

Abstract
Background
We have previously shown that ethanol increases cellular apoptosis to developing neurons via the effects on oxidative stress of neurons directly and via increasing production of microglia-derived factors. In order to study further the mechanism of ethanol action on neuronal apoptosis, we determined the effects of two well known PKA activators, dibutyryl cAMP (dbcAMP) and brain-derived neurotrophic factor (BDNF), on ethanol activated oxidative stress and apoptotic processes in the hypothalamic neurons in the presence and absence of microglial cells influence.
Methods
In enriched neuronal cells from fetal rat hypothalami treated with ethanol or with conditioned medium from ethanol-treated microglia, we measured cellular apoptosis by the free nucleosome assay and the levels of cAMP, BDNF, O2−, reactive oxygen species (ROS), nitrite, glutathione (GSH) and catalase following treatment with ethanol or ethanol-treated microglial culture conditioned medium. Additionally, we tested the effectiveness of dbcAMP and BDNF in preventing ethanol or ethanol-treated microglial conditioned medium on cellular apoptosis and oxidative stress in enriched hypothalamic neuronal cell in primary cultures.
Results
Neuronal cell cultures following treatment with ethanol or ethanol-activated microglial conditioned medium showed decreased production levels of cAMP and BDNF. Ethanol also increased apoptotic death as well as oxidative status, as demonstrated by higher cellular levels of oxidants but lower levels of antioxidants, in neuronal cells. These effects of ethanol on oxidative stress and cell death were enhanced by the presence of microglia. Treatment with BDNF or dbcAMP decreased ethanol or ethanol-activated microglial conditioned medium-induced changes in the levels of intracellular free radicals, ROS and O2, nitrite, GSH and catalase.
Conclusions
These data support the possibility that ethanol by acting directly and via increasing the production of microglial-derived factors reduces cellular levels of cAMP and BDNF to increase cellular oxidative status and apoptosis in hypothalamic neuronal cells in primary cultures.
INTRODUCTION
Induction of apoptosis by ethanol has been implicated in the complications related to fetal alcohol syndrome. Many brain regions are particularly susceptible to ethanol during the prenatal period of development, which represents a dynamic period of growth and differentiation. Prenatal administration of ethanol reduces the number of neurons in various brain regions including the hippocampus, cerebral cortex, cerebellum, olfactory bulb, and hypothalamus (Chen et al., 2006; De et al; 1994; Goodlett et al., 1991; Miller and Potempa, 1990; West et al., 1984). Within the hypothalamus, prenatal ethanol has been shown to produce functional abnormalities of several neuronal populations including β-endorphin (Sarkar et al., 2007), corticotropin releasing hormone (Lee et al., 2000), a-melanocyte stimulating hormone, neuropeptide y, galanin (Barson et al., 2010), orexin 1 (Stettner et al., 2011), arginine vasopressin (Bird et al., 2006), vasoactive intestinal peptide (Rojas et al., 1999) and luteinizing hormone releasing hormone producing neurons (Scott et al., 1995). Many of the functional defects of the hypothalamus in prenatal ethanol-exposed animals are related to the loss of the neuronal cell population (Baker and Shoemaker, 1995; De et al., 1994; Sarkar et al., 2007). Recently, a role of oxidative stress was demonstrated in the mechanism of ethanol activated apoptotic neuronal death in the hypothalamus (Boyadjieva and Sarkar, 2012). Furthermore, it has been shown that ethanol induces oxidative stress in hypothalamic neurons by increasing the cellular production of O2−, ROS and nitrite while decreasing the level of GSH and the cellular activity of GSH-Px, catalase and SOD activities via activation of microglial-derived factor(s). Tumor necrosis factor-α (TNF-α) is identified as one of microglial-derived factors that may mediate ethanol’s apoptotic action on hypothalamic neuronal cells (Boyadjieva and Sarkar, 2010). One of the mechanisms by which TNF-α induces neuronal demise is by creating an inflammatory environment, which triggers signaling cascade for neuronal apoptotic process. In traumatic brain injury model, it has been demonstrated that the cAMP-PKA signaling cascade is downregulated in association with an increase of TNF-α (Atkins et al., 2007). Additionally, nuclear factor-kappaB (NF-kB), which mediates TNF-α actions on neuronal apoptosis, is suppressed by the PKA activators at the transcriptional levels (Takahashi et al., 2002). Hence, we determined the effects of two well known PKA activators, dbcAMP and BDNF, on ethanol activated oxidative stress and apoptotic processes in the hypothalamic neurons in the presence and absence of microglial cells in primary cultures.
MATERIALS AND METHODS
Animal Use
Pregnant Sprague-Dawley female rats were obtained from Charles River Laboratories (Wilmington, MA) and were used as the source of fetal rat brains for hypothalamic cell cultures. Animal surgery and care were performed in accordance with institutional guidelines and complied with the National Institutes of Health policy.
