

Abstract
Tolfenamic acid lowers the levels of the amyloid precursor protein (APP) and amyloid beta (Aβ) when administered to C57BL/6 mice by lowering their transcriptional regulator specificity protein 1 (SP1). To determine whether changes upstream in the amyloidogenic pathway that forms Aβ plaques would improve cognitive outcomes, we administered tolfenamic acid for 34 days to hemizygous R1.40 transgenic mice. Following the characterization of cognitive deficits in these mice, assessment of spatial learning and memory functions revealed that treatment with tolfenamic acid attenuated long-term memory and working memory deficits, determined using Morris water maze (MWM) and the Y-maze. These improvements occurred within a shorter period of exposure than that seen with clinically approved drugs. Cognitive enhancement was accompanied by reduction in the levels of the SP1 protein (but not mRNA), followed by lowering both the mRNA and protein levels of APP, and subsequent Aβ levels. These findings provide evidence that tolfenamic acid can disrupt the pathological processes associated with AD and are relevant to its scheduled biomarker study in AD patients.
Keywords: Alzheimer’s disease, tolfenamic acid, AD transgenic mouse model, learning and memory, Morris water maze, Y-maze
1. Introduction
Alzheimer’s disease (AD) represents the most prevalent neurodegenerative disease in the elderly. During the course of the disease, memory, cognitive performance, and other daily activities are all impaired as a result of extensive neuronal loss (Berg, et al., 1993; Braak and Braak, 1997; Nelson, et al., 2012). AD is characterized by the presence of neuropathological deposits consisting of extracellular senile plaques of β-amyloid core and intraneuronal neurofibrillary tangles (NFTs), especially in the cerebral cortex, hippocampus, subcortical nuclei and amygdala (Selkoe, 2001; Reddy, et al., 2010; Ballard, et al., 2011; Harrington, 2012)
The amyloid precursor protein (APP) is processed by the beta-site APP-cleaving enzyme 1 (BACE1) and γ-secretase to generate various β-amyloid (Aβ) peptide isoforms that can accumulate resulting in the formation of the insoluble aggregates of amyloid plaques (Shoji, et al., 1992; Urbanc, et al., 1999; Querfurth and LaFerla, 2010; Zhang, et al., 2012). Aβ1-40 and Aβ1-42 are the major generated isoforms with Aβ1-42 found to be more aggregative triggering amyloid plaque formation (Naslund, et al., 2000; Finder and Glockshuber, 2007). Accumulation of Aβ into amyloid plaques initiates a pathological cascade resulting in synaptic dysfunction and neuronal death that contributes to the neurodegeneration observed in AD according to the amyloid hypothesis (Hardy and Higgins, 1992; Selkoe, 2001). However, other studies suggest that Aβ soluble oligomers and aggregates are the toxic species, and that in AD patients, soluble Aβ levels highly correlate with disease severity markers (McLean, et al., 1999; Kroth, et al., 2012).
FDA-approved drugs for treatment of AD include four cholinesterase inhibitors and one N-methyl-D-aspartate receptor antagonist. However, these medications are not disease-modifying and they do not stop the progression of AD (Ozudogru and Lippa, 2012). Current research focuses on interventions that target Aβ production and aggregation, and the production of hyperphosphorylated tau (Roberson and Mucke, 2006; Gotz, et al., 2012; Ozudogru and Lippa, 2012); however no therapeutic strategy has explored more upstream interventions at the transcriptional level. Specifitcity protein 1 (SP1) co-activates the transcription of APP, BACE1 and tau genes (Heicklen-Klein and Ginzburg, 2000; Christensen, et al., 2004; Docagne, et al., 2004), and consequently changes in its levels can alter the downstream pathways related to amyloidogensis (Basha, et al., 2005; Adwan, et al., 2011) and tau (unpublished data) pathology. Tolfenamic acid, a non-steroidal anti-inflammatory drug (NSAID), induces the degradation of SP1 protein (Abdelrahim, et al., 2006) and data from our lab have shown that the treatment of wild type C57BL/6 mice with tolfenamic acid lowered the levels of cerebral SP1 and the expression of AD-related Sp1 target genes such as APP (Adwan, et al., 2011).
Transgenic mouse models of AD are useful for elaborating mechanisms involved in the development and progression of AD. In addition, they allow for testing of new therapies in vivo to provide more accurate data for testing in human clinical trials (Hock and Lamb, 2001; Duff and Suleman, 2004). Our previous studies were conducted in wild type C57BL/6 mice that do not exhibit AD pathology and so we could not assess for cognitive function improvement associated with an amyloidogenic pathway. Thus, we decided to examine the ability of tolfenamic acid to lower AD proteins, including SP1, APP, and Aβ, and to examine whether such reductions are commensurate with improvements in cognitive functions in a mouse model of AD. Since the endogenous APP promoter is largely driven by SP1 (Docagne, et al., 2004), we chose to study the effects of tolfenamic acid (which degrades SP1), in the hemizygous R1.40 mice, a genomic-base transgenic mouse model that harbors the Swedish mutation APPK670N/M671L, driven by the human APP promoter (Lamb, et al., 1993).
Therefore, following the characterization of learning and memory impairment in female hemizygous R1.40 transgenic mice, 5 and 50 mg/kg/day tolfenamic acid was administered to female R1.40 mice aging between 14–21 months via oral gavage for 34 days and learning and memory functions were assessed in the Morris water maze (MWM) and the Y-maze. On Day 35, mice were euthanized and AD associated proteins including SP1, APP and soluble and insoluble Aβ1–40 and Aβ1–42 were assessed in the frontal cortex, which displays extensive AD pathology in this animal model (Kulnane and Lamb, 2001; Lehman, et al., 2003).
2. Methods
2.1. Animal model
The transgenic mouse model R1.40 was utilized for this study. The rational for choosing this transgenic line relies on the fact that the mutant human APP gene is driven by its endogenous human promoter unlike the majority of other transgenic lines, which utilize hamster PrP or murine Thy-1 promoters (Hock and Lamb, 2001). The human promoter region has numerous CpG boxes that SP1 binds to whilst activating gene expression. Thus R1.40 is an ideal mouse model to conduct experiments on, as tolfenamic acid lowers SP1 and thus alters upstream transcriptional pathways of APP. These transgenic mice, B6.129-Tg(APPSw)40btla/J, were obtained from Jackson laboratory (Bar Harbor, ME, USA) and colonies of hemizygous and homozygous strains were established in-house. R1.40 is a genomic-based transgenic mouse model that was developed by Bruce T. Lamb, it utilizes a yeast artificial chromosome (YAC) that contains the full 400 kb human APP gene and flanking sequence of approximately 250 kb to harbor the Swedish mutation APPK670N/M671L, including the human transcriptional regulatory elements needed for proper spatial and temporal expression (Reaume, et al., 1996; Lamb, et al., 1997; Hock and Lamb, 2001). The developed hemizygous R1.40 line shows a significant increase in APP and Aβ production as early as three months of age with Aβ plaque deposition occurring at 24–26 months of age compared to the wild type mice. Furthermore, the mnemonic deficits in R1.40 were similar to those observed in AD (Lamb, et al., 1999; Hock, et al., 2009).
In order to establish R1.40 transgenic mice colonies, mice were bred and genotyped in-house at the University of Rhode Island. To ensure the accuracy of genotyping results, two genotyping techniques were performed: standard PCR followed by gel electrophoresis on 1.5% agarose gel, and the TaqMan® allelic discrimination assay (Applied Biosystems, Foster City, CA, USA, see supplementary data for details). Animals of mixed genotypes were housed in standard mouse cages in the URI animal quarter rooms with a 12:12 hr light-dark cycle (light on 6:00 AM, light off at 6:00 PM). Temperature was maintained at 22±2°C with humidity levels of 55±5% and food and water available for mice ad libitum. The University of Rhode Island Institutional Animal Care and Use Committee (IACUC) approved all protocols including the breeding and genotyping methods. Animals were under continuous supervision by a URI veterinarian during the entire study and during drug administration.
2.2. Assessment of cognitive deficits in hemizygous R1.40 transgenic mice
In order to characterize memory and cognitive deficits in the hemizygous R1.40 transgenic mouse model, behavioral testing in mazes that are reliant on the integrity of the hippocampus and brain cortex was conducted using the MWM and spontaneous alternations in the Y-maze. Preliminary studies showed cognitive deficits in both male and female mice and the drug under study was active in both genders. In this study, we decided to use a single gender to minimize any possible influence or interference that may be created due to the use of mixed genders in the experiments. Thus, female hemizygous APP transgenic (n=19) and female control wild type (n=18) groups of ages ranging between 9–20 months were used.
