

Abstract
Introduction
Diclofenac, a nonsteroidal antiinflammatory drug, is commonly used as antipyretic therapy in intensive care. The purpose of this study was to investigate the effects of parenteral diclofenac infusion on brain homeostasis, including brain-tissue oxygen tension (PbtO2) and brain metabolism after aneurysmal subarachnoid hemorrhage (aSAH).
Methods
We conducted a prospective, observational study with retrospective analysis of 21 consecutive aSAH patients with multimodal neuromonitoring. Cerebral perfusion pressure (CPP), mean arterial pressure (MAP), intracranial pressure (ICP), body temperature, and PbtO2 were analyzed after parenteral diclofenac infusion administered over a 34-minute period (20 to 45 IQR). Data are given as mean ± standard error of mean and median with interquartile range (IQR), as appropriate. Time-series data were analyzed by using a general linear model extended by generalized estimation equations (GEEs).
Results
One-hundred twenty-three interventions were analyzed. Body temperature decreased from 38.3°C ± 0.05°C by 0.8°C ± 0.06°C (P < 0.001). A 10% decrease in MAP and CPP (P < 0.001) necessitated an increase of vasopressors in 26% (n = 32), colloids in 33% (n = 41), and crystalloids in 5% (n = 7) of interventions. PbtO2 decreased by 13% from a baseline value of 28.1 ± 2.2 mm Hg, resulting in brain-tissue hypoxia (PbtO2 <20 mm Hg) in 38% (n = 8) of patients and 35% (n = 43) of interventions. PbtO2 <30 mm Hg before intervention was associated with brain-tissue hypoxia after parenteral diclofenac infusion (likelihood ratio, 40; AUC, 93%; 95% confidence interval (CI), 87% to 99%; P < 0.001). Cerebral metabolism showed no significant changes after parenteral diclofenac infusion.
Conclusions
Parenteral diclofenac infusion after aSAH effectively reduces body temperature, but may lead to CPP decrease and brain-tissue hypoxia, which were both associated with poor outcome after aSAH.
Introduction
Fever is common in patients with aneurysmal subarachnoid hemorrhage (aSAH) and independently associated with poor outcome [1-4]. Brain-temperature elevations correlate with an increase of cerebral metabolic rate of oxygen (CMRO2) [5], intensified brain metabolism [6], aggravation of cerebral edema, and increased intracranial pressure (ICP) [7]. The hypothalamic set-point temperature can be lowered with nonsteroidal antiinflammatory drugs (NSAIDs) through inhibition of prostaglandine-E2 (PGE2) synthesis [8]. The NSAID diclofenac is commonly used in intensive care units for fever treatment [9-11]. Side effects of parenteral diclofenac, such as decrease of mean arterial blood pressure (MAP) [11] and cerebral perfusion pressure (CPP) [10,12], have been previously described. Hemodynamic stability for maintaining adequate cerebral perfusion during vasospasm is of utmost importance after aSAH [13]. The prognostic significance of low CPP and episodes of brain-tissue hypoxia (PbtO2 <20 mm Hg) after aSAH and traumatic brain injury (TBI) has been extensively studied [14-17]. The purpose of this trial was to describe the effects of parenteral diclofenac infusion on CPP, brain-tissue oxygen tension (PbtO2), and brain metabolism in the early period of aSAH.
Materials and methods
Patients
Prospectively collected data of 29 consecutive aSAH patients with brain multimodal neuromonitoring between September 2010 and March 2012 were retrospectively analyzed. Twenty-one patients receiving parenteral diclofenac infusion during the neuromonitoring time were included (see Additional file 1). Criteria for invasive neuromonitoring were approved by the institutional review board of Innsbruck Medical University (UN3898 285/4.8) as follows: (a) GCS ≤8, (b) poor likelihood for regaining consciousness within the following 48 hours, (c) high likelihood for surviving at least 48 hours, and (d) age older than 18 years. Written informed consent was obtained according to federal regulations.