Enriched hypothalamic neuronal cell cultures
Primary cultures of fetal hypothalamic neuronal cell cultures were prepared from the mediobasal part of the hypothalamus (containing neuroendocrine neurons, including beta-endorphin, dopamine, thyrotropin-releasing hormone and growth hormone-releasing hormone; Brown, 1998). In brief, pregnant rats at 18 to 20 days of gestation were sacrificed, fetuses were removed by aseptic surgical procedure, brains from the fetuses were immediately removed, hypothalami were separated and placed in ice-cold Hanks’ balanced salt solution containing antibiotic solution (100 U/ml penicillin, 100 μg/ml streptomycin, and 250 ng/ml amphotericin B), 0.1% bovine serum albumin, and 200 μM ascorbic acid (all from Sigma-Aldrich, St. Louis, MO). The block of mediobasal hypothalamic tissue consisted of approximately 1 mm rostral to the optic chiasma and just caudal to the mammillary bodies, laterally to the hypothalamic sulci, and dorsally to 2 mm deep. The hypothalamic cells were washed and then incubated at 37°C for 5 min using the same medium. After dispersion, the cells were plated at a density of 3.0 × 106 cells per 25-mm2 flask and at a density of 1.0 × 106 cells per well in a 24-well plate. Both the flask and plate were coated with polyornithine at a concentration 100 μg/ml and then incubated for 3 h. The cells were maintained in Dulbecco’s modified Eagle’s medium with 10% fetal calf serum at 37°C and 7.5% CO2 in a humidified water-jacketed incubator for 2 days. On day 2, the medium was replaced with HDME containing 10% fetal calf serum, 33.6 μg/ml uridine and 13.2 μg/ml 5-fluorodeoxiuridine (Sigma-Aldrich, St. Louis, MO) to stop the overgrowth of glial cells. On day 4, the medium was replaced with serum-free, chemically defined HDME medium containing serum supplement (SS; consisting of 30 nM selenium, 20 nM progesterone, 1 μM iron-free human transferrin, 100 μM putrescine, and 5 μg/ml insulin). Cells were maintained for the next two days with this medium. By this time, the cultures were approximately 85 – 90% neurons, as determined by MAP2 positivity (Boyadjieva and Sarkar, 2010). Hence, these cultures were considered enriched neuronal cell cultures.
Microglial cell cultures
Microglia cells were prepared from E19 rat cerebral cortex using a method modified from that described previously (McCarty and de Vellis, 1980). Cortices were dissociated by passing through a 70-μm Nitex mesh and plated at 2 ×105 cells/cm2. Cultures were fed every four days with DMEM/MEM/Hams 12 (DMEMF12) in a 4:5:1 ratio with 10% fetal calf serum (FCS). On day 12, the culture was shaken on a rotary shaker at 800 rpm for 1 h. The suspended cells were plated on uncoated T25 flasks and incubated for 1 h at 37°C. Then the medium containing suspended cells was discarded and adherent cells were fed with DMEMF12 with 10% FCS for 3 days to develop the microglial culture prior to experimentation. To confirm the microglia positive cells, the culture was labeled with microglial marker (antibody Annexin 1) or the astrocyte marker glial fibrillary acidic protein (GFAP). The cultures with positive cells for Annexin 1 were considered microglial cells. Microglial cells were maintained in DMEMF12 with serum supplement in 24 well plates (1×105) and treated for 24 h with 50 mM (0.3% v/v) or vehicle. Then, the medium from cultured microglia cells was collected and centrifuged and the supernatants were used to treat enriched neuronal cells in cultures.
Ethanol levels in culture media samples were measured using an AMI Analyser (Analox Instrument; Lunenburg, MA) and aqueous ethanol standards (Analox). Measurement of ethanol level in the conditioned media at various time periods after ethanol treatment indicated very low to undetectable levels of ethanol at 24 h (Fig. 1A).
Figure 1.
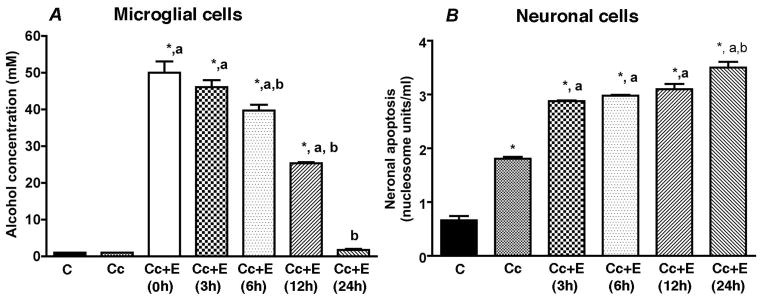
Changes in the ethanol concentration (A) and neurotoxic potency (B) of microglial cell culture conditioned media at different times following a treatment with 50 mM ethanol. Enriched fetal hypothalamic microglial cell cultures (50, 000 cells/well) were exposed to no ethanol (Cc) or a 50 mM dose of ethanol (Cc+E). Media samples were collected at various time points (0, 3, 6, 12 and 24 h) to measure ethanol concentrations or neurotoxic potencies. Neurotoxic potencies of microglial cell conditioned media were determined by adding this media in enriched fetal hypothalamic neuronal cell cultures (50 000 cells/well) and determining their ability to induce cellular apoptosis as measured by Nucleosome assay. N=5. *P<0.05, as compared to C. a, P<0.05, as compared to Cc. b, P<0.05, as compared to rest of Cc+E-treatment groups.
Culture experiments
Previously, we have shown that ethanol dose-dependently increases cellular apoptosis to developing neurons, and that a 50 mM dose of ethanol produced the maximum effect on neuronal apoptosis (Boyadjieva et al., 2010). Hence, we employed the 50 mM dose of ethanol in all the experiments described here. We also used a 24 h treatment period, since this time period was maximally effective in altering oxidative status of neurons and their apoptosis following treatment with 50 mM dose of ethanol (Boyadjieva et al., 2012; Fig. 1b).
In study1, enriched neuronal cells (50,000 cells/well) or microglial cells (50,000 cells/well) in 24 wells were plated for 2 days in cultures and then a group of cultures was exposed to media only (control), 50 mM ethanol only, microglia conditioned medium only without (Cc) or 50 mM ethanol activated microglial conditioned media (Cc+E) and incubated for 24 h. Cellular extracts were prepared to measure cAMP levels and media was used to determine the levels of BDNF using ELISA.
