

Summary
Degradation of the cholesterol side-chain in M. tuberculosis is initiated by two cytochromes P450, CYP125A1 and CYP142A1, that sequentially oxidize C26 to the alcohol, aldehyde and acid metabolites. Here we report characterization of the homologous enzymes CYP125A3 and CYP142A2 from M. smegmatis mc2 155. Heterologously expressed, purified CYP125A3 and CYP142A2 bound cholesterol, 4-cholesten-3-one, and antifungal azole drugs. CYP125A3 or CYP142A2 reconstituted with spinach ferredoxin and ferredoxin reductase efficiently hydroxylated 4-cholesten-3-one to the C-26 alcohol and subsequently to the acid. The X-ray structures of both substrate-free CYP125A3 and CYP142A2 and of cholest-4-en-3-one-bound CYP142A2 reveal significant differences in the substrate binding sites compared with the homologous M. tuberculosis proteins. Deletion of cyp125A3 or cyp142A2 does not impair growth of M. smegmatis mc2 155 on cholesterol. However, deletion only of cyp125A3 causes a reduction of both the alcohol and acid metabolites and a strong induction of cyp142 at the mRNA and protein levels, indicating that CYP142A2 serves as a functionally redundant back up enzyme for CYP125A3. In contrast to M. tuberculosis, the M. smegmatis Δcyp125Δcyp142 double mutant retains its ability to grow on cholesterol albeit with a diminished capacity, indicating an additional level of redundancy within its genome.
Introduction
Cytochromes P450 (CYPs) form a widely distributed class of heme-containing monooxygenases that are present in all domains of life. Actinobacteria genomes possess an unusually high number of CYPs (e. g., 20 CYPs in Mycobacterium tuberculosis H37Rv, 29 CYPs in Rhodococcus jostii RHA1 and 40 CYPs in Mycobacterium smegmatis), in contrast to the absence of CYPs in Escherichia coli and in comparison to the 57 CYPs in the 3.3 Gb human genome (Ouellet et al., 2010a; Hudson et al., 2012). The typical function of CYPs is to catalyze the oxidation of organic substrates via their heme prosthetic group. This mono-oxygenation reaction involves the insertion of one oxygen atom from molecular oxygen into the substrate, while the second oxygen undergoes reduction to water (Ouellet et al., 2010a). The key stages of the catalytic cycle are: (i) substrate entry into the distal active site, which displaces the weakly iron-co-ordinated water present in the CYP resting state, (ii) heme-iron reduction from the ferric to the ferrous state by an electron-transport partner system and binding of molecular oxygen to the ferrous iron, (iii) a further single electron reduction of the oxy-complex and two protonation steps to release one oxygen as water and concomitantly generate a highly reactive intermediate iron(IV)-oxo porphyrin π-radical cation ([FeIV=O]+•), termed Compound I, and finally (iv) incorporation of the oxygen atom from Compound I into the substrate via a radical rebound mechanism (Denisov et al. 2005; Johnston et al. 2011).
Cholesterol and related steroids are ubiquitous throughout the environment due to their presence in cytoplasmic membranes and their role as precursors of vitamin D, the bile acids and all the sterol hormones. The microbial degradation of cholesterol proceeds through two biochemical stages: sterol side-chain elimination and steroid ring opening (Van der Geize and Dijkhuizen, 2004). The first step in cholesterol ring opening is the transformation of cholesterol into cholest-4-en-3-one that can be catalyzed in M. smegmatis by at least two different enzymes encoded by the genes MSMEG_5228 and MSMEG_5233, respectively (Uhía et al., 2011). Both enzymes are 3β-hydroxysteroid dehydrogenases belonging to the short-chain dehydrogenase/reductase (SDR) superfamily that binds NAD(P)(H) with a Rossman fold motif (Oppermann et al., 2003). The protein encoded by the MSMEG_5228 gene is very similar to the cholesterol dehydrogenases from Nocardia sp. and M. tuberculosis (Rv1106c) (Horinouchi et al., 1991; Yang et al., 2007), while the protein encoded by the MSMEG_5233 gene is similar to the AcmA dehydrogenase from Sterolibacterium denitrificans which is an O2 independent hydroxylase that belongs to the dimethyl sulfoxide dehydrogenase molybdoenzyme family (Chiang et al., 2008; Dermer and Fuchs, 2012).
Recent data demonstrate that two key enzymes, CYP125 and CYP142 initiate cholesterol side-chain degradation in M. tuberculosis and R. jostii RHA1 (McLean et al., 2009; Capyk et al., 2009; Rosloniec et al., 2009; Ouellet et al., 2010b). These P450s perform sequential oxidations of the cholesterol side-chain at the C26 position, forming first the terminal alcohol, then the aldehyde, and finally the acid. This activity ultimately enables β-oxidation of the cholesterol side-chain (Ouellet et al., 2011; McLean et al., 2012). CYP125, the major P450 involved in side-chain oxidation in M. tuberculosis, is located in the igr operon, which is also important for M. tuberculosis survival in macrophages (Chang et al., 2009). An essential role of CYP125 for growth on cholesterol and alleviation of the toxicity of the cholest-4-en-3-one intermediate was observed with the CDC1551 strain of M. tuberculosis and in Mycobacterium bovis (BCG), but was not seen with the H37Rv strain, suggesting that the latter possesses one or more compensatory enzymes that allow it to cope in the absence of CYP125 (Ouellet et al., 2010b; Capyk et al., 2009). CYP142 can compensate for a deficiency of CYP125 and, in certain M. tuberculosis strains, cooperates with CYP125 in cholesterol catabolism (Driscoll et al., 2010; Johnston et al., 2010). Both cytochromes are able to oxidize the aliphatic side-chain of cholesterol or cholest-4-en-3-one at C-26 to the carboxylic acid. CYP125 generates oxidized sterols of the (25S) configuration, whereas the opposite (25R) stereochemistry is obtained with CYP142 (Johnston et al., 2010).
The CYP125 orthologue in M. smegmatis, a rapidly-growing mycobacterium originally isolated from human smegma and commonly found in soil and water, is encoded by the MSMEG_5995 gene located within the MSMEG_5995_5990 putative operon. This operon has been suggested to be involved in cholesterol side-chain oxidation based on transcriptomic data and on its similarity with the igr operon from M. tuberculosis (Uhía et al., 2012). The CYP142 orthologue in M. smegmatis is encoded by the MSMEG_5918 gene located within the cholesterol gene cluster 2 that is induced in the presence of cholesterol (Uhía et al., 2012). The KstR regulator negatively controls the expression of both MSMEG_5995 and MSMEG_5918 genes (Kendall et al., 2007).
In this work, we report biochemical and structural characterization of the enzymes CYP125 and CYP142 from M. smegmatis mc2 155 and analyze their role in the metabolism of cholesterol by constructing appropriate deletion mutants. Whereas CYP142 serves as the sole back up for CYP125 in M. tuberculosis for the oxidation of the cholesterol side-chain, the Δcyp125Δcyp142 mutant of M. smegmatis retains its ability to utilize cholesterol as a carbon source, implying the presence of an additional level of redundancy within its genome.
Results
Cyp125 and Cyp142 genome region comparisons in M. tuberculosis and M. smegmatis
The cholesterol degradation pathway of cholesterol is highly conserved within the Actinobacteria, and particularly in the genus Mycobacterium. Bioinformatic analysis (TBLASTN) revealed that most of the mycobacterial genomes that are completely sequenced, except for M. kansasii and M. leprae TN, code for putative CYP125A3 and CYP142A2 orthologues (≥70% of identity), although in strains such as CDC1551 CYP142 is present as a pseudogene.
CYP125A3 (MSMEG_5995) from M. smegmatis shows a high amino acid sequence identity with CYP125A1 from M. tuberculosis (Rv3545c) (77%) (Table 1). MSMEG_5995 is located in the MSMEG_5990-MSMEG_5995 operon (Fig. S1) which shows a high identity (77–84%) with the igr operon (Rv3545c-Rv3540c) of M. tuberculosis that encodes an incomplete β-oxidation pathway for cholesterol metabolism (Table 1) (Miner et al., 2009; Thomas et al., 2011) and that is essential for survival of the pathogen (Sassetti and Rubin, 2003). The rest of the annotated functions within this operon are a lipid transfer protein (lpt2/MSMEG_5990/Rv3540c), two MaoC-like hydratases (MSMEG_5991/Rv3541c and MSMEG_5992/Rv3542c) and two acyl-CoA dehydrogenases (fadE29/MSMEG_5993/Rv3543c and fadE28/MSMEG_5994/Rv3544c). Directly opposite to MSMEG_5995 is located an acetyl-CoA acetyl transferase (fadA5/MSMEG_5996/Rv3546) that has been recently described as essential for utilization of cholesterol as a sole carbon source and for full virulence of M. tuberculosis in the chronic stage of mouse lung infection (Nesbitt et al, 2010).
Table 1.
