 80 | 6–17.8 | 12.4–63.7 | 6.0–14.2 |
| Gender, n(%) | |||||||||
| male | 254 (63) | 109 (60) | 222 (50.0) | 58 (35.2) | 39 (25.2) | 319 (41.8) | 132 (61.4) | 100 (41.49) | 351 (59.3) |
| Wash-out prior to BDR test*, weeks | 4 | 4 | 6 | 4–6 (fluticasone) | 2 | 2–6 | 0–4 | 0–6 | 4 |
| Albuterol puffs (90ug/puff) | 2 | 2 | 2 | 2 | 2 | 2 | 4 | 2–4 | 2 |
| pre-BD FEV1 pp, mean (sd) | 93.4(14.0) | 94.7(13.3) | 61.5 (6.8) | 84.3 (12.3) | 78.8 (17.7) | 69.8 (14.7) | 99.3 (12.6) | 85.9 (13.5) | 99.8 (17.2) |
| BDR, mean (sd) | 11(10) | 12 (11) | 40.15 (20.9) | 6.4 (6.1) | 9.7 (11.1) | 26.7 (23.2) | 9.5 (8.4) | 11.6 (21.8) | 5.7 (9.2) |
Subjects were permitted to use rescue medications as needed during the wash-out period
Initial GWAS Population
A total of 403 non-Hispanic white asthmatic children and their parents from the Childhood Asthma Management Program (CAMP)13,14 were successfully genotyped on the Illumina HumanHap550v3 BeadChip (San Diego, CA).15 BDR at randomization were conducted for each proband upon entry into the trial following 2 inhalations of albuterol. A total of 11 longitudinal BDR values were measured in a subset of 171 asthmatics randomized to inhaled albuterol therapy as needed over four years of this clinical trial. Genome-wide association analysis included 534,290 autosomal SNPs that had passed quality control metrics (see Supplemental Material).
Primary Replication Populations
A total of 1536 SNPs were selected for genotyping, of which 1397 were successful, in three non-Hispanic white adult asthma trials (pooled n=764) using the Illumina GoldenGate Custom Array (Illumina Inc., San Diego, CA). SNP selection criteria are detailed below (Statistical Methodology). These replication populations included: 1) the Asthma Trial (AT, n=444)16,17; 2) the Leukotriene modifier or Corticosteroid or Corticosteroid Salmeterol (LOCCS, n=165) trial18; and 3) the Effectiveness of Low Dose Theophylline as Add-on Treatment in Asthma (LODO, n=155) trial.19 BDR at randomization was conducted for each subject upon entry into these clinical trials.
Secondary Replication Populations
A total of 13 SNPs with one-sided p-values < 0.05 (based on the direction of association in CAMP) in the primary replication analysis were further tested in two additional asthma trials: 1) the Childhood Asthma Research and Education Network (CARE, n=215) and 2) the Asthma Clinical Research Network (ACRN, n=241).20 As these individuals had been genotyped on the Affymetrix Genome-Wide Human SNP Array 6.0 (Santa Clara, CA), imputed data was used that was generated for the HapMap Phase 2 Release 22 SNPs21 by applying the Markov Chain Haplotyping (MaCH) software.22 Finally, eight of these 13 SNPs were further tested in the Genetics of Asthma in Costa Rica Study (GACRS), which were successfully genotyped on the HumanOmniExpress-12v1_A chip.23 BDR at randomization was conducted for each subject upon entry into these clinical trials.
Statistical Methodology
The primary outcome measure of all analyses was BDR to the inhaled s2-agonist albuterol, which was calculated as the percent change in forced expiratory volume in one second (FEV1): BDR=100 × [(postFEV1-preFEV1)/preFEV1], where preFEV1 is the lung function before albuterol treatment (baseline) and postFEV1 is the lung function following albuterol treatment. The overall analysis strategy is presented in Figure 1. To compensate for the limited statistical power given the small sample size of the CAMP trial, we used five statistical models to identify the most robust genetic associations: generalized linear model of BDR in 403 probands, using recessive (1) and additive (2) models; mixed model of 11 repeated measures of BDR over four years in 171 individuals randomized to as-needed inhaled β2-agonist, using recessive (3) and additive (4) models; and family-based association test (FBAT) of BDR at randomization in 403 CAMP parent-offspring trios (5). All models were adjusted for age, sex, and baseline preFEV1 and model 5 was additionally adjusted for height. Each SNP was given a score of 0 to 5 based on the total number of p-values below 0.05 from all five association tests. All SNPs scoring 5 (n = 437) were carried forward for genotyping in the primary replication cohort but those scoring 4 were then ranked according to their p-values from the FBAT analysis as this model is robust against population stratification. No SNPs scoring below 4 were included for replication. All tests using generalized linear (additive and recessive) models were performed in PLINK (http://pngu.mgh.harvard.edu/purcell/plink/)24 and included SNPs with minor allele frequencies (MAF) ≥ 0.05. FBAT applied a pedigree-based analysis tool (PBAT) previously described.25 All replication analyses used a single measure of BDR at randomization, with adjustments for age, sex, height and baseline preFEV1. Multiple comparisons were adjusted using the Liptak weighted Z method.26 Additional details are available in the Supplemental Material.
Figure 1.