In study 2, enriched neuronal cells (50 000 cells/well) in 24 wells were plated for 2 days in cultures and then a group of cultures was exposed to media only (control), 50 mM ethanol only, 50 mM ethanol and various doses of dbcAMP (0.1 μM, 1.0 μM or 10.0 μM) or BDNF (0.1, 1.0 or 10 ng/ml) for a period of 24 h. These cultures were used to determine the dose-response effects of cAMP and BDNF on cellular apoptosis, as measured by nucleosome assays. Data of these experiments revealed that a 1.0 μM dose of dbcAMP and a 10 ng/ml of BDNF were able to completely block ethanol neurotoxic effects (Fig. 1). Hence, we used these doses of cAMP and BDNF in the following studies.
In studies 3 and 4, we treated enriched neuronal cells with vehicle, ethanol (E; 50 mM) and dbcAMP (cAMP; 1.0 μM) or BDNF (10 ng/ml) with or without ethanol (50 mM) for a period of 24 h, and then measured the cell contents of nucleosome (cellular markers of apoptosis), O2−, ROS and nitrite (oxidative molecules), GSH (antioxidative molecule) and catalase (enzymes produces antioxidative molecules).
In studies 5 and 6, a group of enriched neuronal cultures was exposed to vehicle (C), microglia conditioned medium only without (Cc) or with 1.0 μM cAMP (Cc + cAMP) or 10 ng/ml BDNF (Cc + BDNF), ethanol (50 mM)-activated microglia conditioned medium without (Cc + E) or with 1.0 μM cAMP (Cc +E + cAMP) or with 10 ng/ml BDNF (Cc + E + BDNF). The cell extracts were collected and used for the measurement of cell contents of nucleosome, O2−, ROS and nitrite, GSH and catalase.
We have used 2 to 3 cultures/treatment and repeated each experiment 3 times. Cultures with any cell number abnormality or assay errors were not included in the experiments
Measurement of apoptosis
The cellular apoptosis was measured using nucleosome ELISA (Calbiochem, USA). The ELISA is a specific for nucleosomes containing single- or double-stranded DNA. It is onestep sandwich ELISA, colorimetric assay. Cell lysis, followed by immunochemical determination of histone-complexed DNA fragments in a microplate wells are basic steps in the method. After incubating cells with or without ethanol and with or without BDNF or dbcAMP, the cells were incubated with lysis buffer (1.0 ml) for 10 min and the intact nuclei were pelleted by centrifugation. The aliquot of the supernatant (cell lysate) was taken to determine the amount of apoptotic nucleosomes by following the instruction of manufacturer.
Determination of intracellular O2− production
Intracellular production of O2− was determined by the reduction of nitroblue tetrazolium (Calbiochem, Billerica, MA) as described previously (McDonald et al., 1997) with some modifications. Enriched hypothalamic neurons in culture (50 000 cells/well in 24 wells) were treated with or without ethanol (25, 50 or 100 mM) for a period of 24 or 48 h. At the final point of the experiment, the nitroblue-tetrazolium (1μg/ml) was added and the cells were incubated for 60 min. After treatment, microscopic examination verified the generation of insoluble formazan as dark purple granules. The medium was removed, and the formazan was dissolved in dimethyl-sulfoxide. The lysates were transferred to a 96-well plate, and the absorbance at 570 nm was measured with a spectrophotometer. Each sample was run in duplicates in the assay.
Intracellular reactive oxygen species assay
The production of intracellular reactive oxygen species (ROS) was measured by 2′,7′-dichlorofluorescin (DCF) oxidation method and using the 2′,7′-dichlorohydrofluorescein diacetate (H2DCFH-DA) assay kit and methods provided by Molecular Probes (Eugene, OR). Molecular Probes offers derivatives of reduced fluorescein and calcein as cell-permeant indicators for ROS. Chemically reduced and acetylated forms of 2′,7′-dichlorofluorescein (DCF) and calcein are nonfluorescent until the acetate groups are removed by intracellular esterases and oxidation occurs within the cell. The carboxy derivative of fluorescein, carboxy-H2DCFDA (C400), carries additional negative charges that improve its retention compared to noncarboxylated forms. H2DCF-DA is a cell-permeable probe converted into DCF-DA by intracellular esterases, and its oxidation results in fluorescent DCF. The final concentration of H2DCF-DA ranges between 2–10 μM. Enriched hypothalamic cells (50 000 cells/well/24 wells plate) were incubated with ethanol for a period of 24 or 48 h and the intracellular reactive oxygen species were determined.
Nitrite assay
As an indicator of nitric oxide production, the amount of nitrite accumulated in the cultures were determined with a colorimetric assay using Griess reagent (1% sulfanilamide, 2.5% H3PO4, 0.1% N-(1-naphthyl) ethylenediamine dihydrochloride). Briefly, 50 μl of Gries reagent and 50 μl of the culture supernatant were incubated in the dark at room temperature for 10 min. After incubation, color intensity was measured at 540 nM using the Spectra Max Plus microplate spectrophotometer. The sample nitrite concentrations were determined from a sodium nitrite standard curve.
Glutathione (GSH) assay
The cellular levels of glutathione were determined by Glutation ASSAY (Calbiochem-EMD Chemicals, USA). Briefly, cells were washed and taken for determination of glutathione levels by following procedures provided by the manufacturer. The kit provides all reagents for simple and quick assay to measure the level of total glutathione (GSSG + GSH) in cells. The sample is first deproteinized with the 5% 5-sulfosalicylic acid solution. Glutathione content of the sample is then assayed using a kinetic assay in which catalytic amounts of glutathione cause a continuous reduction of 5,5′-dithiobis-(2-nitrobenzoic) acid (DTNB) to TNB. The oxidized glutathione formed is recycled by glutathione reductase and NADPH. The product, TNB, is assayed colorimetrically at 412 nm.