Protein comparison in M. tuberculosis and M. smegmatis.
| M. smegmatis | MTB | % ID | Name | Annotation | References |
|---|---|---|---|---|---|
| MSMEG_5914 | Rv3515c | 83 | FadD19 | Acyl-CoA synthetase | Cole et al., 1998 |
| MSMEG_5915 | Rv3516 | 82 | EchA19 | Enoyl-CoA hydratase | Cole et al., 1998 |
| MSMEG_5916 | ---- | ---- | ---- | ---- | -- |
| MSMEG_5917 | Rv3517 | 59 | ---- | Hypothetical protein | -- |
| MSMEG_5918 | Rv3518c | 78 | CYP142A1 | Cytochrome P450 monooxygenase 142 |
Driscoll et al., 2010
Johnston et al., 2010 |
| MSMEG_5919 | Rv3519 | 66 | ---- | Hypothetical protein | -- |
| MSMEG_5920 | Rv3520 | 85 | ---- | Coenzyme F420-dependent oxidoreductase | Gu et al., 2003 |
| MSMEG_5921 | Rv3521 | 76 | ---- | Hypothetical protein | -- |
| MSMEG_5922 | Rv3522 | 80 | Lpt4 | Lipid-transfer protein | Cole et al., 1998 |
| MSMEG_5923 | Rv3523 | 87 | Lpt3 | Acetyl-CoA acetyltransferase | Cole et al., 1998 |
|
| |||||
| MSMEG_5990 | Rv3540c | 84 | Lpt2 | Lipid-transfer protein | Cole et al., 1998 |
| MSMEG_5991 | Rv3541c | 76 | ---- | Putatie enoyl-CoA hydratase | -- |
| MSMEG_5992 | Rv3542c | 74 | ---- | Putative enoyl-CoA hydratase | -- |
| MSMEG_5993 | Rv3543c | 77 | FadE29 | Acyl-CoA dehydrogenase | Thomas et al., 2011 |
| MSMEG_5994 | Rv3544c | 73 | FadE28 | Acyl-CoA dehydrogenase | Thomas et al., 2011 |
| MSMEG_5995 | Rv3545c | 77 | CYP125A1 | Cytochrome P450 125 |
McLean et al., 2009
Capyk et al., 2009 Ouellet et al., 2010b |
| MSMEG_5996 | Rv3546 | 84 | FadA5 | Acetyl-CoA acetyltransferase | Nesbitt et al., 2010 |
MSMEG_5918 coding for CYP142A2 is located in the M. smegmatis cholesterol regulon outside the igr like region (Uhía et al., 2012) and shares high amino acid sequence identity with CYP142A1 from M. tuberculosis (Rv3518c) (78%) (Table 1). The annotated functions of the genes within this genomic region are an acyl-CoA synthetase (fadD19/MSMEG_5914/Rv3515c) , anenoyl-CoA hydratase (echA19/MSMEG_5915/Rv3516c), three hypothetical proteins (MSMEG_5917/Rv3517c, MSMEG_5919/Rv3519 and MSMEG_5921/Rv3521), a coenzyme F420-dependent oxidoreductase (MSMEG_5920/Rv3520c), a lipid-transfer protein (lpt4/MSMEG_5922/Rv3522) and an acetyl-CoA acetyltransferase (lpt3/MSMEG_5923/Rv3523) (Table 1). The organization of the genomic region encoding CYP142 in M. smegmatis is very similar to that of M. tuberculosis (Fig. S1). The only difference detected is the transcriptional direction of the MSMEG_5919 gene, which is opposite for the orthologue in M. tuberculosis. This change could modify the transcriptional regulation of the cyp142 gene since there is a KstR1 operator sequence in the MSMEG_5919-MSMEG_5920 intergenic region. The gene products around the cyp142 gene have been suggested to be involved in cholesterol side degradation in M. tuberculosis. Recent data demonstrated that FadD19 is a steroid CoA ligase essential for degradation of C-24 branched sterol side-chains in Rhodococcus rhodochrous DSM43269 (Wilbrink et al., 2011).
Purification and spectral features of CYP125A3 and CYP142A2
Recombinant CYP125A3 and CYP142A2 from M. smegmatis were heterologously expressed in E. coli DH5α using the pCWOri+ vector. As stated in Experimental Procedures, they were purified to homogeneity by immobilized metal ion affinity chromatography followed by two steps of ion exchange chromatography yielding 35 and 43 mg purified protein per liter of harvested culture, respectively. SDS-PAGE analysis indicated that both CYP125A3 and CYP142A2 constituted >99% of the protein in the purified sample. CYP125A3 displayed spectral properties typical for a ferric P450 with most of the heme-iron in a high spin (HS) state with a Soret band at 393 nm and a small shoulder at 415 nm (corresponding to a low spin state) (Fig. 1A, solid line). This is consistent with the spectroscopic properties of CYP125 found by Ouellet et al., 2010b; Capyk et al, 2009 and McLean et al., 2009. In contrast, CYP142 has all its heme in the low spin state (LS), exhibiting a Soret band at 418 nm and the smaller α and β bands at 567 nm and 536 nm respectively, as it was described for CYP142 from M. tuberculosis (Driscoll et al., 2010) (Fig. 1B, solid line).
Fig. 1.
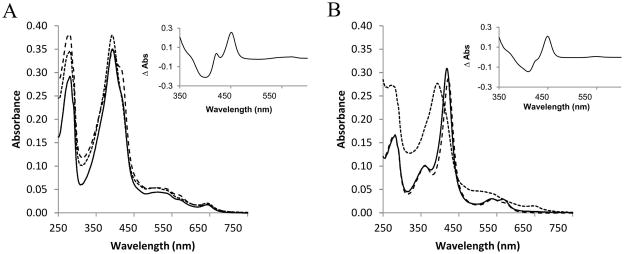
Absolute Soret region absorption spectra of CYP125 (A) and CYP142 (B) in their resting (solid line), cholesterol-bound (dotted line) and econazole-bound (dashed line) forms. The protein concentration was always 3 μM. Cholesterol (in 10% of MβCD) and econazole (in methanol) were added at 50 μM. The insets show difference spectra generated by subtracting the spectra for the dithionite-treated enzymes from the ferrous-carbon monoxide complexes.
Reduction with sodium dithionite in the presence of carbon monoxide results in the formation of Fe2+-CO complexes giving CO-difference spectra with peaks at 451 nm and 449 nm for CYP125A3 and CYP142A2, respectively (Fig. 1A and 1B, insets). Both proteins have a secondary peak at 422 nm, which is more evident in the case of CYP125, revealing the population of the P420 form of each isozyme.
CYP125A3 and CYP142A2 bind cholesterol, cholest-4-en-3-one and antifungal azole drugs
Binding of steroid ligands and antifungal azole drugs to CYP125A3 and CYP142A2 was studied by measuring the changes in the optical absorption spectra. Fig. 1 shows the absolute Soret region absorption spectra of CYP125 (Fig. 1A) and CYP142 (Fig. 1B) after incubation with cholesterol 50 μM (dotted line) and econazole 50 μM (dashed line). In contrast to the case of CYP125 and CYP142 from M. tuberculosis (Ouellet et al., 2010b; Johnston et al., 2010), the addition of cholesterol to CYP125A3 and CYP142A2 results in complete conversion to the HS form as a result of the displacement of the water molecule coordinated to the heme iron atom. Due to the predominantly HS resting state of CYP125A3 only small changes are observed in the optical spectrum when cholesterol is added, notably an increase of the Soret band at 393 nm and a strong decrease of the shoulder at 415 nm (Fig. 1A). In the case of CYP142A2, the addition of cholesterol results in a clear Type I shift of the Soret band from 418 nm to 393 nm (Fig. 1B). As observed previously for M. tuberculosis CYP125 and CYP142 (Driscoll et al., 2010; Ouellet et al., 2010a), several antifungal azoles bind to CYP125 and CYP142, inducing a Type II spectral shift. Coordination of econazole to each protein causes a partial conversion of CYP125 to the LS state, resulting in a Type II shift of the Soret band to 415 nm (Fig. 1A), and a complete conversion of CYP142, inducing a Soret shift to 422 nm (Fig. 1B).
The KD values of CYP125A3 ad CYP142A2 for cholesterol, cholest-4-en-3-one and several antifungal azoles (miconazole, econazole and clotrimazole) were obtained from the spectral titration curves. The plots of the induced spectral change versus the steroid concentration (Fig. 2) and the azole concentration (Fig. S2) were fitted to a quadratic tight binding equation (Equation 1, see “Experimental procedures”) to generate the KD values that are listed in Table 2. For comparison, the literature KD values for CYP125 and CYP142 from M. tuberculosis are also included.
Fig. 2.

Binding of cholest-4-en-3-one and cholesterol to CYP125A3 (A) and CYP142A2 (B). Ligand binding was monitored by the concentration dependent difference spectra observed in the Soret region for the substrates cholest-4-en-3-one (top) and cholesterol (bottom).
Table 2.
Apparent dissociation constant (KD) for steroids and antifungal azole drugs.
|
KD (μM)
|
||||
|---|---|---|---|---|
| CYP125Msm | CYP142Msm | CYP125Mtb | CYP142 Mtb | |
| Substrate | ||||
| Cholesterol | 1.1±0.5 | 0.14±0.07 | 0.11±0.06a | 0.018±0.005b |
| Cholest-4-en-3-one | 2.3±1.4 | 0.52±0.26 | 1.18±0.11a | 0.114±0.017 b |
|
| ||||
| Inhibitor | ||||
| Miconazole | 1.66±0.21 | 6.58±0.70 | 4.6±0.4c | 4.0±0.5d |
| Econazole | 7.38±0.71 | 7.40±0.52 | 11.7±0.7 c | 4.6±0.2d |
| Clotrimazole | 14.53±1.58 | 16.20±0.75 | 5.3±0.6 c | 3.8±0.9 d |
Values from Ouellet et al., 2010b
Values from Johnston et al., 2010
Values from McLean et al., 2009
Values from Driscoll et al., 2010
CYP125A3 and CYP142A2 catalyze the monooxygenation of C-26 steroids
The enzymatic activities of the two M. smegmatis P450s in this study were examined in vitro using the heterologous electron donor partners, spinach ferredoxin and ferredoxin reductase, and an NADPH regenerating system. We observed the oxidation of cholest-3-en-4-one after 5 and 20 min of incubation with both CYP125A3 and CYP142A2, as judged from the appearance of new peaks in the HPLC chromatograms. The relative retention times (Rt) and mass spectra were consistent with production of 26-hydroxycholest-4-en-3-one (Rt 3.14 min, M+ 401) by both enzymes. The subsequent oxidation to cholest-4-en-3-one-26-oic acid (Rt 2.15 min, M+ 415) via the aldehyde cholest-4-en-3-one-26-al (Rt 4.53 min, M+ 399) was observed in assays with both enzymes, although it was more prevalent in assays using CYP142A2. The assignments of these products were based on their relative retention times and analysis of their mass spectra, which exhibit diagnostic peaks that match earlier assignments (Fig. S3) (Ouellet et al, 2010b; Johnston et al. 2010).
Steady-state kinetics were measured and the parameters fit to the Michaelis-Menten equation (see Experimental Procedures), for the oxidation of cholent-4-en-3-one to 26-hydroxycholest-4-en-3-one. CYP125A3 and CYP142A2 showed similar KM values of 14.0 and 10.3 μM, respectively; however, the overall rate of catalysis by CYP142A2 was approximately twice that of CYP125A3 (Fig. 3).
Fig. 3.