An overview of the genome-wide analyses methods and replication strategies used. The initial GWAS in CAMP applied five statistical models (linear regression of BDR at randomization in 403 asthmatics using additive and recessive models, longitudinal mixed models of 11 repeated BDR measures in 171 probands using additive and recessive models, and a family-based association test of BDR at randomization in 403 trios). A total of 1536 SNPs providing p-values < 0.05 from five or four of these models (the latter rankd by FBAT p-values) were selected for genotyping and replication in LOCCS/LODO/AT (n=764). The 13 replicated SNPs (one sided p-values < 0.05) were further tested in ACRN, CARE and GACRS.
Expression Quantitative Trait Analysis
Microarray data from immortalized lymphoblastoid cell lines of 117 asthmatics (non-Hispanic white CAMP subjects), spotted on the Illumina HumanRef8v2 microarray BeadChips, were used to test the correlation of genetic variants with gene expression. These cells were cultured and treated with ethanol (sham) as a control for differential analysis with corticosteroid (dexamethasone) treated cells for a separate pharmacogenetic investigation (unpublished data). The microarray data from the sham arm of this experiment was vst-transformed and quantile normalized using the Lumi package in Bioconductor.27 A cis-expression quantitative trait locus (eQTL) was defined as a SNP that was correlated with the expression of a gene within 50 kb. A trans-eQTL was a SNP correlation with a transcript located more than 50 kb away or on a separate chromosome entirely.
Results
The baseline characteristics of all asthma populations including CAMP, the three primary replication trials and the three secondary replication populations are shown in Table 1. Whereas the initial GWAS using CAMP consisted of childhood asthmatics, the replication populations included both childhood (CARE and GACRS) and adult asthmatics (pooled AT/LOCCS/LODO and ACRN). It is also notable that the adult asthma populations had fewer males and lower pre-bronchodilator FEV1 percent predicted (Pre-BD FEV1pp) values, which was previously correlated with higher BDR.28 To compensate for the variability in baseline preFEV1 across populations, we adjusted each association test for this variable in addition to accounting for it in our phenotype definition (BDR=100×[(postFEV1-preFEV1)/preFEV1]).
Genome-wide analysis in CAMP
A plot of the –Log10(p-values) against the chromosomal location of each SNP from the family-based association (FBAT) analysis is shown in Figure 2. A quantile-quantile plot of the expected p-values of the FBAT analysis under the null hypothesis and the actual observed p-values illustrates that the majority of p-values were greater than expected by chance, suggesting that the test was conservative [Supplemental Figure 1]. However, there are several p-values less than what was expected by chance. For example, the lowest p-value was 5.28×10−7 for rs8112048 located 3′ of the zinc finger protein 14 (ZNF14) gene but this did not meet genome-wide significance. In addition, we noted that many SNPs in previously implicated genes (ARG1, ADRB2, CRHR-2, and AC9)5–11 were absent from our GWAS due to differences in genotyping platforms. Of the four markers included in our GWAS (rs1042713 in ADRB2, rs4723002 and rs226716 in CRHR2, and rs2230739 in AC9), nominal association was found for rs1042713 in ADRB2 (p < 0.02), which is the most investigated locus for BDR. Finally, the genomic inflation factor estimate was 1.01, demonstrating minimal population stratification.
Figure 2.

The distribution of BDR at randomization across all asthma trial populations. BDR is defined as a percent change in lung function (FEV1) in response to inhaled albuterol across all asthma trial populations.
Replication Analyses
Data for the 1397 replication SNPs from the three adult asthma trials were pooled for analysis to maximize the statistical power for detecting associations. A total of 13 SNPs replicated in the same direction as the initial GWAS population (CAMP) and were carried forward for analysis in the secondary replication phase (Table 2). The intergenic SNP, rs11252394, with a p-value of 0.0099 (beta = 3.1) from the additive model in CAMP, had a one-sided p-value of 1.21×10−6 in the primary replication phase, which remained significant following Bonferroni correction for multiple comparisons. However, this SNP did not replicate in the secondary replication phase. Next, nominal association signals (p-values < 0.05) were derived for an intronic SNP, rs6988229, in the collagen type XXII alpha 1 (COL22A1) gene in CAMP (recessive p-value = 0.004, beta = 3.26). This SNP further replicated across all asthma populations except for CARE (Liptak combined p = 8.51E-06). Finally, five additional SNPs showed marginal association (p < 0.05) in the primary replication and one of the three secondary replication populations: rs166330, rs166332, rs17495520, rs6002674, and rs1522113. The latter marker (additive p-value = 0.014 and beta = 3.23 in CAMP), is located in intron 8 of CLOCK and in perfect linkage disequilibrium (correlation coefficient (r2) of 1.0 in CAMP) with a non-synonymous variant (rs34897046; Serine208Cysteine (S208C)) in exon 9 of the same gene.29 The top 13 SNPs explain 23.8% of the overall genetic variance in BDR, based on the correlation coefficient for each analysis. This calculation assumed that the genetic contribution of each SNP is independent of the other genetic associations.
Table 2.