Catalase assay
The catalase activity was determined by a colorimetric assay for the catalase activity in neuronal cells using a kit (Cat# CAT 100; Sigma). At the end of experiments, cells were washed with PBS twice, lysed by lyses buffer and used in the assay (according to the instructions of the kit). The catalase activity was determined by following the instructions of the manufacturer. To determine the specificity of the reaction, catalase activity was inhibited by the addition of azide. The activity was determined in units/ml.
Statistical analysis
The data shown in the figures and text are mean ± S.E.M. Data comparisons among multiple groups were made using one-way analysis of variance (ANOVA). Post hoc tests involved the Student-Newman-Keuls test. Two-way ANOVA tests were used to compare ethanol dose-response curves between 24 h and 48h. Post hoc tests involved the Bonferroni test. A value of P < 0.05 was considered significant.
RESULTS
Determination of ethanol concentrations and neurotoxic potencies of microglial cell culture conditioned media at different times following a treatment with 50 mM ethanol
In this study we have chosen to use a 50 mM dose to study the drug effect of ethanol on microglial cells, since this dose of ethanolwas considered moderately effective in altering oxidative status and neurotoxicity (Boyadjieva and Sarkar, 2010; 2012). The concentration of ethanol in microglial cell culture medium was determined at different time periods after addition of a 50 mM dose of ethanol. As shown in Figure 1a, the level of ethanol in culture medium was decreased with time and reached at 80% added level at 6h, 50% added level at 12h and at 4% range at 24h. Microglial cell conditioned media samples were collected at different time periods and tested for their ability to induce neuronal apoptosis. As shown in Fig 1b, the ability of ethanol-treated microglial cell conditioned medium to induce neuronal apoptosis was increased with time and reached the peak at 24h. These data suggests that, once activated, microglial cells require about 24h period to fully response to ethanol.
Effects of ethanol or ethanol-activated microglial conditioned media on cAMP and BDNF production in hypothalamic neurons
In this study we tested the ability of microglial cell conditioned medium to alter the neuronal production of cell signaling molecules cAMP and BDNF. lyTreatment of enriched neuronal cells with the 50 mM dose of ethanol markedly reduced the media levels of BDNF and cellular levels of cAMP (Fig. 2A and B). Incubation of neuronal cells with conditioned medium from untreated or ethanol-treated (50 mM, 24 h) microglial cell cultures was also able to decrease BDNF (Fig. 2A) and cAMP production (Fig. 2B) from neuronal cells. When the suppressive effects of ethanol, microglial conditioned medium and ethanol-activated microglial conditioned medium were compared, ethanol-activated microglial conditioned medium showed the highest suppression.
Figure 2.

Effect of ethanol, microglia conditioned medium or ethanol-treated microglia conditioned medium on cellular levels of BDNF (A) and cAMP (B) in enriched neuronal cells in primary cultures. Enriched fetal hypothalamic neuronal cell cultures (50 000 cells/well) were exposed to no ethanol (Control) or a 50 mM dose of ethanol for a period of 24 h. Enriched fetal hypothalamic neuronal cell cultures (50 000 cells/well) were exposed to no ethanol (C), 50 mM ethanol (E), the conditioned medium from non-ethanol-treated microglial cells (Cc), the conditioned medium collected from microglial cells treated with 50 mM ethanol (Cc+E) for a period of 24 h. The cell extracts were used to measure cAMP and media levels was used to determine the levels of BDNF using ELISA. N=8. *P<0. 001, as compared to C. aP<0. 05, as compared to E. bP<0. 05, as compared to Cc.
Effects of dbcAMP or BDNF on ethanol-activated cellular apoptosis in neuronal cells
Since ethanol reduces cAMP and BDNF levels in neuronal cultures, we tested whether replacement of these cell signaling molecules protect these cells from ethanol toxicity. To test this, we treated neuronal cells with vehicle (C), ethanol (E; 50 mM) alone or with various doses of dbcAMP (A1-0.1 μM; A2-1.0 μM; A3-10.0 μM) or various doses of BDNF (B1-0.1 ng/ml; B2-1.0 ng/ml; B3-10.0 ng/ml) for a period of 24 h and then measured the cell contents of nucleosome (cellular markers of apoptosis). As shown in Fig. 3, ethanol increased about 3-fold level of nucleosome in neuronal cells. Addition of cAMP or BDNF dose-dependently reduced ethanol action on cellular levels of nucleosome in neuronal cells. The minimum dose of dbcAMP or BDNF required to block ethanol action was 1 μM or 10 ng/ml, respectively.
Figure 3.
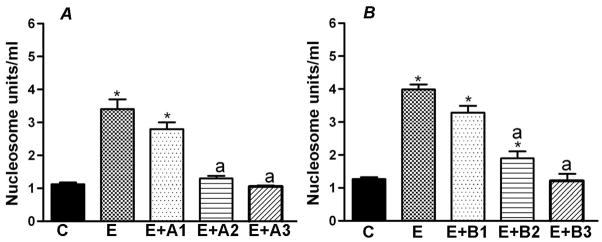
Effects of cAMP (A) or BDNF (B) on ethanol-induced apoptosis of hypothalamic neuronal cells in primary cultures. Enriched fetal hypothalamic neuronal cells (50 000 cells/well) in 24 wells were plated for 2 days in cultures and then a group of cultures was exposed to media only (control), 50 mM ethanol only, 50 mM ethanol and various doses of dbcAMP (A1, 0.1 μM; B1, 1.0 μM: C1, 10.0 μM) or BDNF (B1, 0.1; B2, 1.0; B3, 10 ng/ml) for a period of 24 h. These cultures were used to determine the dose-response effects of cAMP and BDNF on cellular apoptosis, as measured by nucleosome assays. N=8. *P<0. 001, as compared to C. a, P<0.01, as compared to E or E+A1 or E+B1.