General management
In general, patient care conformed to guidelines of the American Heart Association [18]. Patients were clinically graded with the Hunt and Hess scale and radiologically assessed with the modified Fisher scale [19] of the first available cerebral computed tomography (CT) scan. Prophylactic, parenteral continuous nimodipine (1 to 2 mg/hour) was routinely given in all patients. A mean flow velocity >200 cm/second of the basal cerebral arteries assessed with transcranial Doppler sonography (TCD, DWL Doppler-Box system; Compumedics, Singen, Germany) was considered as TCD flow-velocity acceleration. Catheter angiography was performed in patients with TCD flow-velocity acceleration. Patients with angiographic vasospasm were treated with intraarterial nimodipine and vasopressors. All patients included in this study had continuous invasive blood pressure monitoring. Fluid therapy (colloids or crystalloids) were used as first-line, and vasopressors (noradrenaline, phenylephrine, or dobutamine) were used as second-line therapy for maintaining MAP and CPP. Whenever a critical CPP <50 mm Hg was reached, vasopressors were started/increased immediately. In case of unexpected blood pressure decreases, the parenteral nimodipine dose was decreased or stopped.
We did not perform wake-up trials on the patients, following our institutional guidelines. Therefore, delayed cerebral infarction (DCI) was defined as any new infarct appearing on cerebral CT scan that was judged by an independent radiologist to be attributable to vasospasm. Pneumonia was defined as radiologic infiltrate and elevated white blood cell count. Start and finish time of any pharmacologic intervention, including dosage, were exactly documented in the electronic patient data-management system (PDMS; Centricity Critical Care 7.0, General Electric Healthcare Company, IL, USA).
Fever definitions and management
Body temperature was measured with the temperature sensor of the bladder catheter. Fever was defined as a body temperature >38.4°C because this was the threshold for fever treatment. In certain cases, fever therapy was initiated at lower body temperatures at the discretion of the intensive care physician. Parenteral diclofenac (75 mg; maximum dosage, 150 mg/day) or parenteral acetaminophen (1,000 mg; maximum dosage, 3,000 mg/day) was used as first-line antipyretic therapy. The choice of treatment was at the discretion of the treating intensivist. In case of ineffectiveness or contraindications against the first-line antipyretic treatment (meperidine; Pethidine; 100 mg; maximum, 400 mg/day) as second-line therapy was given. If temperature still remained above 38.4°C, normothermia (36.5°C) was maintained in four patients with an invasive cooling device (CoolGard 3000 or ThermoGard XP; Alsius Corporation, Zoll Medical Corporation, MA, USA) as rescue therapy [20].
Interventions
For fever treatment, 75 mg diclofenac-sodium diluted in 100 ml normal saline (Ratiopharm, parenteral diclofenac (Diclobene), 75 mg, drug approval number: 1-19719) was administered intravenously. Starting and ending time points of intervention were exactly documented in the PDMS. Interventions with administration of other antipyretics or invasive cooling 4 hours before or after parenteral diclofenac were excluded (details on the inclusion algorithm are shown in Appendix Figure 1).
Figure 1.
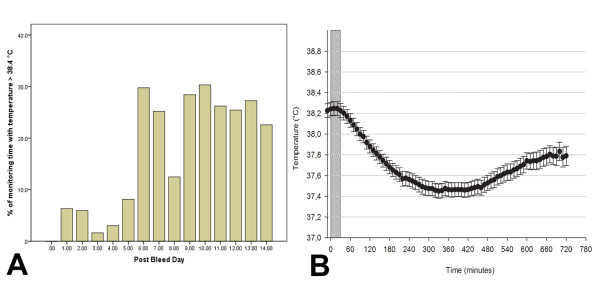
Fever burden during monitoring time and body temperature after parenteral diclofenac. (A) Cumulative fever burden in percentage of total monitoring time, fragmented on days after initial hemorrhage (post-bleed day 0-14). Time in fever (temperature >38.4°C) during monitoring time reached a maximum on post-bleed day 10. (B) demonstrating mean body temperature (●) after parenteral diclofenac (zero on X-axis is the first 10-minute-average-interval during intervention, n = 123; P < 0.001). The width of the dotted bar indicates median infusion time (34 minutes; 20 to 45 IQR). The maximum effect on core body temperature was reached after 330 minutes (median, 260 to 470, IQR). Values are illustrated in mean ± SEM.