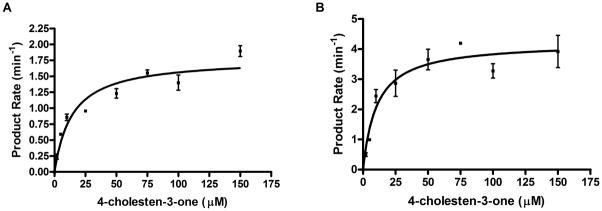
CYP125A3 (A) and CYP142A2 (B) oxidation of cholest-4-en-3-one. Fitting results to the Michaelis-Menten kinetic equation (see Experimental Procedures) for substrate oxidation assays. Reactions were run for 5 to 20 min, the products were separated by HPLC, and the rate of 26-hydroxycholest-4-en-3-one formation was determined. Minor amounts of the aldehyde and acid products were observed in some reactions, but were not included in the analysis.
Overall structure of CYP125
Consistent with the 77% sequence identity between two proteins, the 2.0 Å X-ray structure of M. smegmatis CYP125A3 determined in this work is rather similar to that of the M. tuberculosis homolog, particularly when the substrate-free structures are compared (Fig. 4A). Although the substrate – bound form of CYP125A3 has not yet been characterized, the substrate position can be reasonably inferred from that of the M. tuberculosis CYP125A1-cholest-4-en-3-one complex (Ouellet et al, 2010b). Based on this approximation, residues contacting the cholest-4-en-3-one aliphatic side-chain are invariant. The conserved amino acid substitutions near the ring system include W83, M87 and L94 in M. smegmatis, compared with F100, I104 and V111 in M. tuberculosis, respectively. All three residues are bulkier in M. smegmatis and are situated on the flat side of the ring system. The most notable difference between the two binding sites is the lack of the D108-K214 salt-bridge interaction guarding the entrance to the active site at van der Waals distance to the substrate keto group in M. tuberculosis. Both residues are represented by alanine in M. smegmatis.
Fig. 4.
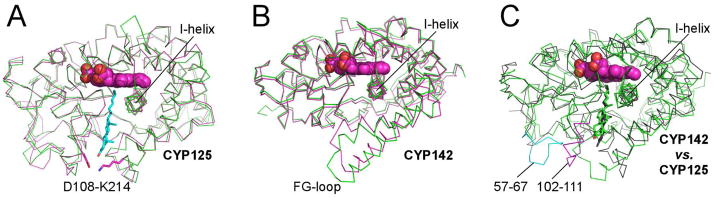
Overall structures of CYP125 and CYP142. (A) Cα traces for substrate-free CYP125A3 of M. smegmatis (green) and CYP125A1 of M. tuberculosis (magenta) are shown overlapped. The D108-K214 salt-bridge, missing from M. smegmatis, is shown in magenta sticks. Cholest-4-en-3-one (cyan sticks) is shown in the orientation observed in CYP125A1 of M. tuberculosis (PDB ID 2X5W). (B) Cα traces for substrate-bound CYP142A2 of M. smegmatis (green) and CYP142A1 of M. tuberculosis (magenta) are shown overlapped. The F-G-loop is in a more open conformation in the M. smegmatis structure. (C) Cα traces for substrate-bound CYP142A2 of M. smegmatis (green) and CYP125A1 of M. tuberculosis (grey) are shown overlapped. Highlighted in magenta and cyan are two amino acid inserts in CYP125 sequence that are missing from CYP142. Overlapped substrate molecules demonstrate the tilted position of cholest-4-en-3-one in CYP142A2 (green) compared to that of CYP125A1 (grey).
Overall structure of CYP142
Although sharing a sequence of 78%, differences have also been observed between the CYP142 counterparts from M. tuberculosis and M. smegmatis. The substrate-free M. smegmatis CYP142A2 is captured in a more “open” conformation than the M. tuberculosis enzyme due to repositioning of the F- and G-helices (Fig. 4B). Each protein chain in substrate-free CYP142A2 is associated with a molecule of β-methyl cyclodextrin used to deliver the steroid substrate to the active site. Although the substrate failed to enter the active site in this crystal form, β-methyl cyclodextrin molecules bound in the symmetry- related or special positions in the crystal, were unambiguously distinguished by donut-shaped electron densities. Due to lack of true lateral symmetry, all six are represented by alternative conformations, one flipped relative to the other. The untraceable bulk of electron density in the middle of each “donut” may belong to cholest-4-en-3-one cargo molecules.
The cholest-4-en-3-one-bound CYP142A2 crystals were obtained from an alternative set of crystallization conditions (Table 3). In the 1.69 Å CYP142A2 structure, the substrate was unambiguously defined by the electron density in a single binding orientation (Fig. 5) in all four molecules in an asymmetric unit. Substrate-bound CYP142A2 was in a more open conformation than the substrate-free form, with the G-helix positioned further from the protein core to provide support for the substrate molecule. As in the CYP125 counterpart, the residues contacting the cholest-4-en-3-one aliphatic side-chain are invariant. Three amino acid substitutions, all of bulkier residues, including L72→M75, M74→Y77 and M222→F255, distinguish the substrate binding sites of M. smegmatis CYP142A2 vs M. tuberculosis CYP142A1. Again, all three are clustered along the flat face of the sterol tetracyclic fused ring system (Fig. 5A). To accommodate bulkier side-chains, cholest-4-en-3-one is bent away from the triad (Fig. 5B). Distances between the heme iron and the carbon atoms of the branched methyl groups are 4.1 Å and 5.6 Å, favoring formation of a product with R-configuration at C25.
Table 3.
Data collection and refinement statistics.
| Protein | CYP125 | CYP142 | CYP142 |
|
| |||
| PDB ID | 2APY | 3ZBY | 2YOO |
|
| |||
| Substrate | none | none | Cholest-4-en-3-one |
|
| |||
| Data collection | |||
| Space group | I41 | C2 | P21 |
| Cell dimensions | |||
| a, b, c (Å) | 108.2, 108.2, 120.2 | 94.1, 162.8, 266.4 | 56.7, 106.2, 126.5 |
| a, b, c (°) | 90, 90, 90 | 90, 90, 90 | 90, 90.67, 90 |
| Molecules in AU | 1 | 6 | 4 |
| Wavelength | 1.11587 | 1.11587 | 1.11587 |
| Resolution (Å) | 2.0 | 1.93 | 1.69 |
| Rsym or Rmerge (%) | 7.7 (85.1)1 | 7.9 (54.2) | 8.8 (49.8) |
| I / σI | 11.3 (1.6) | 7.1 (1.5) | 6.8 (1.5) |
| Completeness (%) | 99.9 (100.0) | 94.5 (74.4) | 93.5 (65.8) |
| Redundancy | 4.7 (4.5) | 2.7 (1.8) | 2.2 (1.9) |
| Crystallization conditions | 45% Tacsimate, pH 7.0 | 1.4 M ammonium sulfate | 24% PEG 3350 |
| 2% Glucose | 0.1 M Bis Tris, pH 7.5 | 0.2 M MgCl2 | |
| 0.1 M Na Cacodilate, pH 6.5 | 0.1 M LiCl | 0.1 M MES, pH 6.0 | |
| Refinement | |||
| No. reflections | 43952 | 287620 | 148257 |
| Rwork / Rfree (%) | 16.4/20.6 | 19.0/23.0 | 16.5/21.9 |
| No. atoms | |||
| Protein | 3259 | 19122 | 12429 |
| Heme | 43 | 258 | 172 |
| Substrate | none | none | 112 |
| Solvent | 339 | 2637 | 1794 |
| Mean B value | 39.8 | 25.6 | 16.0 |
| B-factors | |||
| Protein | 39.1 | 24.3 | 14.5 |
| Heme | 28.1 | 13.3 | 8.3 |
| Substrate | n/a | n/a | 11.1 |
| Solvent | 48.8 | 33.5 | 28.0 |
| R.m.s deviations | |||
| Bond lengths (Å) | 0.025 | 0.025 | 0.024 |
| Bond angles (°) | 2.056 | 2.173 | 1.985 |
Values in parentheses are for highest-resolution shell.
Fig. 5.

Cholest-4-en-3-one binding. (A, B) Cholest-4-en-3-one in the CYP142A2 active site. The cholest-4-en-3-one molecule is shown in yellow sticks, with non-invariant residues clustered over the flat surface of the steroid ring system shown in cyan. Views in A and B differ by 90° rotation about the vertical axis. A fragment of the 2Fo-Fc electron density map (blue mesh) is contoured at 1.5σ. The distances between the heme iron and the carbon atoms of the branched methyl groups in cholest-4-en-3-one are in Angstroms. The smooth curvature of the ring system helps accommodate the bulkier side-chains in CYP124A2. (C, D) Surface representation of CYP125A1 (C) and CYP142A2 (D). Protein surfaces are colored by elements with carbon grey, oxygen red, nitrogen blue and sulfur yellow. (C) Amino acid inserts in the CYP125 sequences missing from CYP142 are mapped by magenta (102–111) and cyan (57–67), as indicated in the alignments in Fig. 6. (D) Cholest-4-en-3-one represented by VDW spheres exposes the carbonyl group (red) to the surface of CYP142A2. The exposed carbonyl group is surrounded largely by a hydrophobic area.
Cholest-4-en-3-one binding in CYP125 and CYP142
The most striking observation comes from a comparison of the CYP125A1 and CYP142A2 substrate-bound forms. Mapping the amino acid contacts within 5 Å of the substrate on sequence alignments demonstrated that topologically identical residues constitute substrate binding sites in both proteins (Fig. 6). Yet, a 10-amino acid insert (102–111) after β-strand 3 adopts a helical structure in the crystal and provides extensive contacts that completely shield the substrate from bulk solvent in CYP125A1 (Fig. 5C). The helical structure of this short fragment is stabilized by yet another insert in the CYP125 sequence (57–67), which is missing from CYP142 (Fig. 6). Loss of both inserts in CYP142 results in exposure of the carbonyl group of cholest-4-en-3-one to the surface (Fig. 5D). This remarkable difference in the active site topology suggests that in contrast to CYP125, CYP142 may operate on C3-modified steroids substrates, such as cholesteryl esters of fatty acids or cholesterol sulfate.
Fig. 6.