Summary of GWAS and replication analyses in all asthma clinical trials.
|
|
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Replication #1 | Replication #2 | ||||||||||
|
|
|||||||||||
| SNP | Chr | MA | MAF | Gene | Model | CAMP GWAS (effect estimate) | Pooled AT/LOCCS/LODO | ACRN | CARE | GACRS | Liptak Combined P val |
| rs11252394 | 10 | A | 0.08 | Additive | 0.0099b (+) | 1.21E-06 | 0.4173 | 0.3472 | - | 1.98E-07 | |
| rs6988229 | 8 | T | 0.20 | COL22A1 | Recessive | 0.0004c (+) | 0.0050 | 0.0505 | 0.9283 | 0.0139 | 8.51E-06 |
| rs9552679 | 13 | C | 0.26 | Additive | 0.0007d (−) | 0.0004 | 0.7884 | 0.4410 | - | 3.02E-05 | |
| rs1663330 | 14 | G | 0.33 | Additive | 0.0020b (+) | 0.0028 | 0.0234 | 0.7544 | - | 4.54E-05 | |
| rs1663332 | 14 | T | 0.37 | Additive | 0.0028b(+) | 0.0006 | 0.0374 | 0.6874 | 0.3595 | 8.07E-05 | |
| rs17495520 | 5 | T | 0.14 | Additive | 0.038b (+) | 0.0110 | 0.0030 | 0.3587 | - | 0.0003 | |
| rs10511905 | 9 | G | 0.22 | Additive | 0.006a (−) | 0.0087 | 0.1388 | 0.3149 | - | 0.0003 | |
| rs518350 | 22 | T | 0.12 | Recessive | 0.0065e (+) | 0.0010 | 0.4987 | 0.2090 | 0.3334 | 2.70E-04 | |
| rs17701271 | 4 | A | 0.22 | Recessive | 0.004c (+) | 0.0026 | 0.1371 | 0.8756 | 0.2031 | 0.0003 | |
| rs6002674 | 22 | C | 0.15 | Recessive | 0.0012e (−) | 0.0040 | 0.9808 | 0.0310 | 0.5675 | 0.0027 | |
| rs1419555 | 7 | T | 0.37 | Additive | 0.0043d (+) | 0.0060 | 0.7723 | 0.6879 | 0.4932 | 0.0068 | |
| rs1423515 | 5 | A | 0.05 | Additive | 0.027b (+) | 0.0400 | 0.4104 | 0.4710 | - | 0.0089 | |
| rs1522113 | 4 | A | 0.05 | CLOCK | Additive | 0.014b (+) | 0.0037 | 0.9260 | 0.0427 | 0.8633 | 0.0177 |
Association results for 13 replicated SNPs (p-values < 0.05) from the primary replication phase only are shown, sorted by Liptak Combined P-values. The p-values presented for CAMP are 2-sided while p-values for the replication populations are 1-sided, based on the direction of association (denoted as + or −) relative to the CAMP analysis. MAF: minor allele frequency in CAMP.
Lowest p-value is with the family-based association test (FBAT)
Lowest p-value is with the additive generalized linear model
Lowest p-value is with the recessive generalized linear model
Lowest p-value is with the additive longitudinal analysis
Lowest p-value is with the recessive longitudinal analysis
Analysis of microarray data from lymphoblastoid cell lines from a subset of CAMP subjects determined that the missense variant in CLOCK is associated with variable gene expression of both CLOCK (p-value = 0.05) and one of its downstream effectors Period 2 gene (PER2, p-value = 0.003) [Supplemental Figure 2]. Individuals with one mutant allele (CG genotype, n = 20) had greater expression of both CLOCK and PER2 compared to individuals without this minor allele (GG genotype, n = 94). The SNP rs6988229 in the COL22A1 locus on the other hand did not demonstrate any cis-regulatory effects, however, it is correlated with the expression of multiple other genes (trans-acting effects on gene expression). This includes another member of the G protein-coupled receptor superfamily (GPR110). The top five trans-effects of each of the 13 SNPs from Table 2 are shown in Supplemental Table 1. These results did not suggest a regulatory role for the intergenic SNP on chromosome 10 (rs11252394). While these associations with gene expression suggest functional effects of some of our associated polymorphisms, further investigation is necessary to validate their functional effects and the mechanism by which they might regulate BDR.
Discussion
This manuscript describes a comprehensive GWAS of treatment response to β2-agonists in asthmatics, which identifies novel pharmacogenetic loci associated with clinical response variability. Due to the limited size of the asthma drug trial populations, which is common in pharmacogenetic investigations, we implemented a novel strategy to select SNPs for replication. Specifically, we prioritized SNPs by evaluating p-values from 5 different statistical models, thereby taking advantage of the longitudinal nature of the phenotypic data, the entire sample at randomization, as well as the genotype data from the parents. SNPs with the lowest p-values (< 0.05) across all five statistical models were judged to represent the most robust associations, followed by SNPs yielding p-values < 0.05 in four of the five analyses. The latter were prioritized by FBAT p-values for replication analysis. A total of 1397 were successfully genotyped and tested for replication in three independent clinical trials. The top 13 replicated SNPs were subsequently tested for association with BDR in three secondary asthma populations (Table 2). While only one intergenic SNP significantly replicated in the primary phase, six SNPs provided nominal p-values < 0.05 in both the primary replication phase and in one or more of the secondary replication populations, including intronic SNPs in the COL22A1 and CLOCK genes.