2.2.1. MWM
We have tested the mice in the hidden version of the MWM. In this task, the mice had to locate the hidden platform by learning multiple spatial relationships between the platform and the distal extra-maze cues (Vorhees and Williams, 2006; Gulinello, et al., 2009; Laczo, et al., 2009). The apparatus consisted of a white 48″ diameter pool that is 30″ in height and was filled with water to a depth of 14″. The water was kept opaque by the addition of white non-toxic liquid washable paint. The pool was surrounded by distinct fixed visual cues that the animals used to navigate to reach the escape platform. A clear Plexiglas platform 10 cm2 was kept submerged 0.5 cm below the surface of the water. The temperature of the water was maintained at 25 ±2°C during all experiments in the water maze. On Day 15 of tolfenamic acid administration, mice received a habituation trial in which they were allowed to swim freely for 60 sec. On the following day and for a total of 8 days, mice received training sessions of 3 trials daily. The starting position for each trial was randomly assigned between the four possible positions (one per quadrant) while the platform position was fixed in each trial. Each animal was allowed to swim until they found the immersed hidden platform or for a maximum duration of 60 sec. If the mouse failed to locate the platform, it would be gently guided to sit on the platform for a maximum duration of 30 sec. Mice were also left to sit on the platform for a maximum of 10 sec upon successful trial. After completion of the 8 acquisition sessions, probe trials for up to 60 sec on Day 1 and Day 11 following the last day of training were performed to assess long term memory retention by studying the preference of the mice for the correct quadrant that previously contained the hidden platform. The swim paths and latencies to locate the platform and time spent in quadrants were videotaped and tracked with a computerized video tracking system (ObjectScan, Clever Sys. Inc., Reston, VA, USA) and the resultant data were analyzed.
2.2.2. Spontaneous alternation in the Y-maze
The spontaneous alternation ratio, defined as the percentage of the number of arm entries different from the previous two entries divided by the total arm entries minus two (King, et al., 1999; Hock, et al., 2009), was measured. Testing was conducted in a white Y-maze with arms that were 12″ (long) by 3″ (wide) with 8″ high walls. Mice were placed into one of the Y-maze arms and were left to explore the maze freely for 5 minutes. After each trial, the maze was cleaned with 70% ethanol. Y-maze trials were videotaped and tracked with a computerized video tracking system (ObjectScan, Clever Sys. Inc., Reston, VA, USA) and the data was analyzed.
2.3. Animal exposure to tolfenamic acid and behavioral testing
Tolfenamic acid was obtained from Sigma Aldrich (St. Louis, MO, USA) and female hemizygous APP YAC R1.40 transgenic mice aged between 14–21 months were used to examine the ability of tolfenamic acid to reduce the amyloidogenic load and to improve the learning and memory deficits of the mice. From our initial published studies with tolfenamic acid (Adwan, et al., 2011), we found that 5 and 50 mg/kg/day were the optimum low and high doses capable to produce reductions in amyloidogenic proteins. We also found that a treatment duration of 15 days was the minimum period required to show significant reductions in APP and Aβ. Thus, on day 15 of drug administration, we started conducting behavioral experiments and tolfenamic acid administration continued until the last day of the behavioral studies. The animals were divided into three groups of similar age variation and were administered tolfenamic acid daily via oral gavage for 34 days; one group was administered 5 mg/kg/day tolfenamic acid in corn oil (n=6); the second group was administered 50 mg/kg/day tolfenamic acid in corn oil (n=7) and the third group was administered the corn oil vehicle, (n=6). After 14 days of dosing, mice were tested in the MWM as described in section 2.2.1. On Day 33, mice were tested for spontaneous alternation in the Y-maze as described in section 2.2.2. Finally, on Day 35, mice were euthanized and brain tissue was extracted, dissected and stored at −80°C. Figure 1 provides a timeline for the experimental events involved in drug administration and behavioral assessment.
Fig. 1. A timeline of tolfenamic acid administration and behavioral assessment.
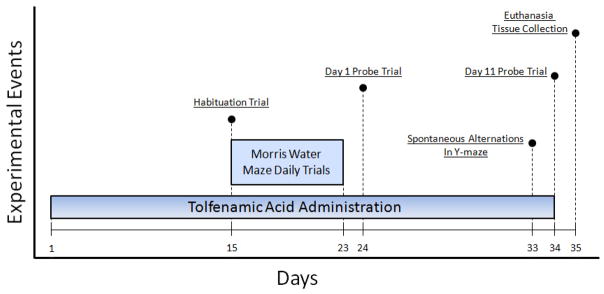
Mice were administered tolfenamic acid daily for 34 days. Behavioral testing in the MWM began on the 15th day of dosing with daily training sessions that lasted until the 23rd day of the study. On the 24th day of dosing, we conducted the first session of probe trials (Day 1) and then we performed a second session of probe trials on the 34th of dosing (Day 11), to assess for long-term memory retention. Trials for the spontaneous alternations in the Y-maze were conducted on the 33rd day of dosing as well. Animals were euthanized on the 35th day of the study and brain tissue was dissected, collected and stored in the −80°C freezer.
2.4. Protein extraction and Western blot analysis
Brain cortices were homogenized with RIPA lysis buffer containing 10 mM Tris-HCl [pH 7.4], 150 mM NaCl, 1% Triton X-100, 0.1% SDS, 1 mM EDTA, and 0.1% protease inhibitor cocktail. The homogenates were centrifuged at 10,000 × g for 10 min at 4°C and supernatants were collected. Protein levels were determined using the BCA kit (Pierce Biotechnology, Inc., Rockford, IL, USA). Forty μg of total protein was separated on 8% SDS polyacrylamide gel and then transferred onto polyvinylidiene diflouride (PVDF) membranes (GE Healthcare, Piscataway, NJ, USA). Membranes were blocked using 5% non-fat milk in TBST (Tris buffered saline + 0.5% Tween-20, pH 7.4) for 1 hr. Membranes were then incubated overnight at 4°C with the specific antibody diluted in TBST for 1 hr (1:2000 dilution of 6E10 for APP (Covance Research Products Inc., Dedham, MA, USA) and 1:500 dilution of 1C6 for SP1 (Santa Cruz Biotechnology, Santa Cruz, CA, USA). After washing membranes 4 times with TBST, membranes were incubated for one hour with the anti-mouse IRDye® 680 (Li-Cor Bioscience, Lincoln, NE) (1:5000) at room temperature. After washing the membranes twice with TBST and once with TBS, the images were developed using the Li-Cor Odyssey infrared imaging system (Li-Cor Bioscience, Lincoln, NE, USA). The membranes were also re-probed for β-actin (Sigma Aldrich, St. Louis, MO, USA) at a dilution of 1:2500 in TBST to obtain the APP/β-Actin ratio. The intensities of the obtained Western blot bands were determined using Odyssey V1.2 software (Li-Cor Bioscience, Lincoln, NE, USA).
2.5. RNA isolation, synthesis of cDNA and quantitative Real-Time PCR
RNA was isolated from cortical tissue following the TRIzol® Reagent method (Invitrogen, Carlsbad, CA, USA), checked for integrity using NanoDrop 2000 Micro-Volume UV-Vis Spectrophotometer (Thermo Scientific, Wilmington, DE, USA) and gel electrophoresis. RNA was reverse transcribed to cDNA using the iScript™ Select cDNA Synthesis Kit following manufacturer’s instructions (Bio-Rad, Hercules, CA, USA). About 1000ng of RNA was diluted to 19.5μl with nuclease free water, then 3μl Oligo (dT) mix, 6μl 5x iScript Select reaction mix, and 1.5μl of iScript reverse transcriptase was added. Samples were incubated at 42°C for 90 min then at 85°C for 5 min to terminate the reaction. All incubations were conducted using MJ Research MiniCycler™ PTC-150 (Bio-Rad, Hercules, CA, USA). Primer pairs for Sp1, APP and β-actin were obtained from Invitrogen (Carlsbad, CA, USA) as follows: Sp1 sense: 5′-CAA GCC CAA ACA ATC ACC TT-3′, and antisense, 5′-CAA TGG GTG TGA GAG TGG TG-3′; APP sense: 5′-TGC AGC AGA ACG GAT ATG AG-3′, and antisense: 5′-ACA CCG ATG GGT AGT GAA GC-3′; β-actin sense: 5′-TGT TAC CAA CTG GGA CGA CA-3′, and antisense: 5′-TCT CAG CTG TGG TGG TGA AG-3′. Each Real-Time PCR reaction mix contained 2μl of cDNA, 1μl of each primer, 8.5μl nuclease free water and 12.5μl SYBR® Green PCR Master Mix (Applied Biosystems, Foster City, CA, USA). Real-Time PCR was conducted using 7500 Real-Time PCR System (Applied Biosystems, Foster City, CA, USA) following the standard protocol: 50°C for 2 min followed by 95°C for 10 min, then 40 cycles of 95°C for 15 sec and 60°C for 1 min. Results were analyzed using SDS software and expression data were reported relative to β-actin mRNA using the 2−ΔΔCt method.