Effects of dbcAMP and BDNF on ethanol-activated cellular apoptosis and cellular levels of antioxidants and oxidants in neuronal cells
In order to determine whether the neuroprotective actions of cAMP and BDNF are associated with alterations in the cellular levels of antioxidants and oxidants, we treated enriched neuronal cells with vehicle, 50 mM ethanol, dbcAMP (cAMP; 1.0 μM) or BDNF (10 ng/ml) with or without ethanol (50 mM) for a period of 24 h, and then measured the cell contents of nucleosome, O2−, ROS and nitrite, GSH and catalase.
Consistent with the data shown in Fig. 3, treatment with ethanol at concentration 50 mM for a period of 24 h increased about 2.5 to 3-fold levels of cellular nucleosomes as compared to control group (Fig. 4A and 5A). In agreement with our previous observations (Boyadjieva and Sarkar, 2012), we show here that 50 mM dose of ethanol significantly increased levels of oxidative molecules O2−, ROS and nitrite but decreased the levels of one of the antioxidative molecules GSH and the level of catalase enzyme that produces these molecules (Figs. 4B–F and 5B–F). Treatments with 1.0 μM dbcAMP in control cultures did not significantly change the levels of these antioxidant and oxidant molecules in cultured neurons. However, 1.0 μM dbcAMP concentration decreased the ethanol-induced levels of O2−, ROS and nitrite and normalized the ethanol suppressive effects on GSH and catalase levels (4B–F).
Figure 4.

Effects of cAMP on ethanol-induced cellular apoptosis and cellular levels of antioxidants and oxidants in hypothalamic neuronal cells in primary cultures. Enriched fetal hypothalamic neuronal cell cultures (50 000 cells/well) were exposed to no ethanol (C), 50 mM ethanol (E), 1.0 μM dbcAMP (cAMP) or 50 mM ethanol and 1.0 μM dbcAMP (E+cAMP) for a period of 24 h. The cell extracts were used to measure nucleosome (A), O2−(B), ROS (C), nitrite (D), GSH (E) and catalase activity (F). N=8. *P<0.05, as compared to C. a, P<0.05, as compared to E. bP<0. 05, as compared to cAMP.
Figure 5.

Effects of BDNF on ethanol-induced cellular apoptosis and cellular levels of antioxidants and oxidants in hypothalamic neuronal cells in primary cultures. Enriched fetal hypothalamic neuronal cell cultures (50 000 cells/well) were exposed to no ethanol (C), 50 mM ethanol (E), 10 ng/ml BDNF or E+BDNF for a period of 24 h. The cell extracts were used to measure nucleosome (A), O2− (B), ROS (C), nitrite (D), GSH (E) and catalase activity (F). N=8. *P<0.05, as compared to C. a, P<0.05, as compared to E.
Treatment with 10 ng BDNF also show strong protective effect against ethanol toxicity since the treatment completely prevented ethanol action on cellular nucleosome without affecting the basal levels of this apoptotic enzyme (Fig. 5A). Treatments with BDNF also decreased the ethanol-induced levels of O2−, ROS and nitrite and normalized the ethanol suppressive effects on GSH and catalase levels (5B–F).
Effects of dbcAMP and BDNF on microglia’s ability to alter ethanol’s effects on cellular apoptosis and cellular levels of antioxidants and oxidant in neuronal cells
Since microglia promoted ethanol actions on cAMP and BDNF production from neuronal cells, we determined the effects of dbcAMP and BDNF on microglia’s ability to alter ethanol’s effects on cellular apoptosis and cellular levels of antioxidants and oxidant in neuronal cells. Both substances were tested on enriched neurons incubated with ethanol-treated (50 mM for a period of 24 h) or control treated microglia cell culture conditioned media and then measured the cell contents of nucleosome, O2−, ROS and nitrite, GSH and catalase. These data are presented in Figs. 6 and 7.
Fig. 6.

Effects of dbcAMP on ethanol-untreated or ethanol-treated microglia conditioned medium on cellular apoptosis and cellular levels of antioxidants and oxidants in hypothalamic neuronal cells in primary cultures. Enriched fetal hypothalamic neuronal cell cultures (50 000 cells/well) were exposed to no ethanol (C), the conditioned medium from non-ethanol-treated microglial cells (Cc), 1.0 μM dbcAMP and Cc (Cc+cAMP), the conditioned medium collected from microglial cells treated with 50 mM ethanol (Cc+E) or Cc+E+cAMP for a period of 24 h. The cell extracts were used to measure nucleosome (A), O2− (B), ROS (C), nitrite (D), GSH (E) and catalase activity (F). N=8. *P<0.05, as compared to C. a, P<0.05, as compared to Cc. b, P<0.05, as compared to Cc+E.
Fig. 7.

Effects of BDNF on ethanol-untreated or ethanol-treated microglia conditioned medium on cellular apoptosis and cellular levels of antioxidants and oxidants in hypothalamic neuronal cells in primary cultures. Enriched fetal hypothalamic neuronal cell cultures (50 000 cells/well) were exposed to no ethanol (C), the conditioned medium from non-ethanol-treated microglial cells (Cc), 10 ng/ml BDNF and Cc (Cc+BDNF), the conditioned medium collected from microglial cells treated with 50 mM ethanol (Cc+E) or Cc+E+BDNF for a period of 24 h. The cell extracts were used to measure nucleosome (A), O2− (B), ROS (C), nitrite (D), GSH (E) and catalase activity (F). N=8. *P<0.05, as compared to C. a, P<0.05, as compared to Cc. b, P<0.05, as compared to Cc+E.