The use of five statistical models in our initial GWAS is an innovative approach for identifying genetic associations for BDR in asthma. As each statistical model has unique strengths and weaknesses, our rationale for ranking SNPs for replication based on p-values from all five models was to identify the most robust associations (i.e. those most likely to replicate and represent true pharmacogenetic associations). For example, population-based tests are more powerful to detect associations by including more individuals than the number of informative families used in the FBAT, but the former is more vulnerable to population stratification. Thus, FBAT allows us to confirm SNP associations that are not influenced by population stratification. In addition, we were able to take advantage of the longitudinal BDR data recorded at 11 time points over the four year clinical trial for a subset of our population to confirm associations that are repeatable within individuals over time. Moreover, we opted to include a recessive model because while an additive genetic model can easily identify dominant transmissions, it does not identify recessive transmissions as easily. We believe that this novel approach reduced the likelihood of false-positive association signals.
The strongest association signal that significantly replicated in the primary replication phase, albeit not associated across the secondary replication populations, was an intergenic SNP rs11252394 (Liptak p-value = 1.98E-07). Despite it being not proximal to a gene within 50 kb, a closer look at this genomic region revealed several excellent biological candidates within 2.5 Mb including Protein Kinase C theta (PRKCQ), inter-leukin receptors (IL15RA, IL2RA) and Krüppel-like factor 6 (KLF6). All four genes have been previously reported to regulate pulmonary inflammation using in vitro cellular and murine models. In fact, a PRKCQ antagonist was investigated by Wyeth Research as a novel treatment for asthma given the role of this gene in airway inflammation and hyper-responsiveness.30–32 Inhibition of IL15RA and IL2RA in mice demonstrated decreased lung inflammation.33,34 Finally, blocking of KLF6 in vitro decreased Transforming Growth Factor β (TGFβ) production that is correlated with airway remodeling and asthma development.35 While rs11252394 is not known to regulate the expression of any of these genes, nor is it known to be in LD with SNPs within these loci, further investigation is warranted to identify the causative variant, if any, in this genomic region which may underlie this association signal.
Another association signal that replicated, albeit only marginally, in the primary replication phase and across two of the secondary replication trials, was an intronic SNP (rs6988229) in the COL22A1 gene. Little is known about this gene other than it encodes a protein that acts as a cell adhesion ligand for skin epithelial cells and fibroblasts, further investigations are necessary to determine how genetic variants at this locus might influence BDR. Cis-eQTL analysis indicates that this SNP does not regulate expression of the COL22A1 transcript (p= 0.86). However, this SNP is significantly correlated with the expression of multiple other genes [Supplemental Table 1]. This includes another member of G protein-coupled receptor superfamily (GPR110), to which the β2-adrenergic receptor also belongs, which is known to regulate smooth muscle contractions and relaxations.36 Multiple splice variants of this gene, like many other members of this large gene family, has been shown to be expressed at significantly higher levels in airway smooth muscle cells.37
While the polymorphism in the CLOCK gene (rs1522113) was only marginally associated with BDR at randomization in CAMP using the additive model (p-value = 0.014), and nominally replicated in AT/LOCCS/LODO and CARE, it is an excellent biological candidate for regulating bronchodilator response in asthmatics. Previous studies suggest that CLOCK expression and β2-agonists affect the expression of circadian rhythm genes, which regulate asthma symptoms. Embryonic fibroblast cells from mice homozygous for mutant CLOCK expressed circadian rhythm genes in a non-cyclic manner, a phenotype that was rescued by ectopic expression of CLOCK.38 DeBruyne et al. reported that the circadian rhythm in peripheral tissues such as the liver and lung are also regulated by CLOCK.39 CLOCK binds to the E-box enhancer located 5′ of circadian genes such as the Periods (PER) 1, 2, and 3 to regulate their expression.40β2-agonists have also been shown to induce the expression of human period 1 (hPER1) gene in bronchial epithelial BEAS-2B cells.41 Furthermore, the administration of β2-agonists, particularly long-acting, reduces nocturnal asthma.42,43β2-agonists have also been shown to regulate the expression of these circadian rhythm genes through the phosphorylation of cAMP responsive element binding (CREB) protein which bind to CRE 5′ of these genes.40 The role of the circadian rhythm in asthma is apparent in that the narrowing of the airways are more severe between midnight and early morning hours.44 In addition, nocturnal asthma exacerbations are commonly experienced between 4 AM and 8 AM,43 which may be the combined effect of the circadian clock and the diminishing effect of asthma medications throughout the night.
In addition to a genetic association between rs1522113 and BDR in asthma, we determined that this intronic SNP is in perfect linkage disequilibrium with a missense variant in exon 9 (rs34897046; S208C), which is predicted to result in the loss of a (Serine) phosphorylation site.45 This coding SNP is predicted to be “deleterious” by SIFT (Sorting Intolerant From Tolerant)46 or “possibly damaging” by PolyPhen2.47 Finally, analysis of microarray data from lymphoblastoid cell lines of CAMP subjects indicates a marginal association between the mutant allele (208C) and increased expression of CLOCK (p value = 0.054), as well as increased expression of a downstream circadian rhythm gene Period 2 (PER2, p value = 0.003) [Supplemental Figure 2]. While this suggests that the associated polymorphism in CLOCK may be functional, further experiments are necessary to investigate the regulatory potential of this variant in the CLOCK pathway and the mechanism by which it modulates BDR.