Monitoring and data acquisition
Monitoring probes were placed into the hemisphere deemed at greatest risk for secondary injury and either tunneled or fixed by using a triple-lumen bolt. CT scan of the brain was used to check the probe location, usually within 24 hours after implantation. PbtO2 was measured with a Clark-type probe (Licox, Integra, Germany), intracranial pressure (ICP) with an intraparenchymal probe (Neurovent, Raumedic, Germany). Brain-tissue hypoxia was defined as brain-tissue oxygen tension <20 mm Hg, based on previous studies [14,21] demonstrating increased odds for poor outcome and metabolic distress below this threshold value. Cerebral microdialysis (CMD) was performed by using a 100-kDa-cutoff microdialysis catheter (CMA-71; CMA/MicrodialysisTM, Stockholm, Sweden) at a perfusion rate of 0.3 μl/min. Samples were collected hourly and frozen at -80°C. Outcome was evaluated prospectively 3 months after aSAH with telephone interview by using the modified Rankin Scale (mRS). Poor neurologic outcome was defined as mRS >4. The study nurse was blinded to the monitoring data and their analysis. Complications (pneumonia, vasospasm, DCI) were stated after adjudication of all relevant clinical information in weekly meetings by the study team (RH, BP, RB, MF, AS, and ES).
Data management and statistics
The PDMS was used to acquire digital data for ICP, MAP, CPP, PbtO2 , and body temperature every 3 minutes from the monitoring device (Carescape B650; General Electric Company, IL, USA). Monitoring data were averaged to 10-minute mean values. A 10-minute mean average before intervention was considered a baseline value for a hemodynamic parameter, and 1 hour before intervention was considered a baseline value for the CMD dataset. Time-series data were analyzed with a generalized linear model by using a normal distribution and identity link function. The model was extended by generalized estimating equations (GEEs) by using time after intervention as a factor and important parameters (CPP, body temperature) as covariates.
An autoregressive matrix of the first order (AR-1) was used to handle repeated observations within subjects [22]. Logarithmic transformation was applied to meet assumptions of normality. Cut-off levels were calculated by using receiver operating characteristics (ROCs).
SPSS 19.0 was used for statistical testing. P < 0.05 was considered statistically significant.
Data are given in median and interquartile range (IQR) or in mean ± one standard error of the mean (SEM).
Results
Study population and intervention
Baseline characteristics are demonstrated in Table 1. Mean age was 55 ± 11 years, and the median Hunt and Hess grade was 4 (3 to 5, IQR). Neuromonitoring was initiated at day 1 (1 to 2, IQR) and maintained for 12 days (8 to 14, IQR); 29% (n = 6) of patients had poor outcomes.
Table 1.
Demographic details of study patients
| Clinical characteristics | |||
|---|---|---|---|
| Age (years) | 56 (47-63) | ||
| Gender (female) | 13 (62) | ||
| Admission H&H | 2-3 | 6 (29) | |
| 4-5 | 15 (71) | ||
| Admission Apache II Score | 17 (14-19) | ||
| Admission radiologic characteristics | |||
| mFisher scale | 1 | 2 (10) | |
| 2 | 1 (4) | ||
| 3 | 9 (43) | ||
| 4 | 9 (43) | ||
| IVH sum score | 3 (0-6) | ||
| Aneurysm size (mm) | 9 (4-11) | ||
| Generalized cerebral edema | 10 (48) | ||
| Intracerebral hematoma | 7 (33) | ||
| Surgical procedures | |||
| Hydrocephalus requiring EVD | 18 (86) | ||
| Clipping | 14 (67) | ||
| Complications | |||
| Pneumonia | 13 (62) | ||
| DCI | 4 (19) | ||
| Moderate to severe angiographic vasospasm | 6 (29) | ||
| Outcome characteristics | |||
| Length of hospital stay (days) | 40 (29-48) | ||
| Three-months mRS | 0-1 | 5 (24) | |
| 2-3 | 7 (33) | ||
| 4-5 | 5 (24) | ||
| 6 | 4 (19) | ||
Apache II score (acute physiology and chronic health score); DCI, delayed cerebral infarction; EVD, external ventricular drainage; H&H, Hunt and Hess grade; IVH, intraventricular hemorrhage; mFisher scale, modified Fisher scale; mRS, modified Rankin scale. Data are presented in median (IQR) and count (%).