2.6. ELISA Aβ1–40 and Aβ1–42 Assay
Enzyme-linked immunosorbent assay (ELISA) using human Aβ1–40 and Aβ1–42 assay IBL kits JP27713 and JP27711 determined Aβ levels, both soluble and insoluble fractions, (Immuno-Biological Laboratories, Gunma, Japan). These kits are solid-phase sandwich ELISA with two types of highly specific antibodies that are 100% reactive with mouse Aβ1-40 with a sensitivity of 5.00 pg/ml (JP27713), and 70% reactive with mouse Aβ1-42 with 4.03 pg/ml sensitivity (JP27711). The assay conditions were followed according to a method described in the literature (Morishima-Kawashima, et al., 2000; Wu, et al., 2008). The levels of Aβ in the test samples were calculated relative to the standard curve generated on each plate.
2.7. Statistical analysis
Biochemical data are expressed as the mean ± the standard error of the mean (SEM). Assessment of performance in MWM daily training sessions between the hemizygous R1.40 and the wild type groups was determined using repeated measure analysis of variance (ANOVA) while probe trials differences between both groups was determined by a two-tailed student t-test. The significance of differences between various treatment groups was determined by repeated measure ANOVA and Tukey-Kramer multiple comparison a posteriori analysis. All statistical analyses were conducted using GraphPad InStat 3 software (GraphPad Software, La Jolla, CA, USA). Probability (p) value of <0.05 was considered statistically significant.
3. Results
3.1. Overall safety and toxicity observations
Overt toxic effects of tolfenamic acid administration were not observed in these animals. No changes in body weight were observed and treatment with tolfenamic acid did not result in any abnormal behavior compared to non-exposed mice. Although the safety of tolfenamic acid has been previously established, as it has been used in Europe for decades for rheumatoid arthritis and for migraine, recent toxicity studies conducted by one of our collaborators showed that chronic administration of tolfenamic acid of 50 mg/kg three times a week for 6 weeks did not cause changes in hematocrit or hemoglobin in tolfenamic acid exposed mice. In addition, histopathological analysis showed that acute and chronic administration of tolfenamic acid did not produce adverse changes in gastric and intestinal epithelia (available unpublished data).
3.2. Hemizygous R1.40 mice exhibit cognitive impairments when tested in the MWM and in the Y-maze
Spatial navigation in the MWM is one type of test used to assess long-term memory functions, which are usually impaired in AD. In the daily sessions which consisted of three trials per day for a total of eight days, repeated measure ANOVA indicated no significant difference in the performance, assessed by measuring escape latency, between the transgenic group and the control wild type mice in this task (F(7,245) = 0.924, p > 0.05) (Fig. 2A). However, probe trials that assessed the percent time in the correct quadrant that contained the hidden platform showed that as the retention delay increased from Day 1 following the last day of the daily training sessions to Day 11, the transgenic mice exhibited memory impairment as they failed to remember the location of the hidden platform. As shown in figure 2B, probe trials in Day 1 following the last day of daily training sessions showed no significant difference between both groups (p > 0.1). However, after a longer delay on Day 11 following the last day of daily trainings sessions, the hemizygous R1.40 transgenic mice significantly spent less time (Mean = 23%; SEM = 1.99%) compared to the control wild type mice (Mean = 29.8%; SEM= 2.58%), p = 0.045 (Fig. 2B). In addition, ANOVA analysis of target preference showed that both WT and R1.40 mice spent significantly more time in the correct quadrant in the first probe trial. However, in the second probe trial, only the WT group retained such significant target preference. The R1.40 mice group failed to maintain target preference indicating memory impairment (See supplementary data).
Fig. 2. Memory impairments in hemizygous APP YAC transgenic mice, line R1.40, assessed by the swimming task in the MWM.
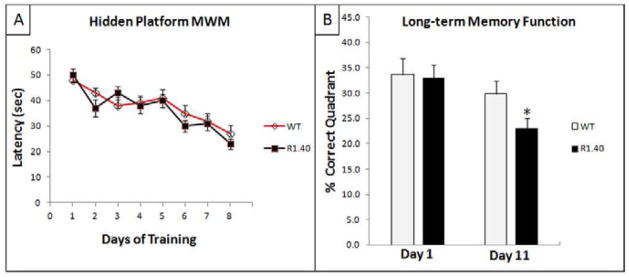
A) Acquisition patterns during daily training; B) Probe trials for long-term memory retention on Days 1 and 11 following acquisition-training trials. “*” indicates that the values are significantly different from control, as determined by a Student’s t-test (p<0.05).
Working memory function was assessed by measuring the spontaneous alternations in the Y-maze. This task is based on the innate nature of the mice to visit arms that have not been recently visited (King, et al., 1999; Hock, et al., 2009). Results revealed that hemizygous transgenic R1.40 mice exhibited impaired working memory function as they had a significantly lower spontaneous alternation ratio (Mean = 0.413; SEM = 0.021) than the control wild type mice (Mean = 0.496; SEM = 0.019), p = 0.0047 (Fig. 3).
Fig. 3. Working memory impairments in hemizygous APP YAC transgenic, line R1.40 mice.

Mice were examined for working memory function assessed by measuring the spontaneous alternation ratio in the Y-maze. “**” Indicates that the values are significantly different from control, as determined by a Student’s t-test (p<0.01)
3.3. Treatment with tolfenamic acid reverses the cognitive deficits in hemizygous R1.40 mouse model
We determined above the presence of impaired behavioral performance in the hemizygous R1.40 mice. This was manifested by a decline in the long-term memory and the working memory functions as identified by poor retention of the spatial location of the platform in the MWM and spontaneous alternations in the Y-maze, respectively.
We further examined the ability of a tolfenamic acid treatment of 5 and 50 mg/kg/day to attenuate these learning and memory deficits in groups of female hemizygous R1.40 transgenic mice aging between 14–21 months. In the MWM task, there was a significant effect of training as ANOVA analysis showed that the difference in escape latency between the first to the last daily training sessions was statistically significant (F(7,144) = 7.833, p < 0.0001). Escape latency between the treatment and the vehicle groups showed no significant differences in daily learning acquisition (Fig. 4A). However, ANOVA analysis indicated an improvement in long-term memory retention in Day 11 probe trials (F(2,15) = 4.145, p = 0.0369) (Fig. 4B). The Tukey-Kramer HSD post-hoc test showed that the mice group treated with 50 mg/kg/day tolfenamic acid exhibited a significant improvement (HSD = 3.12, p < 0.05), while the treatment with 5 mg/kg/day failed to reach significance (HSD = 0.71 p > 0.5) (Fig. 4B). Furthermore, ANOVA analysis of target preference indicated that mice groups treated with 50 mg/kg/day spent significantly more time in the correct quadrant in both probe trials compared to other groups indicating memory improvement (See supplementary data for details).
Fig. 4. The administration of tolfenamic acid to hemizygous APP YAC R1.40 mice improves spatial memory.

Tolfenamic acid was administered by oral gavage daily for 34 days. See the methods section for details. A) Acquisition patterns during daily training trials of 5 mg/kg/day, 50 mg/kg/day and vehicle groups; B) Probe trials assessing the long-term memory retention on Days 1 and 11 following acquisition training trials. “*” Indicates that values are significantly different from vehicle, as determined by one-way ANOVA analysis with Tukey-Kramer post-hoc test to compare all pairs of columns (p<0.05), obtained using GraphPad InStat 3 software. Vehicle n=6; 5 mg/kg/day n=6; 50 mg/kg/day n=7.