Data shown in Fig. 6 indicate that the conditioned medium from controlled microglial cells moderately increased nucleosome levels in neuronal cells (Fig. 6A). In parallel to the increase in nucleosome level, microglial conditioned medium also elevated cellular levels of O2, ROS and nitrite but reduced cellular levels of GSH and catalase in neuronal cultures (Fig. 6B–F). The potency of microglia conditioned medium to alter cellular levels of nucleosome, O2, ROS, nitrite, GSH and catalase was magnified in neuronal cultures when it was obtained from microglia cells that received prior treatment with 50 mM of ethanol for 24 h. Treatment with 1 μM level of cAMP was able to prevent the effects of microglia conditioned media obtained from ethanol treated or untreated microglial cell cultures.
The data on the ability of BDNF (10 ng/ml)to antagonize the effects of ethanol-treated or ethanol-activated microglia on neuronal apoptosis or cellular oxidative status were shown in Fig. 7. BDNF was able to suppress the ability of ethanol untreated or ethanol-activated microglial conditioned medium to increase the level of cellular nucleosome, O2, ROS and nitrite. BDNF also prevented the microglial cell media’s ability to reduce cellular levels of GSH and catalase in neuronal cultures.
DISSCUSION
In this study we demonstrated that ethanol decreases cellular levels of cAMP and BDNF and increases cellular apoptosis in neuronal cells isolated from fetal hypothalami and grown in primary cultures. The effects of ethanol on suppression of cAMP and BDNF and activation of neuronal apoptosis were markedly increased by the presence of microglia. Addition of dbcAMP or BDNF inhibited the ethanol or ethanol-activated microglial conditioned media’s capacity to elevate cellular apoptosis while decreasing the cellular production of O2−, ROS and nitrite but increasing the production of GSH and catalase in neuronal cells. These data suggest the possibility that the neurotoxic effect of ethanol may involve suppression of the cellular production of BDNF/CAMP leading to an increase of cellular oxidative status and the activation of apoptotic process.
Our observation showing the inhibitory effect of ethanol on BDNF production in hypothalamic neuronal cells is in agreement with previous observations made from the studies determining ethanol’s effects on BDNF production in various parts of the brain, including the hypothalamus. For example, ethanol treatment has been shown to decrease the levels of BDNF in cortical cells (Logrip et al., 2009; Climent et al., 2002). Early postnatal ethanol exposures have been shown to induce fluctuation in the expression of BDNF mRNA in the developing rat hippocampus (Miki et al., 2008). Alcohol-preferring rats have innately lower BDNF levels in the nucleus accumbens (NAc) (Yan et al., 2005). Ethanol reduced strial BDNF protein levels in wildtype and BDNF+/− mice (Bosse et al., 2011). Chronic ethanol treatment reduces the tissue levels of BDNF and BDNF mRNA levels in the hypothalamus (Tapia-Arancibia et al., 2001). Thus, chronic ethanol uniformly inhibits BDNF production in most part of the brain. Additionally, our data showing changes of this protein in enriched neuronal cells following ethanol treatment establishes neuron as a major site of ethanol action on BDNF production.
During fetal development BDNF has been shown to participate in processes involving neuronal growth, plasticity, and survival through activation of tyrosine kinase B (TrkB) receptors and its downstream signaling pathways (Davis, 2008; Zucca and Valenzuela, 2010). BDNF down-regulates neurotrophin responsiveness, TrkB protein and TrkB mRNA levels in cultured rat hippocampal neurons. (Frank et al., 1996). Multiple lines of evidence indicate BDNF is neuroprotective by preventing the development from the ethanol neurotoxicity (Gorski, et al., 2003; Jeanblanc et al., 2009; Logrip et al., 2009). Both human and animal studies indicate that BDNF plays a protective role in regulating the reinforcing effects of ethanol (Matsushita et al., 2004; McGough et al., 2004; Yan et al., 2005; Wojnar et al., 2009). Our data demonstrated that BDNF decreased the ethanol-induced apoptotic cell death and ethanol-elevated oxidative status. In addition, our data documented that BDNF increases the ethanol-inhibited antioxidants proteins in developing neurons. BDNF is also known to increase CREB phosphorylation (Hwang et al., 2011). It is known that ethanol affects CREB (Asher et al., 2002; Constantinescu et al., 2002). Moreover, there are data that ROS may control cellular CREB. Taking together, data from the literature and the present results suggest that BDNF may control the ethanol-induced apoptosis in developing neurons by modulating the mechanisms involved the cellular ROS and CREB pathway.
This study also focused on determination of the role of cAMP signaling system in ethanol neurotoxic action. We showed that ethanol reduces the cellular levels of cAMP in hypothalamic neurons and supplementation of these cells with dbcAMP inhibited the elevated oxidative status and apoptosis by ethanol alone or by ethanol activated microglial conditioned media in these neurons. It is known that the cAMP signal transduction pathway inhibits the ROS activity in mitochondria (Hüttemannn et al., 2012) that increases oxidative status and drives various pathological conditions by activating specific signaling pathways (Cataldi, 2010). In addition, the NADPH oxidase enzyme family generating ROS has been shown to be prevented by a cAMP-dependent PKA and Src tyrosine kinase (Gary et al., 2009; Gianni et al., 2008). Hence, alcohol or alcohol-activated microglial-derived factor(s) increases cellular oxidative status and apoptosis possible via inhibiting the cAMP/PKA system.
Taken together, these data provide evidence to support the hypothesis that the neurotoxic effect of ethanol may involve suppression of the cellular production of BDNF and cAMP leading to an increase in cellular oxidative status and the activation of apoptotic process. Our demonstration of the protective role of BDNF and dbcAMP from ethanol neurotoxicity may represent a future approach for understanding and prophylaxis of the fetal alcohol spectrum diseases.
Acknowledgments
This work was supported by National Institute of Health Grants R37 AA08757.