Sequence alignments and analysis. The amino acid sequences for M. smegmatis (Ms) and M. tuberculosis (Mt) CYP125 and CYP142 are aligned using CLUSTALW (Thompson et al., 1994). UniProt database (http://www.uniprot.org/) accession numbers are provided for each protein sequence. Secondary structure elements are assigned based on 2YOO for CYP142A2 (top) and 2X5W for CYP125A1 (bottom). Residues interacting with cholest-4-en-3one within 5 A are highlighted in green. Cross-species differences are highlighted in yellow. Residue numbering on top is according to CYP142A2; residue numbering on bottom is according to CYP125A1. Amino acid inserts in the CYP125 sequences highlighted in cyan and pink are mapped to the CYP125A1 protein surface in Fig. 5 using the same colors.
CYP125 and CYP142 are not essential for the growth of M. smegmatis on cholesterol and cholest-4-en-3-one
The roles of CYP125A3 and CYP142A2 in cholesterol catabolism were also investigated by mutagenesis of their respective coding genes, i.e., MSMEG_5995 and MSMEG_5918. The duplication rates (td) of the single mutant strains ΔCyp125 (20 h) and ΔCyp142 (21 h) were very similar to that the wild-type strain (21 h) when grown on cholesterol. However, the td of strain ΔCyp125 on cholest-4-on-3-one (17 h) increased in 4 h with respect to the wild-type and ΔCyp142 (13 h), presenting an initial lag phase of approximately 48 h. Growing on cholesterol or cholest-4-on-3-one was highly impaired for the double mutant strain ΔCyp125ΔCyp142, with higher duplication rates (25 h on cholesterol and 39 h on cholest-4-on-3-one) and longer lag phases (Fig. 7A and B). When the double mutant strain was complemented with cyp125A3 (pMVCyp125), this strain grew normally on cholesterol and cholest-4-on-3-one, giving duplication rates similar to the wild-type strain (16 h on cholesterol and 13 h on cholest-4-on-3-one) (Fig. 7C). The production of the CYP125A3 protein in the complemented double mutant strain is shown in Fig. 7D. These results suggest that, although CYP125A3 is the main enzyme responsible for the transformation of cholesterol and cholest-4-on-3-one into their oxidized metabolites, CYP142A2 supports the growth in absence of CYP125A3. The capacity of the double mutant to grow on both steroids suggests that at least one other cytochrome P450 encoded in the M. smegmatis genome is able to perform this biochemical step, or that an alternative cholesterol degradation pathway can be induced.
Fig. 7.

Growth analysis of mycobacterial mutant strains. Growth curves of WT (squares), ΔCyp125 (circles), ΔCyp142 (triangles) and ΔCyp125ΔCyp142 (diamonds) on 1.8 mM of cholesterol (A) and 1.8 mM of cholest-4-en-3-one (B) as the sole carbon and energy source. Growth was monitored by measuring the absorbance at 600 nm. Data represent averages of duplicates and error bars indicate ±1 standard deviation.(C) Growth curves of ΔCyp125ΔCyp142 complemented with empty vector pMV261 (empty symbols) and pMVCyp125 (filled symbols) on cholesterol (squares) and cholest-4-en-3-one (circles). (D) Analysis of the production of CYP125A3 in the double mutant ΔCyp125ΔCyp142 complemented with plasmid pMVCyp125 by SDS-PAGE. MW, Broad range molecular markers (Bio-Rad); lane 1, protein extract of strain ΔCyp125ΔCyp142 carrying pMV261; lane 2, protein extract of strain ΔCyp125ΔCyp142 carrying pMVCyp125. As a control, purified CYP125A3 is loaded in lane 4. The narrow indicates the position of the CYP125A3 in the 10% of polyacrylamide gel.
The mutant strains (ΔCyp125 and ΔCyp125ΔCyp142) and the wild-type strain were cultivated on cholest-4-on-3-one to analyze by LC-MS possible differences in the accumulation of intermediate compounds in the culture supernatants. The analysis of the relatives areas for each of the compounds formed during cholest-4-on-3-one catabolism showed lower accumulation of 26-hydroxycholest-4-en-3-one and cholest-4-on-3-one-26-oic acid in the cultures of strains lacking the CYP125A3 protein when compared with the wild-type strain, indicating that the single mutant strain transform cholest-4-on-3-one to 26-hydroxycholest-4-en-3-one and cholest-4-on-3-one-26-oic acid at a slower rate than the wild-type (Fig. 8). Mutation of CYP142A2 produced a slight reduction in the levels of cholest-4-on-3-one-26-oic acid, but the concentration of 26-hydroxycholest-4-en-3-one was not measurably affected. However, this reduction in the levels of the acid did not impair the growth on cholest-4-on-3-one, as shown in Fig. 7B. Finally, the levels of 26-hydroxycholest-4-en-3-one and cholest-4-on-3-one-26-oic acid were drastically reduced in the double mutant strain (ΔCyp125ΔCyp142) even at longer times of culture (>80 h) when the growth of this strain is detected.
Fig. 8.

Analysis of the compounds present in supernatants of different M. smegmatis strains. The panels show LC-MS profiles of the 26-hydroxycholest-4-en-3-one (solid line) and cholest-4-en-3-one-26-oic acid (dashed line) versus time in wild-type and mutant strains grown on 1.8 mM cholest-4-en-3-one. Data represent compound peak area/internal standard peak area.
Expression of native steroid C26-monooxygenase(s) in M. smegmatis cells
In efforts to identify the compensatory C26-monoxygenase activity we examined the endogenous levels of CYP125A3 and CYP142A2 in mutant and wild-type strains growing on cholesterol. Polyclonal antibodies raised against CYP125 and CYP142 from M. tuberculosis were used to detect the presence of these polypeptides by Western Blot analyses. As shown in Fig. 9B, CYP125A3 was detected in the wild-type strain as well as in the ΔCyp142 strain, in both of which it was present at similar levels. In contrast, CYP142A2 levels increased in the ΔCyp125 mutant strain compared with the basal level of this protein in the wild-type strain, indicating that the production of CYP142A2 is induced in the strain lacking the CYP125A3 protein.
Fig. 9.

Analysis of endogenous expression of the CYP125A3 and CYP142A2 enzymes. (A) SDS/PAGE (12.5% gel) of protein extracts (40 μg) from mycobacterial mutants and M. smegmatis cytochromes (20 ng) The positions of wild-type and different mutant strains are indicated. MW, prestained protein molecular mass marker. (B) Western-blot analysis of endogenous production of CYP125 and CYP142 in the different mutant strains. (C) Differential expression of the cyp142 (black bars) and MSMEG_4829 (grey bars) genes in the wild-type, ΔCyp125 and ΔCyp125ΔCyp142 strains cultured in cholesterol or glycerol. Transcription levels were measured using RTq-PCR as described in Experimental procedures. The values indicate the ratios of mRNA levels observed for strains growing on cholesterol relative to glycerol. Data represent averages of triplicates and error bars indicate ±1 standard deviation.
Induction of the expression of the MSMEG_5918 gene in the ΔCyp125 mutant strain was also addressed by RTq-PCR (Fig. 9C). We determined the differential expression using mRNAs from M. smegmatis mc2 155 wild-type and the ΔCyp125 mutant strain grown on cholesterol or glycerol. This analysis showed first, that the expression of the MSMEG_5918 is induced 24-fold when the cells are grown on cholesterol, and second, that expression in the ΔCyp125 mutant strain was induced 13-fold compared with the wild-type strain grown on cholesterol, demonstrating that CYP142A2 acts as a compensatory activity in the strain lacking CYP125A3.
As we have demonstrated, the double mutant strain ΔCyp125ΔCyp142 is able to grow on cholesterol, although the duplication rate is substantially lower. The search for candidate genes responsible for an additional compensatory activity in the double mutant led us to propose the MSMEG_4829 gene, which is induced 10.7 fold in the presence of cholesterol according to the transcriptomic analysis (Uhía et al., 2012). The differential expression of the MSMEG_4829 in the wild-type and ΔCyp125 and ΔCyp125ΔCyp142 mutant strains growing on cholesterol or glycerol was analyzed by RTq-PCR. Fig. 9C shows that MSMEG_4829 expression is slightly induced in the three strains (wild-type (0.8 fold), ΔCyp125 (3.6 fold) and ΔCyp125ΔCyp142 (2.2 fold) suggesting a weak correlation of this gene with cholesterol metabolism.
Discussion
Cholesterol and the related steroids are found throughout the environment and play diverse roles in vivo as precursors to vitamin D, the bile acids, sexual hormones, and as an essential component for the structural integrity of the cell membrane. Microbial degradation of the sterol side-chain has been shown to be initiated by two key cytochromes P450 in M. tuberculosis, CYP125A1 and CYP142A1, that perform sequential oxidations at C26, to form the alcohol, aldehyde and finally acid metabolites, ultimately producing the side-chain required for further degradation via the β-oxidation pathway.
Here we have characterized CYPs 125A3 and 142A2 from M. smegmatis mc2 155, each of which shows >75% identity with its respective M. tuberculosis ortholog. UV-vis spectroscopic analysis of the recombinantly expressed proteins revealed the enzymes were primarily in their catalytically functional P450 forms. CYP125A3, like CYP125A1, is predominantly HS in its resting state with a Soret peak at 393 nm, while CYP142A2, like CYP142A1, is in a LS resting state with a Soret peak at 418 nm. Both CYPs 125A3 and 142A2 show similar low micromolar affinities for type II azole inhibitors such as econazole and miconazole, although the M. tuberculosis orthologs appear to bind clotrimazole approximately three times more tightly.
CYPs 125A3 and 142A2 each bind cholesterol and 4-cholesten-3-one, which perturb the ligand field by liberating the loosely coordinated water, thus inducing a type I spin shift to give a Soret peak at 393 nm. This transition is more pronounced in CYP142A2, since CYP125A3 is already primarily HS in its resting state, which may be one cause for the difference in reported dissociation constants for the CYP125 orthologs.
CYP142A2, also shows a somewhat weaker affinity for its substrates than CYP142A1. When examined spectroscopically, steady state kinetic analysis of cholest-4-en-3-one binding reveals that both sets of orthologous enzymes have similar KM values for this substrate (Johnston et al., 2010). Although the overall rate of catalysis observed was slower for the M. smegmatis orthologs. Both CYP125A3 and CYP142A2 are capable of performing the sequential oxidation of either the cholesterol or cholest-4-en-3-one alkyl side-chain to the corresponding carboxylic acid. CYP142A2 appears to be about twice as catalytically active as CYP125A3 towards cholest-4-en-3-one, again mirroring the observed activity difference in the M. tuberculosis orthologs (Johnston et al., 2010).