While this manuscript represents a comprehensive GWAS of BDR response in asthmatics aimed at identifying the most robust genetic associations for replication in additional asthma trials, there were several limitations. First, our initial GWAS used the phenotype of acute response to a short-acting β2-agonist (BDR at randomization) that was taken in all CAMP probands at the start of the study as well as repeated measures of BDR in a subset of the CAMP probands who were randomized to β2-agonist as needed over the four years of the trial. For the replication cohorts, however, we only used BDR measured upon entry into the respective studies as our replication samples did not have longitudinal data. Therefore, our replication results may not identify BDR associations in asthma patients taking β2-agonist over long periods of time. Second, the mean pre-bronchodilator FEV1 percent predicted values varied across the asthma populations (Table 1). Specifically, those for all childhood asthma trials (CAMP, CARE and GACRS) were noticeably higher than those of adult asthma trials (AT, LOCCS, LODO and ACRN), which was expected. However, pre-bronchodilator FEV1 was adjusted for in our definition of BDR as well in all statistical analyses by including it as a covariate. Finally, baseline medications and recruitment criteria varied across some of the populations. For example, the LOCCS trial had completed a run-in period of 4–6 weeks during which they were administered an inhaled corticosteroid that might have improved their lung function, resulting in reduced BDR. Finally, all participants of the AT trial had a minimum BDR of 15% or greater. We addressed these differences across our trial populations in the pooled analysis of AT/LOCCS/LODO by coding each trial differently. Some of the trials (CAMP, AT, LODO, GACRS) had wash-out periods during which they were taken off their regular asthma therapies but were permitted to use rescue medications as needed. Others such as CARE and ACRN had no wash-out periods. Thus, differences in medical histories may have influenced BDR. However, we believe that these differences further demonstrate the generalizability of our association results.
Although the aim of this GWAS was to identify novel loci for BDR, we noted that this study does not replicate all of the prior associated SNPs.5–11 These results were expected as it is unusual for all of the candidate genes to be significant in any one replication population. For some of these previously associated loci, the genetic effect sizes were very modest, making these genetic variants more difficult to identify. Power simulations, based on our sample sizes (n = 403, 764, and 1,048) and the number of statistical tests, estimated that we had sufficient power (>90%) to identify common SNPs (MAF > 0.1) with effect estimates of 3 percent or greater. In addition, there was not always adequate LD coverage for some of the SNPs that were previously identified at candidate genes. Therefore, it was difficult to assess these genetic associations in our CAMP samples. Specifically, additional variants were genotyped in earlier studies using custom platforms that were not included on the Illumina HapMap550K Beadchip array used for the current GWAS. For example, none of the previously associated SNPs in ARGI were tested in the current GWAS. In fact, only four of the dozen SNPs previously implicated in the remaining three genes (ADRB2, CRHR2 and AC9) were directly tested in our GWAS. However, these did not yield high ranking scores for replication because we had selected SNPs based on p-values across five different statistical models. Furthermore, some of the earlier studies had reported a haplotype effect that was not tested in our study. Finally, our GWAS did not replicate the findings of the previous BDR GWAS, which reported association with SPATS2L.12 A major difference in the current study is the combined analysis of longitudinal BDR measures as well family-based data in addition to BDR at randomization using five statistical models, while the previous GWAS applied only one test of BDR at randomization.
Using a novel genome-wide association analysis method for investigating BDR in asthma, we have identified several genetic loci for further investigation. Among these findings is an intergenic SNP, rs11252394, that is located near multiple genes previously correlated with lung inflammation and therefore, are potential regulators of asthma. Other potentially interesting associations that were marginally associated with our drug response phenotype across multiple trials were intronic SNPs within the COL22A1 and CLOCK loci. While microarray data indicate potential cis- or trans-effects of these SNPs, further investigation is merited to determine their biological significance and potential roles in modulating bronchodilator response to β2-agonists.
Supplementary Material
{kind=link}
{kind=link}
Figure 3.
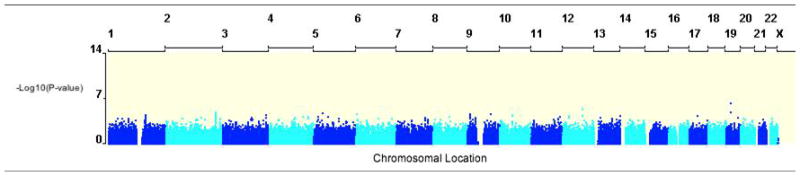
Manhattan plot of –Log10(p-value) for the FBAT analysis of BDR using 403 parent-offspring trios with 534,290 SNPs. Similar plots were generated for the other four statistical models. The analysis was adjusted for age, gender, height, and baseline preFEV1.