One-hundred twenty-three interventions were analyzed. A median of four interventions (two to eight, IQR) per patient were administered over a 34-minute period (20 to 45, IQR). Mean central venous pressure at baseline was 13 ± 0.2 mm Hg. Ten days (7 to 12 days, IQR) after aSAH was the median time point of intervention, when fever was most common (30% of daily monitoring time; Figure 1A).
Effects of parenteral diclofenac infusion on body temperature
Body temperature at baseline (38.3 ± 0.05°C) decreased by 0.8 ± 0.06°C to a minimum value of 37.5 ± 0.05°C within 330 minutes (260 to 470, IQR) after the start of an intervention (P < 0.001; Figure 1B). In 9% (n = 11) of interventions, temperature did not decrease after parenteral diclofenac. We found a significant interaction between body temperature and MAP (P = 0.02), but no interaction between body temperature and PbtO2.
Effects of parenteral diclofenac infusion on MAP, CPP, and ICP
MAP (baseline, 93 ± 1.2 mm Hg) and CPP (baseline, 85 ± 1.4 mm Hg) decreased by 10% after intervention (P < 0.001; Figure 2). Maximum blood pressure decreases were observed 160 minutes (90 to 320, IQR) after intervention. Colloids were administered in 33% (n = 41) and crystalloids in 5% (n = 7) 29 minutes (20 to 86, IQR) after intervention. Vasopressors were increased within 75 minutes (30 to 155 minutes, IQR) in 26% (n = 32) of interventions. A decrease of CPP <70 mm Hg and <50 mm Hg was observed in 71% (n = 87) and 12% (n = 15) of interventions, respectively. ICP (baseline: 8.6 ± 0.4 mm Hg) did not significantly change after intervention.
Figure 2.
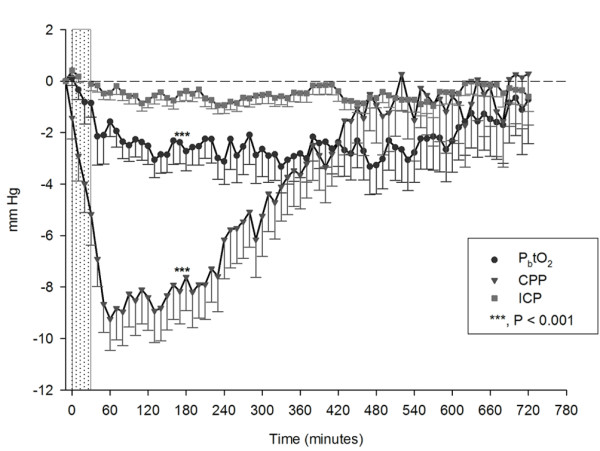
Changes from mean baseline values after parenteral diclofenac (zero on X-axis is the first 10-minute-average interval during interventions; n = 123). CPP (▼, baseline = 85 ± 1.4 mm Hg; P < 0.001), ICP (■, baseline, 8.6 ± 0.4 mm Hg); and PbtO2 (●, baseline = 28.1 ± 2.2 mm Hg; P < 0.001). The dotted bar illustrates the median diclofenac infusion time (34 minutes; IQR, 20 to 45). Values are presented in mean ± SEM. CPP, cerebral perfusion pressure; ICP, intracranial pressure; IQR, interquartile range; PbtO2, brain-tissue oxygen tension. Initial CPP decrease after parenteral diclofenac occurred within the first 10 minutes, indicated as point zero at the X-axis.