Unlike the CYP125 ortholog whose crystal structures reveal an active site entirely enclosed within the protein interior, the carbonyl group of cholest-4-en-3-one in CYP142A2 is exposed to the bulk solvent. Based on this difference in topology of the active sites, we speculate that the compensatory or auxiliary roles attributed to CYP142 in the literature may be a byproduct of its distinct physiologic function of targeting a pool of sterol derivatives inaccessible to CYP125. For instance, cholesteryl esters of fatty acids, the intracellular storage and intravascular transport form of cholesterol, are abundant in human macrophages infected by mycobacteria (Kondo and Murohashi, 1971; Kondo and Kanai, 1974; 1976; Kupur and Mahadevan, 1982). We hypothesize that cholesteryl esters might be potential substrates for M. tuberculosis CYP142A1.
Compared to the M. tuberculosis counterparts, both CYP125A3 and CYP142A2 of M. smegmatis each contain three amino acid substitutions in direct proximity to the substrate in the active site (Fig. 6). The substitutions are all of bulkier residues and are clustered along the flat side of the cholest-4-en-3-one ring system in both CYP125 and CYP142. The larger size of the side-chains may cause the cholest-4-en-3-one distortion observed in CYP142A2 and perhaps explains the difference in substrate affinity between the two CYP142 homologs. Lack of conservation in these critical positions may indicate that M. smegmatis and M. tuberculosis enzymes evolved to convert steroid substrates with different structures of the tetracyclic ring system. In this regard, cholesterol, the major sterol in vertebrates, has a relatively flat and linear structure, while the additional double bond in ergosterol, the major component of cellular membranes in lower eukaryotes such as fungi and protozoa, leads to “puckering” of the ring system. Subtle differences in sterol chemical structure lead to important modifications of molecular organization in biolayers which are associated with dramatic effects in the biological function of the membranes (Dufourc, 2008). Hypothetically, these differences could have been driven by diversification of the active sites of CYP125 and CYP142 orthologs in pathogenic and environmental Mycobacterial strains. M. tuberculosis infecting human cells would have no access to natural steroids different from cholesterol or cholestenone, whereas M. smegmatis would find in the soil phytosterols and many other esters of steroids that could be used as substrates. For this reason the CYPs of M. smegmatis might be more flexible and accept a larger variety of substrates than the orthologous CYPs of M. tuberculosis.
As in M. tuberculosis, deletion of either cyp125A3 or cyp142A2 does not inhibit growth of M. smegmatis on cholesterol, and deletion of cyp125A3 causes a strong induction of cyp142A2 at both the mRNA and protein level. While the deletion of cyp125A3 leads to the reduction of both the alcohol and acid metabolites, removal of cyp142A2 only causes a modest reduction in the acid metabolite, indicating that CYP142A2 serves as a functionally redundant back up enzyme for CYP125A3, just as its ortholog does in M. tuberculosis.
Unlike M. tuberculosis, however, the Δcyp125Δcyp142 double mutant of M. smegmatis, retains its ability to grow on cholesterol, albeit with a diminished capacity. Neither the acid nor the alcohol intermediates accumulate in the double mutant, suggesting that there is an alternative cholesterol degradation pathway or that a tertiary cholesterol side-chain oxidation enzyme is encoded within the M. smegmatis genome. One possibility is the cytochrome encoded by MSMEG_4829 gene (CYP189A1), which is slightly induced in both the wild type and mutant strains of M. smegmatis grown on cholesterol, but it does not appear to have a ortholog in M. tuberculosis. Further investigation of this enzyme for its potential role in understanding the differences in cholesterol metabolism between these two species is currently underway.
In summary, we have identified a highly conserved cholesterol side-chain metabolizing pathway in M. smegmatis. Biochemical and biophysical characterization show remarkable similarities in the orthologous pairs of CYPs 125 and 142 of M. tuberculosis and M. smegmatis. However, M. smegmatis apparently contains an additional system for cholesterol metabolism when these two enzymes are absence, which M. tuberculosis does not. Given the importance of cholesterol in establishing and maintaining host infection for M. tuberculosis, understanding the subtle differences in the M. smegmatis cholesterol metabolic pathway will facilitate the use of this species as a model system for this important human pathogen.
Experimental procedures
Chemicals
Cholesterol, cholest-4-en-3-one, pregnenolone, spinach ferredoxin, spinach ferredoxin-NADP+-reductase, bovine liver catalase, glucose-6-phosphate, glucose-6-phosphate dehydrogenase, Tyloxapol, Tween 80, Tween 20 and β-methyl cyclodextrin, sucrose and catechol were purchased from Sigma-Aldrich (St. Louis, MO). Cholesta-1,4-diene-3-one was obtained from Research plus (Barnegat).
Bacterial strains, plasmids and culture conditions
The bacterial strains and plasmids used in this study are listed in Table S1. M. smegmatis mc2 155 strain was grown in 7H9 medium (Difco) containing 10% albumin-dextrose-catalase supplement (Becton Dickinson), 0.2% glycerol and 0.05% Tween-80 or on 7H10 solid agar medium with the same supplements without Tween-80. For growth in cholesterol and cholest-4-en-3-one, 7H9 minimal medium without supplement, glycerol or Tween-80 was used and both steroids were added at 1.8 mM. Stocks of 5 mM cholesterol and 5 mM cholest-4-en-3-one were dissolved in 10% of tyloxapol with magnetic agitation at 80 °C and then autoclaved. M. smegmatis was always grown at 37 °C in an orbital shaker at 250 r.p.m. E. coli strains were grown in Luria-Bertani (LB) or in Terrific Broth medium (tryptone 12 g l−1, yeast extract 24 g l−1, K2HPO4 12.5 g l−1, KH2PO4 2.3 g l−1 supplemented with 0.4% of glycerol and 2 g l−1 of bactopeptone) at 37 °C in an orbital shaker. Antibiotics were used if indicated at the following concentrations: gentamycin (10 μg ml−1 for E. coli and 5 μg ml−1 for M. smegmatis), kanamycin (50 μg ml−1 for E. coli and 20 μg ml−1 for M. smegmatis) and ampicillin (100 μg ml−1).
DNA extraction
For M. smegmatis genomic DNA extraction, 15 ml of culture was centrifuged; the pellet was resuspended in 400 μl of TE (Tris-EDTA; 10 mM Tris-HCl pH 7.5 and 1 mM EDTA) and incubated 20 min at 80 °C. After cooling to room temperature, 50 μl of 10 mg ml−1 lysozyme was added and the mixture was incubated 1 h at 37 °C. 75 μl of 10% SDS containing proteinase K (10 mg ml−1) was added before incubation for 10 min at 65 °C. 100 μl of 5 M NaCl and 100 μl of CTAB/NaCl preheated at 65 °C was added followed by 10 min incubation at 65 °C. 750 μl of chloroform/isoamyl alcohol (24:1) was added, the mixture was vortexed and centrifuged 5 min at 14,000 g. The aqueous phase was transferred to a new tube and an equal volume of phenol/chloroform/isoamyl alcohol (25:24:1) was added. The mixture was vortexed and centrifuged 5 min at 14,000 g. The aqueous phase was transferred to a new tube and the DNA was precipitated with a 0.6 volume of isopropanol. The DNA was centrifuged 15 min at 14,000 g at 4 °C, washed with 70% ethanol, centrifuged 2 min at 14,000 g and air-dried or dried using a miVac DNA concentrator (GeneVac). Finally, the DNA was resuspended in 40–100 μl of TE. Plasmidic DNA from E. coli strains was extracted using the High Pure Plasmid Purification Kit (Roche), according to the manufacturer’s instructions.
Cyp125 and cyp142 gene deletion by homologous recombination
Mutant strains ΔCyp125 and ΔCyp142 of M. smegmatis mc2155 were constructed by homologous recombination using the plasmid pJQ200x, a derivative of the suicide vector pJQ200, which does not replicate in Mycobacterium (Table S1) (Jackson et al., 2001). The strategy consists of generating two fragments of ~600 bp each, the first one containing the upstream region and a few bp of the 5’end of the gene and the second one containing the downstream region and a few bp of the 3’end of the gene, that are subsequently amplified by PCR using the primers listed in Table S2. The two fragments generated were digested with the corresponding enzymes and cloned into the plasmid pJQ200x. The resulting plasmids pJQUD5995 and pJQUD5918 were electroporated into competent M. smegmatis mc2155. Single crossovers were selected on 7H10 agar plates containing gentamycin and the presence of the xylE gene encoded in pJQ200x was confirmed by spreading catechol over the single colonies of electroporated M. smegmatis. The appearance of a yellow coloration indicates the presence of the xylE gene. Colonies were also contra-selected in 10% sucrose. A single colony was grown in 10 ml of 7H9 medium with 5 μg ml−1 gentamycin up to an optical density of 0.8–0.9. A sample of 20 μl of a 1:2 dilution was plated onto 7H10 agar plates with 10% sucrose to select for double crossovers. Potential double crossovers (sucrose-resistant colonies) were screened for gentamycin sensitivity and the absence of the xylE gene. Gentamycin-sensitives colonies were analysed by PCR to confirm the deletion of the genes MSMEG_5995 and MSMEG_5918. To construct the double knock-out strain ΔCyp125ΔCyp142, the plasmid pJQUD5918 was electroporated into competent M. smegmatis mc2155 ΔCyp125. The mutant strain was selected as explained above.
Complementation of the Δcyp125Δcyp142 mutant with cyp125A3
The coding sequence of cyp125A3 was amplified using the primers listed in Table S2 and the EcoRI-HindIII digested fragment was cloned into pMV261 (Table S1) (Stover et al., 1991) under control of the constitutive hsp60 promoter. The resulting plasmid pMVCyp125 was electroporated into competent M. smegmatis mc2155 ΔCyp125ΔCyp142 and transformants were selected at 37 °C on 7H10 plates containing 20 μg ml−1 kanamycin. The double knock-out strain carrying the plasmid pMV261 was used as a control.