References
- Atkins CM, Oliva AA, Jr, Alonso OF, Pearse DD, Bramlett HM, Dietrich WD. Modulation of the cAMP signaling pathway after traumatic brain injury. Exp Neurol. 2007;208:145–158. doi: 10.1016/j.expneurol.2007.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barson JR, Morganstern I, Leibowitz SF. Galanin and consummatory behavior: special relationship with dietary fat, alcohol and circulating lipids. EXS. 2010;102:87–111. doi: 10.1007/978-3-0346-0228-0_8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird DN, Sato AK, Knee DS, Uyehara CF, Person DA, Claybaugh JR. Effects of prenatal ethanol exposure and sex on the arginine vasopressin response to hemorrhage in the rat. Am J Physiol Regul Integr Comp Physiol. 2006;291:R77–82. doi: 10.1152/ajpregu.00740.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosse KE, Mathews TA, Boss KE. Ethanol-induced increases in extracellular dopamine are blunted in brain-derived neurotrophic factor heterozygous mice. Neurosci Lett. 2011;489:172–176. doi: 10.1016/j.neulet.2010.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyadjieva NI, Sarkar DK. Role of microglia in ethanol’s apoptotic action on hypothalamic neuronal cells in primary cultures. Alcohol Clin Exp Res. 2010;34:1835–1842. doi: 10.1111/j.1530-0277.2010.01271.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyadjieva NI, Sarkar DK. Microglia plays a role in ethanol-induced oxidative stress and apoptosis in developing hypothalamic neurons. Alcohol Clin Exp Res. 2012 Jul 23; doi: 10.1111/j.1530-0277.2012.01889.x. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown GC, Neher JJ. Inflammatory neurodegeneration and mechanisms of microglial killing of neurons. Mol Neurobiol. 2010;41:242–247. doi: 10.1007/s12035-010-8105-9. [DOI] [PubMed] [Google Scholar]
- Cataldi A. Cell responses to oxidative stressors. Curr Phar Des. 2010;16:1387–1395. doi: 10.2174/138161210791033969. [DOI] [PubMed] [Google Scholar]
- Chen CP, Kuhn P, Advis JP, Sarkar DK. Prenatal ethanol exposure alters the expression of period genes governing the circadian function of beta-endorphin neurons in the hypothalamus. J Neurochem. 2006;97:1026–1033. doi: 10.1111/j.1471-4159.2006.03839.x. [DOI] [PubMed] [Google Scholar]
- Climent E, Pascual M, Renau-Piqueras J, Guerri C. Ethanol exposure enhances cell death in the developing cerebral cortex: role of brain-derived neurotrophic factor and its signaling pathways. J Neurosci Res. 2002;68:213–225. doi: 10.1002/jnr.10208. [DOI] [PubMed] [Google Scholar]
- Constantinescu A, Gordon AS, Diamond I. cAMP-dependent protein kinase types I and II differentially regulate cAMP response element-mediated gene expression: implications for neuronal responses to ethanol. J Biol Chem. 2002;277:18810–18816. doi: 10.1074/jbc.M112107200. [DOI] [PubMed] [Google Scholar]
- De A, Boyadjieva NI, Pastorcic M, Reddy BV, Sarkar DK. Cyclic AMP and ethanol interact to control apoptosis and differentiation in hypothalamic beta-endorphin neurons. J Biol Chem. 1994;269:26697–26705. [PubMed] [Google Scholar]
- Davis MI. Ethanol-BDNF interactions: still more questions than answers. Pharmacol Ther. 2008;118:36–57. doi: 10.1016/j.pharmthera.2008.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank L, Ventimiglia R, Anderson K, Lindsay RM, Rudge JS. BDNF down-regulates neurotrophin responsiveness, TrkB protein and TrkB mRNA levels in cultured rat hippocampal neurons. Eur J Neurosci. 1996;8:1220–1230. doi: 10.1111/j.1460-9568.1996.tb01290.x. [DOI] [PubMed] [Google Scholar]
- Gianni D, Bohl B, Courtneidge SA, Bokoch G. The involvement of the tyrosine kinase c-Src in the regulation of reactive oxygen species (ROS) generation mediated by the NADPH oxidase-1. Mol Biol Cell. 2008;19:2984–2994. doi: 10.1091/mbc.E08-02-0138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodlett CR, Thomas JD, West JR. Long-term deficits in cerebellar growth and rotarod performance of rats following “binge-like” alcohol exposure during the neonatal brain growth spurt. Neurotoxicol Teratol. 1991;13:69–74. doi: 10.1016/0892-0362(91)90029-v. [DOI] [PubMed] [Google Scholar]
- Gorski JA, Balogh SA, Wehner JM, Jones KR. Learning deficits in forebrain-restricted brain-derived neurotrophic factor mutant mice. Neuroscience. 2003;121:341–354. doi: 10.1016/s0306-4522(03)00426-3. [DOI] [PubMed] [Google Scholar]