Effects of parenteral diclofenac infusion on PbtO2
PbtO2 decreased by 13% from a baseline value of 28.1 ± 2.2 mm Hg to 24.5 ± 2.1 mm Hg (P < 0.001; Figure 2), resulting in brain-tissue hypoxia (PbtO2 <20 mm Hg) in 35% (n = 43) of all interventions and in 38% (n = 8) of patients. This finding remained significant (P < 0.01) after adjusting for CPP and body temperature, indicating an intrinsic effect of diclofenac on PbtO2. An interaction occurred between PbtO2 and CPP (P = 0.02), which is shown in Figure 3. Baseline PbtO2 <30 mm Hg (Figure 4) was associated with brain-tissue hypoxia after parenteral diclofenac (likelihood ratio, 45; specificity, 81%; sensitivity, 88%; P < 0.001; OR 85.1; 95% CI, 13 to 550), with an area under the curve (AUC) of 93% (95% CI, 87% to 99%). Brain-tissue hypoxia was observed 3 hours (1.5 to 3 hours, IQR) after intervention and persisted for 25 minutes (18 to 180 minutes, IQR). A CPP decrease <70 mm Hg was associated with brain-tissue hypoxia (P < 0.01).
Figure 3.

Time-locked changes in PbtO2 from baseline of four different CPP groups (quartiles) representing maximum CPP decrease observed during the study period.
Figure 4.

Correlation between PbtO2 (x) reaching minimum values after parenteral diclofenac and PbtO2 values before intervention. Lines represent PbtO2 threshold of 20 mm Hg. The right lower quadrant indicates PbtO2 values ≥20 mm Hg at baseline reaching hypoxic values after intervention.
The percentage of total monitoring time with PbtO2 <20 mm Hg (32 ± 9.4% in patients with good outcome versus 66% ± 12% in patients with poor outcome: OR = 1.04; 95% CI, 1.001 to 1.08; P < 0.05), but not the absolute time (48 ± 20 hours versus 56 ± 27 hours; P = 0.07) was independently associated with poor outcome after adjusting for disease severity.
Effects of parenteral diclofenac infusion on cerebral metabolism
Cerebral microdialysis (CMD) was available in 48% (n = 10) of patients and 28% (n = 34) of interventions. No significant change in brain metabolism was noted after parenteral diclofenac (Figure 5) when compared with baseline values (CMD-lactate, 4 ± 0.3 mM; CMD-pyruvate, 141 ± 7 μM; CMD-LPR, 28 ± 1.5; CMD-glucose, 2 ± 0.2 mM; and CMD-glutamate, 22 ± 8 μM).
Figure 5.

Changes in brain metabolism time locked to parenteral diclofenac therapy (hours from intervention). Values presented in mean ± SEM.
Discussion
Our main findings are that parenteral diclofenac infusion after aSAH is associated with a significant decrease of CPP and brain-tissue oxygen tension. PbtO2 values <30 mm Hg before intervention were highly predictive of consecutive brain-tissue hypoxia (PbtO2 <20 mm Hg). The percentage of time in brain-tissue hypoxia was independently associated with poor outcome.
PbtO2 reflects the balance between oxygen supply and demand and can be used as a surrogate marker for cerebral blood flow (CBF) [23,24]. The ability to maintain adequate CBF relatively independent of changes in CPP is termed normal cerebral autoregulation. Impaired cerebral autoregulation frequently occurs after aSAH and may result in a direct dependence of PbtO2 on CPP [25]. We did not assess cerebral autoregulation, but found a decrease in CPP related to the decrease in PbtO2. This mechanism may well explain the PbtO2 decrease in our patients; especially, a CPP <70 mm Hg was associated with brain-tissue hypoxia after intervention. This finding is in line with a recent study indicating a higher risk for brain-tissue hypoxia at CPP values <70 mm Hg [14].