Cloning, expression and purification of CYP125A3 and CYP142A2
MSMEG_5995 (CYP125A3) and MSMEG_5918 (CYP142A2) were amplified by PCR using Pfu Turbo DNA polymerase (New England BioLabs), primers listed in Table S2 and genomic DNA from M. smegmatis mc2155 as a template. The resulting DNA fragments were NdeI-HindIII digested and cloned into the pCWOri+ vector (Table S1) (Barnes et al., 1991) delivering plasmids pCW125SM and pCW142SM. E. coli DH5α cells expressing recombinant proteins were grown at 37 °C and 250 rpm in TB medium containing ampicillin 100 μg ml−1 until OD600 = 0.7–0.8. Then, expression of CYP125A3 and CYP142A2 was induced with 1 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) and 0.5 mM δ-aminolevulinic acid (δ-ALA) and the culture continued for 36 h at 25 °C and 180 rpm. Cultures were harvested by centrifugation (30 min, 5,000 x g, 4 °C) and stored frozen at -80 °C until used. Cell pellets from 4 l of culture were thawed on ice and resuspended in 400 ml of buffer A (50 mM Tris-HCl pH 7.5, 0.5 M NaCl, 0.1 mM EDTA, 20 mM imidazole) with 1 mM phenylmethylsulfonyl fluoride (PMSF). The cell suspension was incubated at 4 °C with agitation for 30 min after addition of lysozyme 0.5 mg ml−1 and DNase 0.1 mg ml−1. The cells were disrupted by sonication using a Branson sonicator (six times with 1 min bursts at 80% power, with 30 s cooling on ice between each burst). Cell debris was removed by centrifugation at 100,000 x g for 45 min at 4 °C and the soluble extract was loaded onto a Ni-NTA column previously equilibrated with buffer A. The column was washed with 400 ml of buffer B (50 mM Tris-HCl pH 7.5, 0.1 mM EDTA and 20 mM imidazole) and the proteins were eluted with 200 ml of buffer C (50 mM Tris-HCl pH 7.5, 0.1 mM EDTA and 250 mM imidazole). All the fractions eluted from the Ni-NTA column were purified by flow-through chromatography on SP-Sepharose Fast-Flow (Amersham Biosciences) and subsequent binding to Q-Sepharose Fast-Flow (Amersham Biosciences), both equilibrated with buffer 50 mM Tris-HCl pH 7.5. After washing the column with 500 ml of the same buffer, the proteins were eluted with 200 ml of 500 mM of NaCl in 50 mM Tris-HCl pH 7.5. The fractions containing P450 were pooled and concentrated to 1 mM using an AmiconUltra concentrating device (Millipore). After concentration, both CYP125A3 and CYP142A2 were dialyzed against 50 mM TrisHCl (pH 7.5) 0.1 mM EDTA buffer.
Optical absorption spectroscopy
UV-visible absorption spectra of the purified CYP125A3 and CYP142A2 proteins were recorded on a Cary UV-visible scanning spectrophotometer (Varian) using a 1-cm path-length quartz cuvette at 23 °C in 50 mM potassium phosphate buffer, pH 7.4, containing 150 mM NaCl. Formation of the ferrous carbon monoxide complex was achieved by bubbling CO gas (Airgas, San Francisco, CA) into the ferric enzyme solution for 30 s through a septum-sealed cuvette prior to the injection of 1 mM sodium dithionite using a gas tight syringe (Hamilton, Reno, NV). Difference spectra were generated by subtracting the spectrum of the ferrous deoxy form from that of its carbon monoxide complex. The concentration of P450 was determined from difference spectra using the extinction coefficient 91,000 M−1 cm−1 (Omura and Sato, 1964). Absolute absorption spectra of CYP125A3 and CYP142A2 were measured in their resting, cholesterol-bound and econazole-bound forms. The protein concentration was 3 μM and cholesterol and econazole were added at 50 μM.
Spectrophotometric binding assays
All ligand binding assays for CYP125A3 and CYP142A2 were performed by spectrophotometric titration in 50 mM potassium phosphate buffer (pH 7.4) containing 150 mM NaCl using a Cary UV-visible scanning spectrophotometer. Stock solutions of the steroids cholesterol and cholest-4-en-3-one were prepared at 10 mM in 10% (w/v) methyl-β-cyclodextrin (MβCD). To measure changes of the optical spectrum caused by the binding of the steroids, 1 ml of protein (3 μM) in buffer was placed into both chambers. After background scanning, 1 μl aliquots of ligands diluted to either 1 mM or 5 mM in 10% of MβCD were titrated into the first chamber and the same volume of MβCD was added to the second chamber to correct for carrier effects. The final concentration of MβCD was never more than 0.1%. Difference spectra were recorded from 350 to 750 nm with a scanning rate of 120 nm/min. To study the binding of antifungal azole inhibitors, stock solutions of econazole, miconazole and clotrimazole were prepared at 10 mM in methanol. 1 ml of protein (2.5 μM) in buffer was placed into both chambers. After background scanning, 0.5–2.0 μl aliquots of inhibitors diluted at 0.5–5 mM in methanol were titrated into the first chamber and the same volume of methanol was added into the second chamber to correct for solvent effects. The final concentration of methanol was always less than 1.5% v/v. Difference spectra were recorded from 350 to 750 nm with a scanning rate of 120 nm/min. To determine the KD values, titration data points were fitted to the quadratic equation using GraphPad Prism. In Equation 1, Aobs is the absorption shift determined at any ligand concentration; Amax is the maximal absorption shift obtained at saturation; KD is the apparent dissociation constant for the inhibitor-enzyme complex; [Et] is the total enzyme concentration used; [S] is the ligand concentration.
| (1) |
Steady-state kinetic studies and product analysis
Reactions were carried out in glass tubes at ambient temperature in a volume of 0.15 ml. Choles-4-en-3-one 10 mM stock solution was prepared in 10% MβCD. CYP125A3 and CYP142A2 (0.5 μM) were preincubated 5 min with substrate in 50 mM potassium phosphate (pH 7.5) containing 0.45% (w/v) MβCD, 150 mM NaCl, 10 mM MgCl2. Reactions were initiated by adding 0.3 mM NADPH, 1 μM spinach ferredoxin, 0.2 U ml−1 spinach ferredoxin-NADP+ reductase, 0.1 mg ml−1 bovine liver catalase and an NADPH-regenerating system consisting of 0.4 U ml−1 glucose-6-phosphate dehydrogenase and 5 mM glucose-6-phosphate. Aliquots of 50 μl were taken at 0, 5 and 20 min and quenched with 150 μl of acetonitrile containing 0.1% formic acid (FA) and 10 μM 1,4-cholestadiene-3-one as an internal standard. The reaction mixtures were centrifuged at 10,000 x g for 4 min. For identification of the metabolites the supernatants were directly analyzed by LC MS using a Waters Micromass ZQ coupled to a Waters Alliance HPLC system equipped with a 2695 separations module, a Waters 2487 Dual λ Absorbance detector, and a reverse phase C18 column (Waters Xterra C18 column, 3.5 μm, 2.1 × 50 mm). The column was eluted isocratically at a flow rate of 0.5 ml/min (solvent A, H2O + 0.1% formic acid; solvent B, CH3CN + 0.1% formic acid) with a gradient starting at 70% B up to 1 min and the solvents ramped up to 100% B over 12 min. The elution was maintained at 100% B up to 14 min and then ramped back to 70% B within 1 min, followed by equilibration at the same composition for 2 min before the next run. The elution was monitored at 240 nm. The mass spectrometer settings were as follows: mode, ES+; capillary voltage, 3.5 kV; cone voltage, 25 V; desolvation temperature, 250 °C. The HPLC peaks of the individual metabolites were collected separately and analyzed by MS using a quadrupole instrument in the positive ion mode. The MS/ MS analysis was performed at an Orbitrap XL instrument in the Higher Energy Collision Dissociation (HCD) mode.
For quantification of the relative amounts of products, the reactions were analyzed by HPLC using an Agilent Series 1200 HPLC system and the same reverse phase C18 column. The samples were eluted isocratically at a flow rate of 0.5 ml/min (solvent A, H2O + 0.1% formic acid; solvent B, CH3CN + 0.1% formic acid) with a gradient starting at 70% B up to 1 min and the solvents ramped up to 100% B over 12 min. The elution was maintained at 100% B up to 14 min and then ramped back to 70% B within 1 min, followed by equilibration at the same composition for 2 min before the next run. The elution was monitored at 240 nm. To determine the KM values, the data points were fitted to the quadratic equation using GraphPad Prism. In Equation 2, Kobs is the product forming rate determined at any ligand concentration; Kmax is the maximal rate; KM is the substrate concentration at which the half maximal rate is achieved; [Et] is the total enzyme concentration used; [S] is the ligand concentration.
| (2) |
Crystallization, data collection and structure determination
Prior to crystallization, the CYP125A3 and CYP142A2 proteins were diluted to 0.2 mM in 10 mM Tris-HCl, pH 7.5 buffer. Crystallization conditions in each case were determined using commercial high-throughput screening kits available in deep-well format (Hampton Research), a nanoliter drop-setting Mosquito robot (TTP LabTech) operating with 96-well plates, and a hanging drop crystallization protocol. Crystals were further optimized in 96-well or 24-well plates for diffraction data collection. Prior to data collection, all crystals were cryo-protected by plunging them into a drop of reservoir solution supplemented with 20–25% ethylene glycol, then flash frozen in liquid nitrogen. In both substrate-free structures determined in this work, cryo-protecting agent ethylene glycol was bound as an iron sixth ligand instead of naturally occurring ligand, a water molecule. Similarly, binding of tetraethylene glycol to the heme iron was reported for M. tuberculosis CYP142A1 (Ouellet, et al., 2010b).
Diffraction data were collected at 100–110 K at beamline 8.3.1, Advanced Light Source, Lawrence Berkeley National Laboratory, USA. Data indexing, integration, and scaling were conducted using MOSFLM (Leslie, 1992) and the programs implemented in the ELVES software suite (Holton and Alber, 2004). The crystal structures were initially determined by molecular replacement using the structures of M. tuberculosis CYP125A1 (PDB ID 2X5W) or CYP142A1 (PDB ID 2XKR) as search models. The M. smegmatis structures were built using COOT (Emsley and Cowtan, 2004) and refined using REFMAC5 (Collaborative Computational Project, 1994; Murshudov et al, 1997) software. Data collection and refinement statistics are shown in Table 3.