- Hwang IK, Yoo KY, Yoo DY, Choi JW, Lee CH, Choi JH, Yoon YS, Won MH. Time-course of changes in phosphorylated CREB in neuroblasts and BDNF in the mouse dentate gyrus at early postnatal stages. Cell Mol Neurobiol. 2011;31:669–674. doi: 10.1007/s10571-011-9686-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hüttemann M, Helling S, Sanderson TH, Sinkler C, Samavati L, Mahapatra G, Varughese A, Lu G, Liu J, Ramzan R, Vogt S, Grossman LI, Doan JW, Marcus K, Lee I. Regulation of mitochondrial respiration and apoptosis through cell signaling: cytochrome c oxidase and cytochrome c in ischemia/reperfusion injury and inflammation. Biochim Biophys Acta. 2012;1817:598–609. doi: 10.1016/j.bbabio.2011.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeanblanc J, He DY, Carnicella S, Kharazia V, Janak PH, Ron D. Endogenous BDNF in the dorsolateral striatum gates alcohol drinking. J Neurosci. 2009;29:13494–13502. doi: 10.1523/JNEUROSCI.2243-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Schmidt D, Tilders F, Rivier C. Increased activity of the hypothalamic-pituitary-adrenal axis of rats exposed to alcohol in utero: role of altered pituitary and hypothalamic function. Mol Cell Neurosci. 2000;16:515–528. doi: 10.1006/mcne.2000.0890. [DOI] [PubMed] [Google Scholar]
- Logrip ML, Janak PH, Ron D. Escalating ethanol intake is associated with altered corticostriatal BDNF expression. J Neurochem. 2009;109:1459–1468. doi: 10.1111/j.1471-4159.2009.06073.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsushita S, Kimura M, Miyakawa T, Yoshino A, Murayama M, Masaki T, Higuchi S. Association study of brain-derived neurotrophic factor gene polymorphism and alcoholism. Alcohol Clin Exp Res. 2004;28:1609–1612. doi: 10.1097/01.alc.0000145697.81741.d2. [DOI] [PubMed] [Google Scholar]
- McGough NN, He DY, Logrip ML, Jeanblanc J, Phamluong K, Luong K, Kharazia V, Janak PH, Ron D. RACK1 and brain-derived neurotrophic factor: a homeostatic pathway that regulates alcohol addiction. J Neurosci. 2004;24:10542–10552. doi: 10.1523/JNEUROSCI.3714-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miki T, Kuma H, Yokoyama T, Sumitani K, Matsumoto Y, Kusaka T, Warita K, Wang ZY, Hosomi N, Imagawa T, Bedi SK, Itoh S, Nakamura Y, Takeuchi Y. Early postnatal ethanol exposure induces fluctuation in the expression of BDNF mRNA in the developing rat hippocampus. Acta Neurobiol Exp (Wars) 2008;68:484–493. doi: 10.55782/ane-2008-1714. [DOI] [PubMed] [Google Scholar]
- Miller MW, Potempa G. Numbers of neurons and glia in mature rat somatosensory cortex: effects of prenatal exposure to ethanol. J Comp Neurol. 1990;293:92–102. doi: 10.1002/cne.902930108. [DOI] [PubMed] [Google Scholar]
- Rojas JC, Vigueras RM, Reyes G, Rojas P, Cintra L, Aguilar-Roblero R. Morphological changes produced by acute prenatal exposure to ethanol on the immunoreactive vasoactive intestinal polypeptide cells of the suprachiasmatic nucleus of the rat. Proc West Pharmacol Soc. 1999;42:75–76. [PubMed] [Google Scholar]
- Sarkar DK, Kuhn P, Marano J, Chen C, Boyadjieva N. Alcohol exposure during the developmental period induces beta-endorphin neuronal death and causes alteration in the opioid control of stress axis function. Endocrinology. 2007;148:2828–2834. doi: 10.1210/en.2006-1606. [DOI] [PubMed] [Google Scholar]
- Scott HC, Zoeller RT, Rudeen PK. Acute prenatal ethanol exposure and luteinizing hormone-releasing hormone messenger RNA expression in the fetal mouse brain. Alcohol Clin Exp Res. 1995;19:153–159. doi: 10.1111/j.1530-0277.1995.tb01484.x. [DOI] [PubMed] [Google Scholar]
- Stettner GM, Kubin L, Volgin DV. Antagonism of orexin 1 receptors eliminates motor hyperactivity and improves homing response acquisition in juvenile rats exposed to alcohol during early postnatal period. Behav Brain Res. 2011;221:324–328. doi: 10.1016/j.bbr.2011.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapia-Arancibia L, Rage F, Givalois L, Dingeon P, Arancibia S, Beaugé F. Effects of alcohol on brain-derived neurotrophic factor mRNA expression in discrete regions of the rat hippocampus and hypothalamus. J Neurosci Res. 2001;63:200–208. doi: 10.1002/1097-4547(20010115)63:2<200::AID-JNR1012>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- Takahashi N, Tetsuka T, Uranishi H, Okamoto T. Inhibition of the NF-kappaB transcriptional activity by protein kinase A. Eur J Biochem. 2002;269:4559–4565. doi: 10.1046/j.1432-1033.2002.03157.x. [DOI] [PubMed] [Google Scholar]
- West JR, Dewey SL, Pierce DR, Black AC., Jr Prenatal and early postnatal exposure to ethanol permanently alters the rat hippocampus. Ciba Found Symp. 1984;105:8–25. doi: 10.1002/9780470720868.ch2. [DOI] [PubMed] [Google Scholar]
- Wojnar M, Brower KJ, Strobbe S, Ilgen M, Matsumoto H, Nowosad I, Sliwerska E, Burmeister M. Association between Val66Met brain-derived neurotrophic factor (BDNF) gene polymorphism and post-treatment relapse in alcohol dependence. Alcohol Clin Exp Res. 2009;33:693–702. doi: 10.1111/j.1530-0277.2008.00886.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan QS, Feng MJ, Yan SE. Different expression of brain-derived neurotrophic factor in the nucleus accumbens of alcohol-preferring (P) and -nonpreferring (NP) rats. Brain Res. 2005;1035:215–218. doi: 10.1016/j.brainres.2004.12.039. [DOI] [PubMed] [Google Scholar]
- Zucca S, Valenzuela CF. Low concentrations of alcohol inhibit BDNF-dependent GABAergic plasticity via L-type Ca2+ channel inhibition in developing CA3 hippocampal pyramidal neurons. J Neurosci. 2010;30:6776–6781. doi: 10.1523/JNEUROSCI.5405-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]