PbtO2 is dependent not only on oxygen delivery, but also on cerebral oxygen consumption [23]. Cerebral metabolic rate of oxygen (CMRO2) as an indicator of cerebral oxygen consumption is temperature dependent [5]. Experimental studies showed a reduction of CMRO2 after parenteral NSAID injection [5]. Therefore, one might expect an increase of PbtO2 due to reduced CMRO2 after temperature reduction.
Interestingly, PbtO2 remained decreased even after CPP had recovered to baseline values. One explanation for this observation could be the inhibitory effect of diclofenac on the cyclooxygenase (COX) [26]. PGE2-syntheses is COX dependent and plays an important role in maintaining CBF and cerebral autoregulation [27]. Inhibition of PGE2 synthesis by diclofenac may reduce CBF and therefore PbtO2. Another explanation for this prolonged PbtO2 decrease may be neurovascular coupling, a mechanism of the brain adapting the CBF to the cerebral ener-gy demand [27]. A decrease in body temperature might reduce cerebral energy demand and therefore CBF [5,28]. This hypothesis is furthermore supported by the lack of metabolic changes despite a decrease in PbtO2.
We found a significant interaction between mean arterial blood pressure and body temperature after diclofenac intervention. This is in line with previously described hemodynamic changes related to physiological mechanisms of temperature regulation, including vasodilatation, sweating, and inhibition of muscle activity [29].
Hemodynamic changes after parenteral diclofenac necessitated increased use of vasopressors and parenteral fluid therapy. Adrenergic stress can promote the development of myocardial stunning [30], which may increase the risk of cerebral infarction from vasospasm, hypotension, and pulmonary edema [31]. Diclofenac infusion time was not standardized, which could have influenced hemodynamic side effects in this study. A randomized controlled trial by Cormio et al. [12] investigated continuous low-dose diclofenac infusion (0.004 to 0.08 mg/kg BW/h) for fever management after TBI and aSAH. The authors showed that this treatment regimen effectively decreases body temperature without the occurrence of hemodynamic side effects. In their control group, antipyretic therapy (0.2 mg/kg diclofenac, 1 g acetaminophen) was administered over a 30-minute period. This infusion rate was associated with a significant CPP decrease (P = 0.03) and is comparable to our results. These findings suggest an association of hemodynamic side effects with the application time of antipyretic therapy. Further studies are needed to investigate whether continuous low-dose diclofenac should preferably be used in aSAH patients.
We found an effective reduction of body temperature after parenteral diclofenac, whereas the degree of temperature reduction and the duration to maximum efficacy were in line with the literature [10,12] and pharmacologic properties of diclofenac (half-life, 1 to 2 hours) [32].
This study has several limitations. First, a causal relation between parenteral diclofenac and PbtO2 decrease cannot be certainly proven based on our data. Importantly, all continuous physiologic and pharmacologic data as well as intervention time points were exactly recorded with an electronic chart and patient-management system. Second, this observational study has no power to compare the effects of different antipyretic drugs on PbtO2, which can be demonstrated only by randomized controlled trials. Third, the infusion rate was not standardized, which might bias the study results. Fourth, we did not analyze cerebral autoregulation or CBF, and microdialysis data must be interpreted cautiously because of limited sample size with simultaneous PbtO2 recordings available during interventions.
Nonetheless, this is the first study investigating brain-tissue oxygen tension and cerebral metabolism after parenteral diclofenac in poor-grade aSAH patients.
Conclusions
This study indicates that parenteral diclofenac effectively reduces body temperature, but may lead to CPP decline and brain-tissue hypoxia, which are both associated with poor outcome after SAH. Tight monitoring and awareness about possible hemodynamic side effects seem mandatory when using parenteral diclofenac in patients with poor-grade aSAH.
Key messages
• Tight monitoring of hemodynamic side effects is mandatory when using parenteral diclofenac in patients with poor-grade aSAH.
• Parenteral diclofenac after poor-grade aSAH effectively reduces body temperature, but may decrease MAP, CPP, and brain-tissue oxygen tension.
• PbtO2 <30 mm Hg before parenteral diclofenac was highly predictive of brain-tissue hypoxia (PbtO2 <20 mm Hg) after intervention.