Results from the ANOVA analysis of the spontaneous alternation ratio in the Y-maze showed a significant improvement in working memory (F(2,15) = 4.479, p = 0.0298) (Fig. 5). The Tukey-Kramer HSD post-hoc test revealed a significant improvement with tolfenamic acid treatment of 50 mg/kg/day (HSD = 4.139, p < 0.05), however, treatment with 5 mg/kg/day did not reach significance (HSD = 1.30, p > 0.05) (Fig. 5)
Fig. 5. Working memory improvement in hemizygous APP YAC transgenic mice, line R1.40, after administration of tolfenamic acid assessed by spontaneous alternations in the Y-maze.

Tolfenamic acid was administered by oral gavage daily for 34 days. See the methods section for details. “*” Indicates that values are significantly different from vehicle control, as determined by one-way ANOVA analysis with Tukey-Kramer post-hoc test to compare all pairs of columns (p<0.05), obtained using GraphPad InStat 3 software. Vehicle n=6; 5 mg/kg/day n=6; 50 mg/kg/day n=7.
3.4. Tolfenamic acid treatment lowers SP1 protein without altering Sp1 gene expression
The protein levels of cortical SP1 normalized to β-actin were analyzed by Western blot following treatment with 0, 5 and 50 mg/kg/day for 34 days. ANOVA analysis revealed a significant difference in SP1 protein levels (F(2.11) = 8.747, p = 0.0053) (Fig. 6A). The Tukey-Kramer HSD post-hoc test indicated a significant decrease of SP1 after treatment with 5 mg/kg/day tolfenamic acid (HSD = 4.145, p < 0.05, % change = − 43%) and after treatment with 50 mg/kg/day tolfenamic acid (HSD = 5.712, p < 0.01, % change = − 52%) (Fig. 6A).
Fig. 6. Reduction of SP1 protein levels in the brain after treatment with tolfenamic acid.

Tolfenamic acid was administered by oral gavage daily for 34 days. See the methods section for details. A) Western blot analysis of cortical SP1 levels as a ratio of the house-keeping protein β-actin after treatment with 5 and 50 mg/kg/day of tolfenamic acid for 34 days. B) Cortical Sp1 mRNA levels determined by Real-Time PCR. “*” Indicates that values are significantly different from vehicle, as determined by one-way ANOVA analysis with Tukey-Kramer post-hoc test to compare all pairs of columns (*p<0.05, **p<0.01), obtained using GraphPad InStat 3 software.
Sp1 mRNA expression was evaluated for the different treatment groups. Analysis of Sp1 mRNA levels showed that treatment with tolfenamic acid did not alter the gene expression of Sp1 (F(2,16) = 0.082, p = 0.922) (Fig. 6B).
3.5. Reduction of APP protein and mRNA following treatment with tolfenamic acid
Cortical APP normalized to β-actin was analyzed by Western blot following treatment with 5 and 50 mg/kg/day for 34 days or with vehicle. ANOVA revealed that APP levels were lowered significantly (F(2,9) = 8.377, p = 0.009) (Fig. 7A). The Tukey-Kramer HSD post-hoc test indicated a significant decrease in APP after treatment with 5 mg/kg/day tolfenamic acid (HSD = 4.877, p < 0.05, % change = − 30.3%) and 50 mg/kg/day tolfenamic acid (HSD = 5.139, p < 0.05, % change = − 32.5%) (Fig. 7A).
Fig. 7. Reduction of amyloid precursor protein (APP) levels in the brain after treatment with tolfenamic acid.

Tolfenamic acid was administered by oral gavage daily for 34 days. See the methods section for details. A) Western blot analysis of cortical APP levels as a ratio of the house keeping protein β-actin after treatment with 5 and 50 mg/kg/day of tolfenamic acid for 34 days; B) Cortical APP mRNA determined by Real-Time PCR. “*” Indicates that values are significantly different from vehicle, as determined by one-way ANOVA analysis with Tukey-Kramer post-hoc test to compare all pairs of columns (p<0.05), obtained using GraphPad InStat 3 software.
In addition to analysis of APP protein levels, APP gene expression was evaluated from the different treatment groups by Real-Time PCR. ANOVA analysis of APP mRNA levels revealed a significant decrease in APP gene expression (F(2,15) = 5.238, p = 0.019) (Fig. 7B). The Tukey-Kramer HSD post-hoc test indicated a significant decrease of APP mRNA after treatment with 5 mg/kg/day tolfenamic acid (HSD = 3.885, p < 0.05, % change = − 50.3%) and after treatment with 50 mg/kg/day tolfenamic acid (HSD = 4.039, p < 0.05, % change = − 52.3%) (Fig. 7B). The lowering of APP was not associated with adverse effects (Adwan, et al., 2011)
3.6. Tolfenamic acid lowers both soluble and insoluble Aβ1–40 and Aβ1–42 levels
We measured the levels of cortical Aβ1–40 and Aβ1–42 levels in the Tris-saline-soluble and in the insoluble fraction using IBL ELISA kits as described in the methods section (Morishima-Kawashima, et al., 2000). ANOVA analysis showed that there was a significant reduction in soluble Aβ1–40 fraction (F(2,14) = 3.89, p = 0.04) (Fig. 8A) and the insoluble Aβ1–40 fraction (F(2,14) = 4.95, p = 0.02) (Fig. 8B). The Tukey-Kramer HSD post-hoc test indicated a significant decrease in soluble Aβ1–40 after treatment with 50 mg/kg/day (HSD = 3.91, p < 0.05, % change = − 30.1%), however, the reduction in soluble Aβ1–40 after treatment with 5 mg/kg/day did not reach significance (HSD = 2.55, p > 0.05, % change = − 21.3%) (Fig. 8A). Multiple group comparison showed a significant decrease in the insoluble Aβ1–40 fraction after treatment with both 5 mg/kg/day (HSD = 3.73, p < 0.05, % change = − 35.2%) and 50 mg/kg/day tolfenamic acid (HSD = 3.97, < 0.05, % change = − 37.4%) (Fig. 8B).
Fig. 8. Reduction of soluble and insoluble Aβ1-40 and Aβ1-42 levels in the brain after treatment with tolfenamic acid.

Tolfenamic acid was administered by oral gavage daily for 34 days. See the methods section for details. ELISA quantified Aβ levels in the cortices of mice treated with 5 and 50 mg/kg/day of tolfenamic acid for 34 days. A) Soluble Aβ1-40; B) Insoluble Aβ1-40; C) Soluble Aβ1-42 and D) Insoluble Aβ1-42 levels. “*” Indicates that values are significantly different from vehicle, as determined one-way ANOVA analysis with Tukey-Kramer post-hoc test to compare all pairs of columns (p<0.05), obtained using GraphPad InStat 3 software.
Also, ANOVA analysis showed a significant decrease in soluble Aβ1–42 fraction (F(2,14) = 6.174, p = 0.012) (Fig. 8C) and the insoluble Aβ1–42 fraction (F(2,14) = 6.343, p = 0.011) (Fig. 8D). The Tukey-Kramer HSD post-hoc test indicated a significant decrease in soluble Aβ1–42 after tolfenamic acid treatment with 5 mg/kg/day (HSD = 4.18, p < 0.05, % change = − 22.3%) and after treatment with 50 mg/kg/day (HSD = 4.518, p < 0.05, % change = − 24.9%) (Fig. 8C). Similarly, multiple group comparison showed a significant decrease in the insoluble Aβ1–42 fraction after treatment with 5 mg/kg/day (HSD = 4.72, p < 0.05, % change = − 31.8%) and 50 mg/kg/day (HSD = 4.79, < 0.05, % change = − 37.7%) (Fig. 8D).
We also observed that 5 and 50 mg/kg/day tolfenamic acid treatment reduced total Aβ1-42/Aβ1-40 ratio −16.3% and −19.9%, respectively. However, this reduction in Aβ1-42/Aβ1-40 did not reach statistical significance (F(2.14) = 1.788, p = 0.203).