Acknowledgments
This work was supported by U01 HL65899 and P01 HL083069 from the National Heart, Lung, and Blood Institute (NHLBI). We thank all families for their enthusiastic participation in the CAMP Genetics Ancillary Study and the CAMP investigators and research teams, who were supported by the NHLBI N01 HR16049. Additional support for this research came from NHLBI grants N01 HR16044, HR16045, HR16046, HR16047, HR16048, HR16049, HR16050, HR16051, and HR16052. All data collection from the CAMP Genetic Ancillary Study was conducted at the Channing Laboratory of the Brigham and Women’s Hospital under appropriate CAMP policies and human subject’s protections. The CAMP Genetics Ancillary Study is supported by U01 HL075419, U01 HL65899, P01 HL083069, R01 HL086601, and T32 HL07427 from the NIH/NHLBI. Collection of microarray data from immortalized lymphoblastoid cell lines of CAMP subjects was supported by K23 HG003983 and R01 HL092197 from the NIH/NHGRI. We acknowledge the American Lung Association (ALA) and the ALA’s Asthma Clinical Research Centers investigators and research teams for use of LOCCS and LODO data, with additional funding from HL071394 and HL074755 from the NHLBI, and Nemours Children’s’ Clinic. GlaxoSmithKline supported the conduct of the LOCCS Trial by an unrestricted grant to the ALA. We acknowledge Sepracor, Inc. for use of the Asthma Trial data. The Single-Nucleotide Polymorphism Health Association Asthma Resource Project (SHARP) was funded by grants from the NHLBI U01 HL51510, U01 HL51834, U01 HL51831, U01 HL51845, U01 HL51843, M01 RR00079, and M01 RR03186 and was carried out by researchers from the Asthma Clinical Research Network (ACRN), CAMP, and Childhood Asthma Research and Education (CARE) Network. Details are available in the Online Repository and on the dbGaP (database of Genotypes and Phenotypes) website: www.ncbi.nlm.nih.gov/sites/entrez?Db=gap. The GACRS was supported by HL04370 and HL66289 from the NIH.
Footnotes
Conflict of Interest
None declared.
References
- 1.Masoli M, Fabian D, Holt S, Beasley R. The global burden of asthma: executive summary of the GINA Dissemination Committee report. Allergy. 2004;59(5):469–478. doi: 10.1111/j.1398-9995.2004.00526.x. [DOI] [PubMed] [Google Scholar]
- 2.Sears MR, Lotvall J. Past, present and future--beta2-adrenoceptor agonists in asthma management. Respir Med. 2005;99(2):152–170. doi: 10.1016/j.rmed.2004.07.003. [DOI] [PubMed] [Google Scholar]
- 3.Drazen JM, Silverman EK, Lee TH. Heterogeneity of therapeutic responses in asthma. Br Med Bull. 2000;56(4):1054–1070. doi: 10.1258/0007142001903535. [DOI] [PubMed] [Google Scholar]
- 4.Barnett SB, Nurmagambetov TA. Costs of asthma in the United States: 2002–2007. J Allergy Clin Immunol. 127(1):145–152. doi: 10.1016/j.jaci.2010.10.020. [DOI] [PubMed] [Google Scholar]
- 5.Litonjua AA, Lasky-Su J, Schneiter K, Tantisira KG, Lazarus R, Klanderman B, et al. ARG1 is a novel bronchodilator response gene: screening and replication in four asthma cohorts. Am J Respir Crit Care Med. 2008;178(7):688–694. doi: 10.1164/rccm.200709-1363OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Martinez FD, Graves PE, Baldini M, Solomon S, Erickson R. Association between genetic polymorphisms of the beta2-adrenoceptor and response to albuterol in children with and without a history of wheezing. J Clin Invest. 1997;100(12):3184–3188. doi: 10.1172/JCI119874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Green SA, Turki J, Innis M, Liggett SB. Amino-terminal polymorphisms of the human beta 2-adrenergic receptor impart distinct agonist-promoted regulatory properties. Biochemistry. 1994;33(32):9414–9419. doi: 10.1021/bi00198a006. [DOI] [PubMed] [Google Scholar]
- 8.Lima JJ, Thomason DB, Mohamed MH, Eberle LV, Self TH, Johnson JA. Impact of genetic polymorphisms of the beta2-adrenergic receptor on albuterol bronchodilator pharmacodynamics. Clin Pharmacol Ther. 1999;65(5):519–525. doi: 10.1016/S0009-9236(99)70071-8. [DOI] [PubMed] [Google Scholar]
- 9.Martin AC, Zhang G, Rueter K, Khoo SK, Bizzintino J, Hayden CM, et al. Beta2-adrenoceptor polymorphisms predict response to beta2-agonists in children with acute asthma. J Asthma. 2008;45(5):383–388. doi: 10.1080/02770900801971792. [DOI] [PubMed] [Google Scholar]