In vivo enzymatic assays
To analyze the differences in the accumulation of intermediate compounds between wild-type and knock-out strains, culture supernatants were analyzed by LC-MS. M. smegmatis strains were grown on cholest-4-en-3-one at 37 °C for 120 h. Enzyme assay aliquots (2 ml) were extracted twice at various extents of reaction (0, 24, 48, 72, 96 and 120 h) with an equal volume of chloroform. This chloroform fraction was subsequently dried under vacuum. Prior to extraction with chloroform, 50 μl of a 20 mg ml−1 pregnenolone chloroform solution was added to the aliquots as an internal standard. The dried fractions were dissolved in 300 μl of acetonitrile and 25 μl were subjected to chromatographic analysis by LC-MS. Experiments were carried out using an LXQ Ion Trap Mass Spectrometer, equipped with an atmospheric pressure chemical ionization source, and interfaced to a Surveyor Plus LC system (all from Thermo Electron, San Jose, CA, USA). Data were acquired with a Surveyor Autosampler and MS Pump and analysed with Xcalibur Software (from Thermo-Fisher Scientific, San Jose, CA, USA). All the experiments were carried out with the following interface parameters: capillary temperature 275 °C, 425 °C for gas temperature in the vaporizer, capillary voltage 39 V, corona discharge needle voltage 6 kV, source current 6 mA and 20 eV for the collision induced dissociation. High-purity nitrogen was used as nebulizer, sheath and auxiliary gas. MS analysis was performed both in full scan (by scanning from m/z 50 to m/z 690) and in selected ion monitoring (SIM) mode by scanning the all the daughter ions of the products involved in cholest-4-en-3-one degradation in positive ionization mode. The quantification was performed from parent mass of cholestenone (m/z 385.4). The specificity was obtained by following the specific fragmentations of all compounds. Chromatographic separation was performed on a Tracer Excel 120 ODSB C18 (4.6 mm x 150 mm, particle size 5 mm) column (Teknokroma, Barcelona, Spain). The chromatography was performed using acetonitrile/water (90/10, v/v) and acetonitrile/isopropanol (85/15, v/v) as mobile phases A and B respectively (flow 1 ml min−1). Gradient was as follows: 100% A for 5 min, increasing to 40% B in 10 min, reaching to 100% B at minute 35, hold for 1 min and return 100% A in 1 min. The HPLC column was re-equilibrated for 6 min in initial conditions. The valve was set to direct LC flow to the mass spectrometer from 1.2 to 42 min, with the remaining LC eluent diverted to waste. Calibration standards from 0.1 mM up 2.5 mM for cholest-4-en-3-one were prepared in 10% tyloxapol. Extraction of analytes was carried out in the same way described below in sample preparation.
Detection of endogenous expression of CYP125A3 and CYP142A2 in wild-type and knockout strains
Western blots were carried out to detect CYP125A3 and CYP142A2 proteins from the whole cell lysate of wild-type, simple and double knock-out strains. Cells were precultured in 7H9 medium for 48 h and then washed in NaCl 0.85% plus Tween 80 0.05%. Cells were synchronized to an A600 of 0.05 and cultured in the presence of cholesterol (1.8 mM) as the sole carbon and energy source until they reached an absorbance of A600 = 1–1.2. Cells (3 ml) were harvested by centrifugation and kept frozen at -80 °C until used. The pellets were resuspended on ice with 0.5 mL of 50 mM Tris-HCl (pH 7.5) and were disrupted by sonication. Soluble extracts were obtained by centrifugation at 14,000 g for 15 min at 4 °C. Total protein concentration was determined by using the Bradford method (Bradford, 1976) with bovine serum albumin as standard. Western blot analysis was performed according to standard protocols. One gel was stained with Coomassie blue to ensure the equal loading in the different wells. Two identical membranes were probed with polyclonal antibodies against CYP125A1 and CYP142A1 from M. tuberculosis H37Rv (Johnston et al., 2010). Both antibodies were previously incubated with double knockout ΔCyp125ΔCyp142 cell extract (v/v) overnight at 4 °C in rotational agitation to avoid interaction with unspecific proteins.
RNA extraction
RNA for RTq-PCR was extracted from 15 ml of cultured M. smegmatis mc2155 wild-type, ΔCyp125 and ΔCyp125ΔCyp142 strains growing in 1.8 mM of cholesterol-tyloxapol or glycerol-tyloxapol media as described previously (Uhía et al., 2011). RNA Quantity was measured using a NanoPhotometerRPearl. Implen, GmbH (Munich, Germany)
Real-time quantitative PCR (RTq-PCR)
RNA was treated with the Dnase I and Removal treatment kit (Ambion) according to manufacturer’s instructions. For reverse transcription, a volume of 20 μl of reaction containing 1 μg of purified RNA, 10 mM DTT, 0.5 mM dNTPs, 200 units of SuperScript II Reverse Transcriptase (Invitrogen) and 5 mM pd(N)6 random hexamer 5′ phosphate (Amersham Biosciences) was used. RNA and random primers were first heated to 65 °C for 5 min for primer annealing and after snap cooling on ice the remaining components were added and then incubated at 42 °C for 2 h. Finally, the reactions were incubated at 70 °C for 15 min. To hydrolyze the remaining RNA after the reverse transcription, 7 μl of 1 M NaOH and 7 μl of 0.5 M EDTA were added and the reactions were incubated at 65 °C for 15 min. 17 μl of 1 M HEPES pH 7.5 was then added to neutralize the solution. The cDNA obtained was purified using the Geneclean Turbo kit (MP Biomedicals) and the concentration was measured using a NanoPhotometerRPearl. Implen, GmbH (Munich, Germany). Real-time quantitative polymerase chain reactions for the analysis of the expression of single genes were performed using an iQ5 Multicolor Real-Time PCR Detection System (Bio-Rad). Samples containing 5 ng of cDNA, 0.2 mM of each primer (the oligonucleotides used are shown in Table S2) and 12.5 μl of SYBRR® Green PCR Master Mix (Applied Biosystems) in 25 μl of total volume were used. The reactions were denatured at 95 °C for 30 s before cycling for 40 cycles of 95 °C for 30 s, 60 °C for 30 s and 72 °C for 30 s. Each gene was measured in triplicate in three independent. Data were obtained and analyzed with the iQ5 Optical System Software (2.0) (Bio-Rad). The relative amount of mRNA for each gene was determined following the 2-Δ ΔCt method (Livak and Schmittgen, 2001), using the mRNA levels of sigA (MSMEG_2758) as internal control (Gomez and Smith, 2000).
Bionformatic analyses
Nucleotide and protein sequence data were obtained from the Comprehensive Microbial Resource server (http://cmr.jcvi.org/tigr-scripts/CMR/CmrHomePage.cgi) from Craig Venter’s Institute, and sequence analysis was performed using the BLAST package at NCBI (National Center for Biotechnology Information server http://blast.ncbi.nlm.nih.gov). Sequence alignments were carried out using ClustalW2 (Thompson et al., 1994), at the EMBL-EBI server (http://www.ebi.ac.uk/Tools/).
Supplementary Material
Acknowledgments
We thank the staff members of beamline 8.3.1, James Holton, George Meigs and Jane Tanamachi, at the Advanced Light Source at Lawrence Berkeley National Laboratory, for assistance with data collection. We also want to thank Dr José Antonio Aínsa and Dr Carlos Martín from the Universidad de Zaragoza for the kind gift of the M. smegmatis mc2155 strain and the plasmid pMV261, and Dr Iria Uhía and Dr Santhosh Sivaramakrishnan for their help and advice during the development of this work. This work was supported by grants from the Spanish Ministry of Science and Innovation BFU2006-15214-C03-01 and BFU2009-11545-C03-03 (to J.L.G), an FPU predoctoral fellowship from the Spanish Ministry of Education and Science (to E.G.F.), NIH grants AI074824 (to P.O.M.), and AI095437 and GM078553 (to L.M.P.). The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231.