4. Discussion
Over the last two decades, more evidence has been garnered regarding the association between chronic NSAIDs intake and up to 80% reduction in risk of AD incidence, slowing of disease progression, and reduction of microglial activation (Andersen, et al., 1995; McGeer, et al., 1996; Stewart, et al., 1997; in t’ Veld, et al., 2001; Lindsay, et al., 2002; Zandi, et al., 2002; Etminan, et al., 2003; Vlad, et al., 2008; Cote, et al., 2012). Several mechanisms have been proposed to explain how NSAIDs could exert their effects on AD independent of their COX inhibitory pathways. Certain NSAIDs including indomethacin, fulfinamic acid, ibuprofen and fenoprofen are agonists for the nuclear transcriptional regulator peroxisome proliferator-activated receptor-γ (PPARγ) (Lehmann, et al., 1997; Heneka, et al., 2011) which has been reported to be reduced in AD brains (Sastre, et al., 2006). PPARγ activation results in a reduction in the levels of Aβ-induced proinflammatory response of microglia and monocytes, and promotes Aβ clearance by astrocytes and microglia (Matsuo, et al., 1996; Combs, et al., 2000; Daynes and Jones, 2002; Yue and Mazzone, 2009; Mandrekar-Colucci, et al., 2012). In addition, a PPARγ responsive element (PPRE) has been identified in the BACE1 gene promoter that is repressed by PPARγ binding resulting in a decrease in BACE1 expression (Sastre, et al., 2003; Sastre, et al., 2006). Weggen et al., reported that a subset of NSAIDs specifically lower Aβ1-42 that was accompanied by a parallel increase in Aβ1-38 suggesting a minor γ-secretase modulatory effect without alteration of the Notch pathway (Weggen, et al., 2001). The specific Aβ1-42 inhibition by that subset of NSAIDs was further studied by Zhou et al., who concluded that the involvement of Rho-Rock pathway in regulating APP processing, and inhibition of Rho activity by specific NSAIDs could preferentially reduce Aβ1-42 (Zhou, et al., 2003).
Tolfenamic acid, of all other NSAIDs, has the unique mechanism of inducing the proteasome dependent degradation of SP transcription factors including SP1, SP3 and SP4 and thereby, decreasing the expression of vascular endothelial growth factors (VEGF) (Abdelrahim, et al., 2006), which is involved in tumor progression and metastases. Because of its SP degradation effects, tolfenamic acid has been studied as a therapeutic agent for cancer and research has confirmed its antitumor effects (Abdelrahim, et al., 2006; Konduri, et al., 2009; Basha, et al., 2011; Colon, et al., 2011).
Sp1 involvement in AD has been researched and it was found that its expression was elevated in the frontal cortex of AD brains as well as in the brains of transgenic mouse models of AD (Santpere, et al., 2006; Citron, et al., 2008). The elevation of Sp1 was accompanied by an increase in COX-2, another Sp1 target gene, suggesting interplay between inflammatory processes and Sp1 driven gene expression (Citron, et al., 2008). Sp1 binds to 5′-GGGGCGGGGC-rich promoter genes such as APP and BACE1 genes, which are involved in AD pathology, and Sp1 overexpression leads to induction of their protein expression (Cai, et al., 2001; Christensen, et al., 2004; Citron, et al., 2008). We have reported that silencing of Sp1 gene by siRNA resulted in 70% decrease in the responsiveness of the human APP promoter (Basha, et al., 2005). Also, we have demonstrated the co-localization of APP, Sp1 and Aβ in various regions of the brain (Brock, et al., 2008). Thus, we proposed the hypothesis that tolfenamic acid could interrupt the de novo synthesis of APP and consequently, alter the downstream levels of Aβ. This hypothesis was confirmed when oral treatment with tolfenamic acid resulted in degradation of SP1 which was accompanied by a reduction in cortical and hippocampal APP gene expression and protein, and Aβ peptide levels (Adwan, et al., 2011). This was further supported by measurements which demonstrated the presence of tolfenamic acid in the brain following IV administration (Subaiea, et al., 2011).
In the present study, we demonstrate that hemizygous R1.40 transgenic mice exhibit cognitive deficits upon assessment of their working memory and long-term memory retention even before the reported age of 24–26 months at which hemizygous R1.40 start to suffer extensive β-amyloid deposition (Lamb et al., 1999). Hemizygous R1.40 mice start to exhibit 7–8 times more Aβ1-42 levels compared to wild types as early as 3–4 months of age (Lamb, et al., 1999). Thus, mice were studied at ages where there is a great increase in Aβ levels in the brain prior to plaque depositions. Spatial learning was assessed in the hidden version of the MWM and we found that the transgenic mice were able to learn to escape to the hidden platform in the daily training sessions (Fig. 2A). However, they exhibited memory retention impairments in the probe trials as they failed to retain the spatial location of the hidden platform with increased delay (Fig. 2B, Day 11). Working memory assessed in the Y-maze was impaired in these mice as demonstrated by numerous alternation errors and a lower alternation ratio compared to the wild type control group (Fig. 3). It has been proposed that recent spatial memory is hippocampal-dependent, while remote spatial memory is dependent on the integrity of the medial prefrontal cortex (King, et al., 1999; Frankland and Bontempi, 2005; Teixeira, et al., 2006). In turn, working memory assessed in the Y-maze is hippocampal and cortical-dependent (Lalonde, 2002). That, together with the findings that the R1.40 mouse model exhibits extensive amyloid deposition in the cortical region compared to that observed in the hippocampal formation (Hock, et al., 2009), may explain the observed results.
We report here that short-term tolfenamic acid administration was able to improve the mnemonic deficits observed in hemizygous R1.40. We found that (Lamb, et al., 1999) 50 mg/kg/day tolfenamic acid was able to reverse the long-term memory retention deficits seen in delayed probe trials of the MWM experiment (Fig. 4B). In addition, the 50 mg/kg/day treatment group showed significant improvement in the Y-maze with less error in spontaneous alternations (Fig. 5). However, the short-term treatment with 5 mg/kg/day showed only modest effects in reversing the memory deficits in these mice. Analysis of amyloid pathology markers indicated that both treatment regimens, 5 and 50 mg/kg/day, were able to significantly lower the protein level of SP1 without altering its gene expression indicating that the decrease in SP1 protein levels is not due to a change in its de novo synthesis but is rather at the post-translational level (Fig. 6A and 6B). Those results were similar to our previous findings in the C57BL/6 treated with tolfenamic acid (Adwan, et al., 2011). In addition, both tolfenamic acid doses significantly reduced the protein levels of APP and its gene expression (Fig. 7A and 7B). Furthermore, levels of the soluble and insoluble Aβ1–40 and Aβ1–42 were reduced by tolfenamic acid with a trend toward lowering the insoluble fractions of Aβ more than that of the soluble fraction (Fig. 8). It is possible that tolfenamic acid may aid in clearance of insoluble Aβ by a PPARγ activation mechanism, however, it is not clear yet if tolfenamic acid activates PPARγ as certain other NSAIDs do.
While tolfenamic acid was effective in lowering the amyloidogenic proteins with both doses, we observed dose-dependent improvement in the cognitive deficits. Whether longer period of treatment with low dose tolfenamic acid could result in significant improvements in cognitive function is yet to be studied. However, it is essential to mention that tolfenamic acid produced behavioral improvements after a rather short-term treatment period compared to other FDA approved AD drugs and NSAIDs. For example, APP23 transgenic mice treated with 0.58 mg/kg/day donepezil, an FDA-approved anticholinesterase drug for AD, showed improved performance in MWM after two months of treatment (Van Dam, et al., 2008). In addition, memantine, another FDA-approved drug for use in moderate to severe AD, has shown to improve cognition in 3x TG-AD mice after three months of treatment (Martinez-Coria, et al., 2010). Also, triple transgenic mice (APPswe, PS1M146V and tauP301L) exhibited improved cognitive functions after treatment with ibuprofen for six months (McKee, et al., 2008).
Many of the anti amyloid drugs that failed in clinical trials were anti-Aβ monoclonal antibodies, γ-secretase inhibitors, BACE1 inhibitors or certain NSAIDs that do not lower SP1 (Herrmann, et al., 2011; Ozudogru and Lippa, 2012). This might be either due to lack of efficacy or ensuing adverse effects. The main targets of these drugs were proteins involved at the end stage of the amyloidogenic pathway. Tolfenamic acid interferes with transcriptional pathways associated with AD-related genes and thus it acts at upstream pathways that can have impacts on downstream events and is not dependent on protein events at the end of the amyloidogenic pathways. Thus, it may hold better promise for success.
In conclusion, tolfenamic acid is unique among other NSAIDs in its ability to inhibit SP1 and thus interrupt APP de novo synthesis and its Aβ products. In this study, we demonstrated that administration of 50 mg/kg/day for 34 days of tolfenamic acid reversed the cognitive deficits in hemizygous R1.40 transgenic mice. Consistent with our previous published findings in C57BL/6 mice, SP1, the amyloidogenic levels of APP and soluble and insoluble Aβ1–40 and Aβ1–42 were markedly lowered by tolfenamic acid in an AD transgenic mice. These data provide further evidence that tolfenamic acid is a promising therapeutic agent as a repurposed AD drug acting through an alternative mechanism and should benefit the scheduled clinical trials of this drug.