- 10.Poon AH, Tantisira KG, Litonjua AA, Lazarus R, Xu J, Lasky-Su J, et al. Association of corticotropin-releasing hormone receptor-2 genetic variants with acute bronchodilator response in asthma. Pharmacogenet Genomics. 2008;18(5):373–382. doi: 10.1097/FPC.0b013e3282fa760a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tantisira KG, Small KM, Litonjua AA, Weiss ST, Liggett SB. Molecular properties and pharmacogenetics of a polymorphism of adenylyl cyclase type 9 in asthma: interaction between beta-agonist and corticosteroid pathways. Hum Mol Genet. 2005;14(12):1671–1677. doi: 10.1093/hmg/ddi175. [DOI] [PubMed] [Google Scholar]
- 12.Himes BE, Jiang X, Hu R, Wu AC, Lasky-Su J, Klanderman B, et al. Genome-wide Association Analysis in Asthma Subjects Identifies SPATS2L as a Novel Bronchodilator Response Gene. PLoS Genet. doi: 10.1371/journal.pgen.1002824. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Long-term effects of budesonide or nedocromil in children with asthma. The Childhood Asthma Management Program Research Group. N Engl J Med. 2000;343(15):1054–1063. doi: 10.1056/NEJM200010123431501. [DOI] [PubMed] [Google Scholar]
- 14.The Childhood Asthma Management Program (CAMP): design, rationale, and methods. Childhood Asthma Management Program Research Group. Control Clin Trials. 1999;20 (1):91–120. [PubMed] [Google Scholar]
- 15.Himes BE, Hunninghake GM, Baurley JW, Rafaels NM, Sleiman P, Strachan DP, et al. Genome-wide association analysis identifies PDE4D as an asthma-susceptibility gene. Am J Hum Genet. 2009;84(5):581–593. doi: 10.1016/j.ajhg.2009.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Baron RM, Palmer LJ, Tantisira K, Gabriel S, Sonna LA, Le L, et al. DNA sequence variants in epithelium-specific ETS-2 and ETS-3 are not associated with asthma. Am J Respir Crit Care Med. 2002;166(7):927–932. doi: 10.1164/rccm.200201-048OC. [DOI] [PubMed] [Google Scholar]
- 17.Silverman ES, Palmer LJ, Subramaniam V, Hallock A, Mathew S, Vallone J, et al. Transforming growth factor-beta1 promoter polymorphism C-509T is associated with asthma. Am J Respir Crit Care Med. 2004;169(2):214–219. doi: 10.1164/rccm.200307-973OC. [DOI] [PubMed] [Google Scholar]
- 18.Peters SP, Anthonisen N, Castro M, Holbrook JT, Irvin CG, Smith LJ, et al. Randomized comparison of strategies for reducing treatment in mild persistent asthma. N Engl J Med. 2007;356(20):2027–2039. doi: 10.1056/NEJMoa070013. [DOI] [PubMed] [Google Scholar]
- 19.Clinical trial of low-dose theophylline and montelukast in patients with poorly controlled asthma. Am J Respir Crit Care Med. 2007;175(3):235–242. [Google Scholar]
- 20.Israel E, Chinchilli VM, Ford JG, Boushey HA, Cherniack R, Craig TJ, et al. Use of regularly scheduled albuterol treatment in asthma: genotype-stratified, randomised, placebo-controlled cross-over trial. Lancet. 2004;364(9444):1505–1512. doi: 10.1016/S0140-6736(04)17273-5. [DOI] [PubMed] [Google Scholar]
- 21.A haplotype map of the human genome. Nature. 2005;437(7063):1299–1320. doi: 10.1038/nature04226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Willer CJ, Sanna S, Jackson AU, Scuteri A, Bonnycastle LL, Clarke R, et al. Newly identified loci that influence lipid concentrations and risk of coronary artery disease. Nat Genet. 2008;40(2):161–169. doi: 10.1038/ng.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hunninghake GM, Soto-Quiros ME, Avila L, Su J, Murphy A, Demeo DL, et al. Polymorphisms in IL13, total IgE, eosinophilia, and asthma exacerbations in childhood. J Allergy Clin Immunol. 2007;120(1):84–90. doi: 10.1016/j.jaci.2007.04.032. [DOI] [PubMed] [Google Scholar]
- 24.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81(3):559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lange C, DeMeo D, Silverman EK, Weiss ST, Laird NM. PBAT: tools for family-based association studies. Am J Hum Genet. 2004;74(2):367–369. doi: 10.1086/381563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liptak T. On the combination of independent tests. Magyar Tud Akad Mat Kutato Int Kozl. 1958;3:171–197. [Google Scholar]
- 27.Du P, Kibbe WA, Lin SM. lumi: a pipeline for processing Illumina microarray. Bioinformatics. 2008;24(13):1547–1548. doi: 10.1093/bioinformatics/btn224. [DOI] [PubMed] [Google Scholar]
- 28.Tantisira KG, Fuhlbrigge AL, Tonascia J, Van Natta M, Zeiger RS, Strunk RC, et al. Bronchodilation and bronchoconstriction: predictors of future lung function in childhood asthma. J Allergy Clin Immunol. 2006;117(6):1264–1271. doi: 10.1016/j.jaci.2006.01.050. [DOI] [PubMed] [Google Scholar]
- 29.Hawkins GA, Meyers DA, Bleecker ER, Pack AI. Identification of coding polymorphisms in human circadian rhythm genes PER1, PER2, PER3, CLOCK, ARNTL, CRY1, CRY2 and TIMELESS in a multi-ethnic screening panel. DNA Seq. 2008;19(1):44–49. doi: 10.1080/10425170701322197. [DOI] [PubMed] [Google Scholar]