Footnotes
The atomic coordinates and structure factors (PDB ID codes 2APY, 3ZBY, and 2YOO) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
References
- Barnes HJ, Arlotto MP, Waterman MR. Expression and enzymatic activity of recombinant cytochrome P450 17 α-hydroxylase in Escherichia coli. Proc Natl Acad Sci USA. 1991;88:5597–5601. doi: 10.1073/pnas.88.13.5597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Capyk JK, Kalscheuer R, Stewart GR, Liu J, Kwon H, Zhao R, Okamoto S, Jacobs WR, Eltis LD, Mohn WW. Mycobacterial cytochrome P450 125 (Cyp125) catalyzes the terminal hydroxylation of C27 steroids. J Biol Chem. 2009;284:35534–35542. doi: 10.1074/jbc.M109.072132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang JC, Miner MD, Pandey AK, Gill WP, Harik NS, Sassetti CM, Sherman DR. igr genes and Mycobacterium tuberculosis cholesterol metabolism. J Bacteriol. 2009;191:5232–5239. doi: 10.1128/JB.00452-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang YR, Ismail W, Heintz D, Schaeffer C, Van Dorsselaer A, Fuchs G. Study of anoxic and oxic cholesterol metabolism by Sterolibacterium denitrificans. J Bacteriol. 2008;190:905–914. doi: 10.1128/JB.01525-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole ST, Brosch R, Parkhill J, Garnier T, Churcher C, Harris D, Gordon SV, Eiglmeier K, Gas S, Barry CE, III, Tekaia F, Badcock K, Basham D, Brown D, Chillingworth T, Connor R, Davies R, Devlin K, Feltwell T, Gentles S, Hamlin N, Holroyd S, Hornsby T, Jagels K, Krogh A, McLean J, Moule S, Murphy L, Oliver K, Osborne J, Quail MA, Rajandream MA, Rogers J, Rutter S, Seeger K, Skelton J, Squares R, Squares S, Sulston JE, Taylor K, Whitehead S, Barrell BG. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature. 1998;393:537–544. doi: 10.1038/31159. [DOI] [PubMed] [Google Scholar]
- Collaborative Computational Project, Number 4. Acta Crysallogr D. 1994;50:760–763. [Google Scholar]
- Denisov IG, Makris TM, Sligar SG, Schlichting I. Structure and Chemistry of Cytochrome P450. Chem Rev. 2005;105:2253–2278. doi: 10.1021/cr0307143. [DOI] [PubMed] [Google Scholar]
- Dermer J, Fuchs G. Molybdoenzyme that catalyzes the anaerobic hydroxylation of a tertiary carbon atom in the side-chain of cholesterol. J Biol Chem. 2012;287:36905–36916. doi: 10.1074/jbc.M112.407304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driscoll MD, McLean KJ, Levy C, Mast N, Pikuleva IA, Lafite P, Rigby SE, Leys D, Munro AW. Structural and biochemical characterization of Mycobacterium tuberculosis CYP142: evidence for multiple cholesterol 27-hydroxylase activities in a human pathogen. J Biol Chem. 2010;285:38270–38282. doi: 10.1074/jbc.M110.164293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dufourc EJ. Sterols and membrane dynamics. J Chem Biol. 2008;1:63–77. doi: 10.1007/s12154-008-0010-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Gomez M, Smith I. Determinants of mycobacterial gene expression. In: Hatfull GF, Jacobs WRJ, editors. Molecular Genetics of Mycobacteria. Washington, DC: American Society for Microbiology Press; 2000. pp. 111–129. [Google Scholar]
- Gu S, Chen J, Dobos KM, Bradbury EM, Belisle JT, Chen X. Comprehensive proteomic profiling of the membrane constituents of a Mycobacterium tuberculosis strain. Mol Cell Proteomics. 2003;2:1284–1296. doi: 10.1074/mcp.M300060-MCP200. [DOI] [PubMed] [Google Scholar]
- Holton J, Alber T. Automated protein crystal structure determination using ELVES. Proc Natl Acad Sci U S A. 2004;101:1537–1542. doi: 10.1073/pnas.0306241101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horinouchi S, Ishizuka H, Beppu T. Cloning, nucleotide sequence, and transcriptional analysis of the NAD(P)-dependent cholesterol dehydrogenase gene from a Nocardia sp. and its hyperexpression in Streptomyces spp. Appl Environ Microbiol. 1991;57:1386–1393. doi: 10.1128/aem.57.5.1386-1393.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson SA, McLean KJ, Munro AW, Abell C. Mycobacterium tuberculosis cytochrome P450 enzymes: a cohort of novel TB drug targets. Biochem Soc Trans. 2012;40:573–579. doi: 10.1042/BST20120062. [DOI] [PubMed] [Google Scholar]
- Jackson M, Camacho LR, Gicquel B, Guilhot C. Gene replacement and transposon delivery using the negative selection marker sacB. In: Parish T, Stoker NG, editors. Mycobacterium tuberculosisProtocols. Totowa, NJ, USA: Humana Press Inc; 2001. pp. 59–75. [DOI] [PubMed] [Google Scholar]
- Johnston JB, Ouellet H, Ortiz de Montellano PR. Functional redundancy of steroid C26-monooxygenase activity in Mycobacterium tuberculosis revealed by biochemical and genetic analyses. J Biol Chem. 2010;285:36352–36360. doi: 10.1074/jbc.M110.161117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston JB, Ouellet H, Podust LM, Ortiz de Montellano PR. Structural control of cytochrome P450-catalyzed ω-hydroxylation. Arch Biochem Biophys. 2011;507:86–94. doi: 10.1016/j.abb.2010.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kendall SL, Withers M, Soffair CN, Moreland NJ, Gurcha S, Sidders B, Frita R, Ten Bokum A, Besra GS, Lott JS, Stoker NG. A highly conserved transcriptional repressor controls a large regulon involved in lipid degradation in Mycobacterium smegmatis and Mycobacterium tuberculosis. Mol Microbiol. 2007;65:684–99. doi: 10.1111/j.1365-2958.2007.05827.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo E, Kanai K. Further studies on the increase in cholesterol ester content of the lungs of tuberculous mice. Jpn J Med Sci Biol. 1974;27:59–65. doi: 10.7883/yoken1952.27.59. [DOI] [PubMed] [Google Scholar]
- Kondo E, Kanai K. Accumulation of cholesterol esters in macrophages incubated with mycobacteria in vitro. Jpn J Med Sci Biol. 1976;29:123–137. doi: 10.7883/yoken1952.29.123. [DOI] [PubMed] [Google Scholar]
- Kondo E, Murohashi T. Esterification of tissue cholesterol with fatty acids in the lungs of tuberculous mice. Jpn J Med Sci Biol. 1971;24:345–356. doi: 10.7883/yoken1952.24.345. [DOI] [PubMed] [Google Scholar]
- Kupur IG, Mahadevan PR. Cholesterol metabolism of macrophages in relation to the presence of Mycobacterium leprae. J Biosci. 1982;4:307–316. [Google Scholar]
- Leslie AGW. Recent changes to the MOSFLM package for processing film and image plate data. Joint CCP4 ESF-EAMCB Newslett Protein Crystallogr. 1992;26 [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2Δ ΔCt method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- McLean K, Hans M, Munro AW. Cholesterol, an essential molecule: diverse roles involving cytochrome P450 enzymes. Biochem Soc Trans. 2012;40:587–593. doi: 10.1042/BST20120077. [DOI] [PubMed] [Google Scholar]
- McLean KJ, Lafite P, Levy C, Cheesman MR, Mast N, Pikuleva IA, Leys D, Munro AW. The structure of Mycobacterium tuberculosis CYP125: molecular basis for cholesterol binding in a P450 needed for host infection. J Biol Chem. 2009;284:35524–35533. doi: 10.1074/jbc.M109.032706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miner MD, Chang JC, Pandey AK, Sassetti CM, Sherman DR. Role of cholesterol in Mycobacterium tuberculosis infection. Indian J Exp Biol. 2009;47:407–11. [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- Nesbitt NM, Yang X, Fontán P, Kolesnikova I, Smith I, Sampson NS, Dubnau E. A Thiolase of Mycobacterium tuberculosis is required for virulence and production of androstenedione and androstadienedione from cholesterol. Infect Immun. 2010;78:275–282. doi: 10.1128/IAI.00893-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omura T, Sato R. The carbon monoxide-binding pigment of liver microsomes. II. Solubilization, purification, and properties. J Biol Chem. 1964;239:2379–85. [PubMed] [Google Scholar]
- Oppermann U, Filling C, Hult M, Shafqat N, Wu X, Lindh M, Shafqat J, Nordling E, Kalberg Y, Persson B, Jörnvall H. Short-chain dehydrogenases/reductases (SDR): the 2002 update. Chem Biol Interact. 2003;143–144:247–253. doi: 10.1016/s0009-2797(02)00164-3. [DOI] [PubMed] [Google Scholar]
- Ouellet H, Johnston JB, Ortiz de Montellano PR. The Mycobacterium tuberculosis cytochrome P450 system. Arch Biochem Biophys. 2010a;493:82–95. doi: 10.1016/j.abb.2009.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouellet H, Guan S, Johnston JB, Chow ED, Kells PM, Burlingame AL, Cox JS, Podust LM, Ortiz de Montellano PR. Mycobacterium tuberculosis CYP125A1, a steroid C27 monooxygenase that detoxifies intracellularly generated cholest-4-en-3-one. Mol Microbiol. 2010b;77:730–742. doi: 10.1111/j.1365-2958.2010.07243.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouellet H, Johnston JB, Ortiz de Montellano PR. Cholesterol catabolism as a therapeutic target in Mycobacterium tuberculosis. Trends Microbiol. 2011;19:530–539. doi: 10.1016/j.tim.2011.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosłoniec KZ, Wilbrink MH, Capyk JK, Mohn WW, Ostendorf M, van der Geize R, Dijkhuizen L, Eltis LD. Cytochrome P450 125 (CYP125) catalyses C26-hydroxylation to initiate sterol side-chain degradation in Rhodococcus jostii RHA1. Mol Microbiol. 2009;74:1031–43. doi: 10.1111/j.1365-2958.2009.06915.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sassetti CM, Rubin EJ. Genetic requirements for mycobacterial survival during infection. Proc Natl Acad Sci USA. 2003;100:12989–94. doi: 10.1073/pnas.2134250100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sivaramakrishnan S, Ouellet H, Matsumura H, Guan S, Moénne-Loccoz P, Burlingame AL, Ortiz de Montellano PR. Proximal ligand electron donation and reactivity of the cytochrome P450 ferric-peroxo anion. J Am Chem Soc. 2012;134:6673–6684. doi: 10.1021/ja211499q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stover CK, de la Cruz VF, Fuerst TR, Burlein JE, Benson LA, Bennett LT, Bansal GP, Young JF, Lee MH, Hatfull GF, Snapper SB, Barletta RG, Jacobs WR, Bloom BR. New use of BCG for recombinant vaccines. Nature. 1991;351:456–460. doi: 10.1038/351456a0. [DOI] [PubMed] [Google Scholar]
- Thomas ST, Vander Ven BC, Sherman DR, Russell DG, Sampson NS. Pathway profiling in Mycobacterium tuberculosis: elucidation of cholesterol-derived catabolite and enzymes that catalyze its metabolism. J Biol Chem. 2011;286:43668–43678. doi: 10.1074/jbc.M111.313643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhía I, Galán B, Kendall SL, Stoker NG, García JL. Cholesterol metabolism in Mycobacterium smegmatis. Environ Microbiol Reports. 2012;4:168–182. doi: 10.1111/j.1758-2229.2011.00314.x. [DOI] [PubMed] [Google Scholar]
- Uhía I, Galán B, Morales V, García JL. Initial step in the catabolism of cholesterol by Mycobacterium smegmatis mc2155. Environ Microbiol. 2011;13:943–959. doi: 10.1111/j.1462-2920.2010.02398.x. [DOI] [PubMed] [Google Scholar]
- van der Geize R, Dijkhuizen L. Harnessing the catabolic diversity of rhodococci for environmental and biotechnological applications. Curr Opin Microbiol. 2004;7:255–261. doi: 10.1016/j.mib.2004.04.001. [DOI] [PubMed] [Google Scholar]
- Wilbrink MH, Petrusma M, Dijkhuizen L, van der Geize R. FadD19 of Rhodococcus rhodochrous DSM43269, a steroid-coenzyme A ligase essential for degradation of C-24 branched sterol side-chains. Appl Environ Microbiol. 2011;77:4455–4464. doi: 10.1128/AEM.00380-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Dubnau E, Smith I, Sampson NS. Rv 1106c from Mycobacterium tuberculosis is a 3β-hydroxysteroid dehydrogenase. Biochemistry. 2007;46:9058–9067. doi: 10.1021/bi700688x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.