Supplementary Material
Acknowledgments
This work was partially supported by grants ES13022, AG027246, 1R56ES015867-01A1, and 1R01ES015867-01A2 from the National Institutes of Health awarded to N.H.Z. The research was made possible by the use of the RI-INBRE Research Core Facility, supported jointly by NCRR/NIH Grant # P20 RR016457 and the network institutions. Special thanks from the authors to Dr. David Serra and Gail Golomb-Mello for their assistance and contribution in the study.
Footnotes
Disclosure statement
The authors hereby declare they have no disclosures or conflicts of interest. The University of Rhode Island Institutional Animal Care and Use Committee (IACUC) has approved all animal studies conducted in this research.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abdelrahim M, Baker CH, Abbruzzese JL, Safe S. Tolfenamic acid and pancreatic cancer growth, angiogenesis, and Sp protein degradation. J Natl Cancer Inst. 2006;98(12):855–68. doi: 10.1093/jnci/djj232. [DOI] [PubMed] [Google Scholar]
- Adwan LI, Basha R, Abdelrahim M, Subaiea GM, Zawia NH. Tolfenamic acid interrupts the de novo synthesis of the beta-amyloid precursor protein and lowers amyloid beta via a transcriptional pathway. Curr Alzheimer Res. 2011;8(4):385–92. doi: 10.2174/156720511795745285. [DOI] [PubMed] [Google Scholar]
- Andersen K, Launer LJ, Ott A, Hoes AW, Breteler MM, Hofman A. Do nonsteroidal anti-inflammatory drugs decrease the risk for Alzheimer’s disease? The Rotterdam Study. Neurology. 1995;45(8):1441–5. doi: 10.1212/wnl.45.8.1441. [DOI] [PubMed] [Google Scholar]
- Ballard C, Gauthier S, Corbett A, Brayne C, Aarsland D, Jones E. Alzheimer’s disease. Lancet. 2011;377(9770):1019–31. doi: 10.1016/S0140-6736(10)61349-9. [DOI] [PubMed] [Google Scholar]
- Basha MR, Wei W, Bakheet SA, Benitez N, Siddiqi HK, Ge YW, Lahiri DK, Zawia NH. The fetal basis of amyloidogenesis: exposure to lead and latent overexpression of amyloid precursor protein and beta-amyloid in the aging brain. J Neurosci. 2005;25(4):823–9. doi: 10.1523/JNEUROSCI.4335-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basha R, Ingersoll SB, Sankpal UT, Ahmad S, Baker CH, Edwards JR, Holloway RW, Kaja S, Abdelrahim M. Tolfenamic acid inhibits ovarian cancer cell growth and decreases the expression of c-Met and survivin through suppressing specificity protein transcription factors. Gynecol Oncol. 2011;122(1):163–70. doi: 10.1016/j.ygyno.2011.03.014. [DOI] [PubMed] [Google Scholar]
- Berg L, McKeel DW, Jr, Miller JP, Baty J, Morris JC. Neuropathological indexes of Alzheimer’s disease in demented and nondemented persons aged 80 years and older. Arch Neurol. 1993;50(4):349–58. doi: 10.1001/archneur.1993.00540040011008. [DOI] [PubMed] [Google Scholar]
- Braak H, Braak E. Frequency of stages of Alzheimer-related lesions in different age categories. Neurobiol Aging. 1997;18(4):351–7. doi: 10.1016/s0197-4580(97)00056-0. [DOI] [PubMed] [Google Scholar]
- Brock B, Basha R, DiPalma K, Anderson A, Harry GJ, Rice DC, Maloney B, Lahiri DK, Zawia NH. Co-localization and distribution of cerebral APP and SP1 and its relationship to amyloidogenesis. J Alzheimers Dis. 2008;13(1):71–80. doi: 10.3233/jad-2008-13108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai H, Wang Y, McCarthy D, Wen H, Borchelt DR, Price DL, Wong PC. BACE1 is the major beta-secretase for generation of Abeta peptides by neurons. Nat Neurosci. 2001;4(3):233–4. doi: 10.1038/85064. [DOI] [PubMed] [Google Scholar]
- Christensen MA, Zhou W, Qing H, Lehman A, Philipsen S, Song W. Transcriptional regulation of BACE1, the beta-amyloid precursor protein beta-secretase, by Sp1. Mol Cell Biol. 2004;24(2):865–74. doi: 10.1128/MCB.24.2.865-874.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Citron BA, Dennis JS, Zeitlin RS, Echeverria V. Transcription factor Sp1 dysregulation in Alzheimer’s disease. J Neurosci Res. 2008;86(11):2499–504. doi: 10.1002/jnr.21695. [DOI] [PubMed] [Google Scholar]
- Colon J, Basha MR, Madero-Visbal R, Konduri S, Baker CH, Herrera LJ, Safe S, Sheikh-Hamad D, Abudayyeh A, Alvarado B, Abdelrahim M. Tolfenamic acid decreases c-Met expression through Sp proteins degradation and inhibits lung cancer cells growth and tumor formation in orthotopic mice. Invest New Drugs. 2011;29(1):41–51. doi: 10.1007/s10637-009-9331-8. [DOI] [PubMed] [Google Scholar]
- Combs CK, Johnson DE, Karlo JC, Cannady SB, Landreth GE. Inflammatory mechanisms in Alzheimer’s disease: inhibition of beta-amyloid-stimulated proinflammatory responses and neurotoxicity by PPARgamma agonists. J Neurosci. 2000;20(2):558–67. doi: 10.1523/JNEUROSCI.20-02-00558.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cote S, Carmichael PH, Verreault R, Lindsay J, Lefebvre J, Laurin D. Nonsteroidal anti-inflammatory drug use and the risk of cognitive impairment and Alzheimer’s disease. Alzheimers Dement. 2012;8(3):219–26. doi: 10.1016/j.jalz.2011.03.012. [DOI] [PubMed] [Google Scholar]
- Daynes RA, Jones DC. Emerging roles of PPARs in inflammation and immunity. Nat Rev Immunol. 2002;2(10):748–59. doi: 10.1038/nri912. [DOI] [PubMed] [Google Scholar]
- Docagne F, Gabriel C, Lebeurrier N, Lesne S, Hommet Y, Plawinski L, Mackenzie ET, Vivien D. Sp1 and Smad transcription factors co-operate to mediate TGF-beta-dependent activation of amyloid-beta precursor protein gene transcription. Biochem J. 2004;383(Pt 2):393–9. doi: 10.1042/BJ20040682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duff K, Suleman F. Transgenic mouse models of Alzheimer’s disease: how useful have they been for therapeutic development? Brief Funct Genomic Proteomic. 2004;3(1):47–59. doi: 10.1093/bfgp/3.1.47. [DOI] [PubMed] [Google Scholar]
- Etminan M, Gill S, Samii A. Effect of non-steroidal anti-inflammatory drugs on risk of Alzheimer’s disease: systematic review and meta-analysis of observational studies. BMJ. 2003;327(7407):128. doi: 10.1136/bmj.327.7407.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finder VH, Glockshuber R. Amyloid-beta aggregation. Neurodegener Dis. 2007;4(1):13–27. doi: 10.1159/000100355. [DOI] [PubMed] [Google Scholar]
- Frankland PW, Bontempi B. The organization of recent and remote memories. Nat Rev Neurosci. 2005;6(2):119–30. doi: 10.1038/nrn1607. [DOI] [PubMed] [Google Scholar]
- Gotz J, Ittner A, Ittner LM. Tau-targeted treatment strategies in Alzheimer’s disease. Brit J Pharmacol. 2012;165(5):1246–59. doi: 10.1111/j.1476-5381.2011.01713.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulinello M, Gertner M, Mendoza G, Schoenfeld BP, Oddo S, LaFerla F, Choi CH, McBride SM, Faber DS. Validation of a 2-day water maze protocol in mice. Behav Brain Res. 2009;196(2):220–7. doi: 10.1016/j.bbr.2008.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy JA, Higgins GA. Alzheimer’s disease: the amyloid cascade hypothesis. Science. 1992;256(5054):184–5. doi: 10.1126/science.1566067. [DOI] [PubMed] [Google Scholar]
- Harrington CR. The Molecular Pathology of Alzheimer’s Disease. Neuroimag Clin N Am. 2012;22(1):11-+. doi: 10.1016/j.nic.2011.11.003. [DOI] [PubMed] [Google Scholar]