- 30.Chaudhary D, Kasaian M. PKCtheta: A potential therapeutic target for T-cell-mediated diseases. Curr Opin Investig Drugs. 2006;7(5):432–437. [PubMed] [Google Scholar]
- 31.Mosyak L, Xu Z, Joseph-McCarthy D, Brooijmans N, Somers W, Chaudhary D. Structure-based optimization of PKCtheta inhibitors. Biochem Soc Trans. 2007;35(Pt 5):1027–1031. doi: 10.1042/BST0351027. [DOI] [PubMed] [Google Scholar]
- 32.Cole DC, Asselin M, Brennan A, Czerwinski R, Ellingboe JW, Fitz L, et al. Identification, characterization and initial hit-to-lead optimization of a series of 4-arylamino-3-pyridinecarbonitrile as protein kinase C theta (PKCtheta) inhibitors. J Med Chem. 2008;51(19):5958–5963. doi: 10.1021/jm800214a. [DOI] [PubMed] [Google Scholar]
- 33.Ruckert R, Brandt K, Braun A, Hoymann HG, Herz U, Budagian V, et al. Blocking IL-15 prevents the induction of allergen-specific T cells and allergic inflammation in vivo. J Immunol. 2005;174(9):5507–5515. doi: 10.4049/jimmunol.174.9.5507. [DOI] [PubMed] [Google Scholar]
- 34.Doganci A, Karwot R, Maxeiner JH, Scholtes P, Schmitt E, Neurath MF, et al. IL-2 receptor beta-chain signaling controls immunosuppressive CD4+ T cells in the draining lymph nodes and lung during allergic airway inflammation in vivo. J Immunol. 2008;181(3):1917–1926. doi: 10.4049/jimmunol.181.3.1917. [DOI] [PubMed] [Google Scholar]
- 35.Mgbemena V, Segovia J, Chang T, Bose S. Kruppel-like factor 6 regulates transforming growth factor-beta gene expression during human respiratory syncytial virus infection. Virol J. 8:409. doi: 10.1186/1743-422X-8-409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McGraw DW, Elwing JM, Fogel KM, Wang WC, Glinka CB, Mihlbachler KA, et al. Crosstalk between Gi and Gq/Gs pathways in airway smooth muscle regulates bronchial contractility and relaxation. J Clin Invest. 2007;117(5):1391–1398. doi: 10.1172/JCI30489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Einstein R, Jordan H, Zhou W, Brenner M, Moses EG, Liggett SB. Alternative splicing of the G protein-coupled receptor superfamily in human airway smooth muscle diversifies the complement of receptors. Proc Natl Acad Sci U S A. 2008;105(13):5230–5235. doi: 10.1073/pnas.0801319105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pando MP, Morse D, Cermakian N, Sassone-Corsi P. Phenotypic rescue of a peripheral clock genetic defect via SCN hierarchical dominance. Cell. 2002;110(1):107–117. doi: 10.1016/s0092-8674(02)00803-6. [DOI] [PubMed] [Google Scholar]
- 39.DeBruyne JP, Weaver DR, Reppert SM. Peripheral circadian oscillators require CLOCK. Curr Biol. 2007;17(14):R538–539. doi: 10.1016/j.cub.2007.05.067. [DOI] [PubMed] [Google Scholar]
- 40.Travnickova-Bendova Z, Cermakian N, Reppert SM, Sassone-Corsi P. Bimodal regulation of mPeriod promoters by CREB-dependent signaling and CLOCK/BMAL1 activity. Proc Natl Acad Sci U S A. 2002;99(11):7728–7733. doi: 10.1073/pnas.102075599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Takata M, Burioka N, Ohdo S, Fukuoka Y, Miyata M, Endo M, et al. Beta2-adrenoceptor agonists induce the mammalian clock gene, hPer1, mRNA in cultured human bronchial epithelium cells in vitro. Chronobiol Int. 2005;22(4):777–783. doi: 10.1080/07420520500179167. [DOI] [PubMed] [Google Scholar]
- 42.Petrie GR, Chookang JY, Hassan WU, Morrison JF, O’Reilly JF, Pearson SB, et al. Bambuterol: effective in nocturnal asthma. Respir Med. 1993;87(8):581–585. doi: 10.1016/s0954-6111(05)80260-4. [DOI] [PubMed] [Google Scholar]
- 43.Bergholtz B. Nocturnal asthma. Causes and treatment. Tidsskr Nor Laegeforen. 1989;109(16):1796–1797. [PubMed] [Google Scholar]
- 44.Hetzel MR, Clark TJ. Comparison of normal and asthmatic circadian rhythms in peak expiratory flow rate. Thorax. 1980;35(10):732–738. doi: 10.1136/thx.35.10.732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wong YH, Lee TY, Liang HK, Huang CM, Wang TY, Yang YH, et al. KinasePhos 2. 0: a web server for identifying protein kinase-specific phosphorylation sites based on sequences and coupling patterns. Nucleic Acids Res. 2007;35(Web Server issue):W588–594. doi: 10.1093/nar/gkm322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ng PC, Henikoff S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31(13):3812–3814. doi: 10.1093/nar/gkg509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sunyaev S, Ramensky V, Koch I, Lathe W, 3rd, Kondrashov AS, Bork P. Prediction of deleterious human alleles. Hum Mol Genet. 2001;10(6):591–597. doi: 10.1093/hmg/10.6.591. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.