- Heicklen-Klein A, Ginzburg I. Tau promoter confers neuronal specificity and binds Sp1 and AP-2. J Neurochem. 2000;75(4):1408–18. doi: 10.1046/j.1471-4159.2000.0751408.x. [DOI] [PubMed] [Google Scholar]
- Heneka MT, Reyes-Irisarri E, Hull M, Kummer MP. Impact and Therapeutic Potential of PPARs in Alzheimer’s Disease. Curr Neuropharmacol. 2011;9(4):643–50. doi: 10.2174/157015911798376325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrmann N, Chau SA, Kircanski I, Lanctot KL. Current and emerging drug treatment options for Alzheimer’s disease: a systematic review. Drugs. 2011;71(15):2031–65. doi: 10.2165/11595870-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Hock BJ, Jr, Lamb BT. Transgenic mouse models of Alzheimer’s disease. Trends Genet. 2001;17(10):S7–12. doi: 10.1016/s0168-9525(01)02449-0. [DOI] [PubMed] [Google Scholar]
- Hock BJ, Lattal KM, Kulnane LS, Abel T, Lamb BT. Pathology associated memory deficits in Swedish mutant genome-based amyloid precursor protein transgenic mice. Curr Aging Sci. 2009;2(3):205–13. doi: 10.2174/1874609810902030205. [DOI] [PubMed] [Google Scholar]
- in t’ Veld BA, Ruitenberg A, Hofman A, Launer LJ, van Duijn CM, Stijnen T, Breteler MM, Stricker BH. Nonsteroidal antiinflammatory drugs and the risk of Alzheimer’s disease. N Engl J Med. 2001;345(21):1515–21. doi: 10.1056/NEJMoa010178. [DOI] [PubMed] [Google Scholar]
- King DL, Arendash GW, Crawford F, Sterk T, Menendez J, Mullan MJ. Progressive and gender-dependent cognitive impairment in the APP(SW) transgenic mouse model for Alzheimer’s disease. Behav Brain Res. 1999;103(2):145–62. doi: 10.1016/s0166-4328(99)00037-6. [DOI] [PubMed] [Google Scholar]
- Konduri S, Colon J, Baker CH, Safe S, Abbruzzese JL, Abudayyeh A, Basha MR, Abdelrahim M. Tolfenamic acid enhances pancreatic cancer cell and tumor response to radiation therapy by inhibiting survivin protein expression. Mol Cancer Ther. 2009;8(3):533–42. doi: 10.1158/1535-7163.MCT-08-0405. [DOI] [PubMed] [Google Scholar]
- Kroth H, Ansaloni A, Varisco Y, Jan A, Sreenivasachary N, Rezaei-Ghaleh N, Giriens V, Lohmann S, Lopez-Deber MP, Adolfsson O, Pihlgren M, Paganetti P, Froestl W, Nagel-Steger L, Willbold D, Schrader T, Zweckstetter M, Pfeifer A, Lashuel HA, Muhs A. Discovery and structure activity relationship of small molecule inhibitors of toxic beta-amyloid-42 fibril formation. J Biol Chem. 2012;287(41):34786–800. doi: 10.1074/jbc.M112.357665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulnane LS, Lamb BT. Neuropathological characterization of mutant amyloid precursor protein yeast artificial chromosome transgenic mice. Neurobiol Dis. 2001;8(6):982–92. doi: 10.1006/nbdi.2001.0446. [DOI] [PubMed] [Google Scholar]
- Laczo J, Vlcek K, Vyhnalek M, Vajnerova O, Ort M, Holmerova I, Tolar M, Andel R, Bojar M, Hort J. Spatial navigation testing discriminates two types of amnestic mild cognitive impairment. Behav Brain Res. 2009;202(2):252–9. doi: 10.1016/j.bbr.2009.03.041. [DOI] [PubMed] [Google Scholar]
- Lalonde R. The neurobiological basis of spontaneous alternation. Neurosci Biobehav Rev. 2002;26(1):91–104. doi: 10.1016/s0149-7634(01)00041-0. [DOI] [PubMed] [Google Scholar]
- Lamb BT, Bardel KA, Kulnane LS, Anderson JJ, Holtz G, Wagner SL, Sisodia SS, Hoeger EJ. Amyloid production and deposition in mutant amyloid precursor protein and presenilin-1 yeast artificial chromosome transgenic mice. Nat Neurosci. 1999;2(8):695–7. doi: 10.1038/11154. [DOI] [PubMed] [Google Scholar]
- Lamb BT, Call LM, Slunt HH, Bardel KA, Lawler AM, Eckman CB, Younkin SG, Holtz G, Wagner SL, Price DL, Sisodia SS, Gearhart JD. Altered metabolism of familial Alzheimer’s disease-linked amyloid precursor protein variants in yeast artificial chromosome transgenic mice. Hum Mol Genet. 1997;6(9):1535–41. doi: 10.1093/hmg/6.9.1535. [DOI] [PubMed] [Google Scholar]
- Lamb BT, Sisodia SS, Lawler AM, Slunt HH, Kitt CA, Kearns WG, Pearson PL, Price DL, Gearhart JD. Introduction and expression of the 400 kilobase amyloid precursor protein gene in transgenic mice [corrected] Nat Genet. 1993;5(1):22–30. doi: 10.1038/ng0993-22. [DOI] [PubMed] [Google Scholar]
- Lehman EJ, Kulnane LS, Lamb BT. Alterations in beta-amyloid production and deposition in brain regions of two transgenic models. Neurobiol Aging. 2003;24(5):645–53. doi: 10.1016/s0197-4580(02)00153-7. [DOI] [PubMed] [Google Scholar]
- Lehmann JM, Lenhard JM, Oliver BB, Ringold GM, Kliewer SA. Peroxisome proliferator-activated receptors alpha and gamma are activated by indomethacin and other non-steroidal anti-inflammatory drugs. J Biol Chem. 1997;272(6):3406–10. doi: 10.1074/jbc.272.6.3406. [DOI] [PubMed] [Google Scholar]
- Lindsay J, Laurin D, Verreault R, Hebert R, Helliwell B, Hill GB, McDowell I. Risk factors for Alzheimer’s disease: a prospective analysis from the Canadian Study of Health and Aging. Am J Epidemiol. 2002;156(5):445–53. doi: 10.1093/aje/kwf074. [DOI] [PubMed] [Google Scholar]
- Mandrekar-Colucci S, Karlo JC, Landreth GE. Mechanisms Underlying the Rapid Peroxisome Proliferator-Activated Receptor-gamma-Mediated Amyloid Clearance and Reversal of Cognitive Deficits in a Murine Model of Alzheimer’s Disease. J Neurosci. 2012;32(30):10117–28. doi: 10.1523/JNEUROSCI.5268-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Coria H, Green KN, Billings LM, Kitazawa M, Albrecht M, Rammes G, Parsons CG, Gupta S, Banerjee P, LaFerla FM. Memantine improves cognition and reduces Alzheimer’s-like neuropathology in transgenic mice. Am J Pathol. 2010;176(2):870–80. doi: 10.2353/ajpath.2010.090452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuo A, Walker DG, Terai K, McGeer PL. Expression of CD43 in human microglia and its downregulation in Alzheimer’s disease. J Neuroimmunol. 1996;71(1–2):81–6. doi: 10.1016/s0165-5728(96)00134-8. [DOI] [PubMed] [Google Scholar]
- McGeer PL, Schulzer M, McGeer EG. Arthritis and anti-inflammatory agents as possible protective factors for Alzheimer’s disease: a review of 17 epidemiologic studies. Neurology. 1996;47(2):425–32. doi: 10.1212/wnl.47.2.425. [DOI] [PubMed] [Google Scholar]
- McKee AC, Carreras I, Hossain L, Ryu H, Klein WL, Oddo S, LaFerla FM, Jenkins BG, Kowall NW, Dedeoglu A. Ibuprofen reduces Abeta, hyperphosphorylated tau and memory deficits in Alzheimer mice. Brain Res. 2008;1207:225–36. doi: 10.1016/j.brainres.2008.01.095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean CA, Cherny RA, Fraser FW, Fuller SJ, Smith MJ, Beyreuther K, Bush AI, Masters CL. Soluble pool of Abeta amyloid as a determinant of severity of neurodegeneration in Alzheimer’s disease. Ann Neurol. 1999;46(6):860–6. doi: 10.1002/1531-8249(199912)46:6<860::aid-ana8>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- Morishima-Kawashima M, Oshima N, Ogata H, Yamaguchi H, Yoshimura M, Sugihara S, Ihara Y. Effect of apolipoprotein E allele epsilon4 on the initial phase of amyloid beta-protein accumulation in the human brain. Am J Pathol. 2000;157(6):2093–9. doi: 10.1016/s0002-9440(10)64847-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naslund J, Haroutunian V, Mohs R, Davis KL, Davies P, Greengard P, Buxbaum JD. Correlation between elevated levels of amyloid beta-peptide in the brain and cognitive decline. JAMA. 2000;283(12):1571–7. doi: 10.1001/jama.283.12.1571. [DOI] [PubMed] [Google Scholar]
- Nelson PT, Alafuzoff I, Bigio EH, Bouras C, Braak H, Cairns NJ, Castellani RJ, Crain BJ, Davies P, Del Tredici K, Duyckaerts C, Frosch MP, Haroutunian V, Hof PR, Hulette CM, Hyman BT, Iwatsubo T, Jellinger KA, Jicha GA, Kovari E, Kukull WA, Leverenz JB, Love S, Mackenzie IR, Mann DM, Masliah E, McKee AC, Montine TJ, Morris JC, Schneider JA, Sonnen JA, Thal DR, Trojanowski JQ, Troncoso JC, Wisniewski T, Woltjer RL, Beach TG. Correlation of Alzheimer disease neuropathologic changes with cognitive status: a review of the literature. J Neuropathol Exp Neurol. 2012;71(5):362–81. doi: 10.1097/NEN.0b013e31825018f7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozudogru SN, Lippa CF. Disease Modifying Drugs Targeting beta-Amyloid. Am J Alzheimers Dis Other Demen. 2012;27(5):296–300. doi: 10.1177/1533317512452034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Querfurth HW, LaFerla FM. Alzheimer’s disease. N Engl J Med. 2010;362(4):329–44. doi: 10.1056/NEJMra0909142. [DOI] [PubMed] [Google Scholar]
- Reaume AG, Howland DS, Trusko SP, Savage MJ, Lang DM, Greenberg BD, Siman R, Scott RW. Enhanced amyloidogenic processing of the beta-amyloid precursor protein in gene-targeted mice bearing the Swedish familial Alzheimer’s disease mutations and a “humanized” Abeta sequence. J Biol Chem. 1996;271(38):23380–8. doi: 10.1074/jbc.271.38.23380. [DOI] [PubMed] [Google Scholar]
- Reddy PH, Manczak M, Mao P, Calkins MJ, Reddy AP, Shirendeb U. Amyloid-beta and mitochondria in aging and Alzheimer’s disease: implications for synaptic damage and cognitive decline. J Alzheimers Dis. 2010;20(Suppl 2):S499–512. doi: 10.3233/JAD-2010-100504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberson ED, Mucke L. 100 years and counting: prospects for defeating Alzheimer’s disease. Science. 2006;314(5800):781–4. doi: 10.1126/science.1132813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santpere G, Nieto M, Puig B, Ferrer I. Abnormal Sp1 transcription factor expression in Alzheimer disease and tauopathies. Neurosci Lett. 2006;397(1–2):30–4. doi: 10.1016/j.neulet.2005.11.062. [DOI] [PubMed] [Google Scholar]
- Sastre M, Dewachter I, Landreth GE, Willson TM, Klockgether T, van Leuven F, Heneka MT. Nonsteroidal anti-inflammatory drugs and peroxisome proliferator-activated receptor-gamma agonists modulate immunostimulated processing of amyloid precursor protein through regulation of beta-secretase. J Neurosci. 2003;23(30):9796–804. doi: 10.1523/JNEUROSCI.23-30-09796.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sastre M, Dewachter I, Rossner S, Bogdanovic N, Rosen E, Borghgraef P, Evert BO, Dumitrescu-Ozimek L, Thal DR, Landreth G, Walter J, Klockgether T, van Leuven F, Heneka MT. Nonsteroidal anti-inflammatory drugs repress beta-secretase gene promoter activity by the activation of PPARgamma. Proc Natl Acad Sci U S A. 2006;103(2):443–8. doi: 10.1073/pnas.0503839103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer’s disease: genes, proteins, and therapy. Physiol Rev. 2001;81(2):741–66. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- Shoji M, Golde TE, Ghiso J, Cheung TT, Estus S, Shaffer LM, Cai XD, McKay DM, Tintner R, Frangione B, et al. Production of the Alzheimer amyloid beta protein by normal proteolytic processing. Science. 1992;258(5079):126–9. doi: 10.1126/science.1439760. [DOI] [PubMed] [Google Scholar]
- Stewart WF, Kawas C, Corrada M, Metter EJ. Risk of Alzheimer’s disease and duration of NSAID use. Neurology. 1997;48(3):626–32. doi: 10.1212/wnl.48.3.626. [DOI] [PubMed] [Google Scholar]
- Subaiea GM, Alansi BH, Serra DA, Alwan M, Zawia NH. The ability of tolfenamic acid to penetrate the brain: a model for testing the brain disposition of candidate Alzheimer’s drugs using multiple platforms. Curr Alzheimer Res. 2011;8(8):860–7. doi: 10.2174/156720511798192691. [DOI] [PubMed] [Google Scholar]
- Teixeira CM, Pomedli SR, Maei HR, Kee N, Frankland PW. Involvement of the anterior cingulate cortex in the expression of remote spatial memory. J Neurosci. 2006;26(29):7555–64. doi: 10.1523/JNEUROSCI.1068-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbanc B, Cruz L, Buldyrev SV, Havlin S, Irizarry MC, Stanley HE, Hyman BT. Dynamics of plaque formation in Alzheimer’s disease. Biophys J. 1999;76(3):1330–4. doi: 10.1016/S0006-3495(99)77295-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Dam D, Coen K, De Deyn PP. Cognitive evaluation of disease-modifying efficacy of donepezil in the APP23 mouse model for Alzheimer’s disease. Psychopharmacology (Berl) 2008;197(1):37–43. doi: 10.1007/s00213-007-1010-x. [DOI] [PubMed] [Google Scholar]
- Vlad SC, Miller DR, Kowall NW, Felson DT. Protective effects of NSAIDs on the development of Alzheimer disease. Neurology. 2008;70(19):1672–7. doi: 10.1212/01.wnl.0000311269.57716.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vorhees CV, Williams MT. Morris water maze: procedures for assessing spatial and related forms of learning and memory. Nat Protoc. 2006;1(2):848–58. doi: 10.1038/nprot.2006.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weggen S, Eriksen JL, Das P, Sagi SA, Wang R, Pietrzik CU, Findlay KA, Smith TE, Murphy MP, Bulter T, Kang DE, Marquez-Sterling N, Golde TE, Koo EH. A subset of NSAIDs lower amyloidogenic Abeta42 independently of cyclooxygenase activity. Nature. 2001;414(6860):212–6. doi: 10.1038/35102591. [DOI] [PubMed] [Google Scholar]
- Wu J, Basha MR, Brock B, Cox DP, Cardozo-Pelaez F, McPherson CA, Harry J, Rice DC, Maloney B, Chen D, Lahiri DK, Zawia NH. Alzheimer’s disease (AD)-like pathology in aged monkeys after infantile exposure to environmental metal lead (Pb): evidence for a developmental origin and environmental link for AD. J Neurosci. 2008;28(1):3–9. doi: 10.1523/JNEUROSCI.4405-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue L, Mazzone T. Peroxisome proliferator-activated receptor {gamma} stimulation of adipocyte ApoE gene transcription mediated by the liver receptor X pathway. J Biol Chem. 2009;284(16):10453–61. doi: 10.1074/jbc.M808482200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zandi PP, Anthony JC, Hayden KM, Mehta K, Mayer L, Breitner JC. Reduced incidence of AD with NSAID but not H2 receptor antagonists: the Cache County Study. Neurology. 2002;59(6):880–6. doi: 10.1212/wnl.59.6.880. [DOI] [PubMed] [Google Scholar]
- Zhang H, Ma QL, Zhang YW, Xu HX. Proteolytic processing of Alzheimer’s ss-amyloid precursor protein. Journal of Neurochemistry. 2012;120:9–21. doi: 10.1111/j.1471-4159.2011.07519.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Su Y, Li B, Liu F, Ryder JW, Wu X, Gonzalez-DeWhitt PA, Gelfanova V, Hale JE, May PC, Paul SM, Ni B. Nonsteroidal anti-inflammatory drugs can lower amyloidogenic Abeta42 by inhibiting Rho. Science. 2003;302(5648):1215–7. doi: 10.1126/science.1090154. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.