Abbreviations
aSAH: aneurysmal subarachnoid hemorrhage; AR-1: autoregressive matrix of the first order; AUC: area under the curve; CBF: cerebral blood flow; CMD: cerebral microdialysis; CMRO2: cerebral metabolic rate of oxygen; COX: cyclooxygenase; CPP: cerebral perfusion pressure; CT: computed tomography; DCI: delayed cerebral infarction; GCS: Glasgow Coma Scale; GEE: generalized estimation equation; ICP: intracranial pressure; IQR: interquartile range; MAP: mean arterial pressure; mRS: modified Rankin Scale; NSAID: nonsteroidal antiinflammatory drug; OR: odds ratio; PbtO2: brain-tissue oxygen tension; PDMS: patient data-management system; PGE2: prostaglandin-E2; ROC: receiver operating characteristic; SEM: standard error of mean; TBI: traumatic brain injury; TCD: transcranial Doppler sonography.
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
AS was involved in the acquisition of data, statistical analysis, interpretation of data, study design, writing and manuscript drafting. RH was involved in the study design, interpretation of data, statistical analysis, manuscript writing and drafting, and final revision of the manuscript. ES, RB, BP, JS, MF, AD, and VK participated in the acquisition and interpretation of data and in the final revision of the manuscript. FS and CT were involved in the study design and data acquisition. WH was involved in study design, data processing, and statistical analysis. All authors read and approved the final manuscript.
Supplementary Material
Flow chart of patients and interventions included in the study. This file contains a flow chart of patients and interventions included in or excluded from the study.
Contributor Information
Alois J Schiefecker, Email: alois.schiefecker@gmail.com.
Bettina Pfausler, Email: bettina.pfausler@i-med.ac.at.
Ronny Beer, Email: ronny.beer@i-med.ac.at.
Florian Sohm, Email: florian.sohm@i-med.ac.at.
Jan Sabo, Email: jan.sabo@student.i-med.ac.at.
Viktoria Knauseder, Email: viktoria.knauseder@student.i-med.ac.at.
Marlene Fischer, Email: marlene.fischer@i-med.ac.at.
Anelia Dietmann, Email: anelia.dietmann@i-med.ac.at.
Werner O Hackl, Email: w.hackl@umit.at.
Claudius Thomé, Email: claudius.thome@i-med.ac.at.
Erich Schmutzhard, Email: erich.schmutzhard@i-med.ac.at.
Raimund Helbok, Email: raimund.helbok@uki.at.
Acknowledgements
We thank Georg Kemmler, PD, for advice on statistical analysis. Further we thank the nursing staff, all physicians of our neurointensive care unit, Monika Beck, and Alexandra Thoeni, B.Sc., for their overall support of this study. RH and AS are supported by a grant of the Austrian National Bank (OeNB-Nr.: 14082).
References
- Diringer MN, Reaven NL, Funk SE, Uman GC. Elevated body temperature independently contributes to increased length of stay in neurologic intensive care unit patients. Crit Care Med. 2004;32:1489–1495. doi: 10.1097/01.ccm.0000129484.61912.84. [DOI] [PubMed] [Google Scholar]
- Stocchetti N, Rossi S, Zanier ER, Colombo A, Beretta L, Citerio G. Pyrexia in head-injured patients admitted to intensive care. Intensive Care Med. 2002;28:1555–1562. doi: 10.1007/s00134-002-1513-1. [DOI] [PubMed] [Google Scholar]
- Fernandez A, Schmidt JM, Claassen J, Pavlicova M, Huddleston D, Kreiter KT, Ostapkovich ND, Kowalski RG, Parra A, Connolly ES, Mayer SA. Fever after subarachnoid hemorrhage: risk factors and impact on outcome. Neurology. 2007;68:1013–1019. doi: 10.1212/01.wnl.0000258543.45879.f5. [DOI] [PubMed] [Google Scholar]
- Zhang G, Zhang JH, Qin X. Fever increased in-hospital mortality after subarachnoid hemorrhage. Acta Neurochir. 2011;110:239–243. doi: 10.1007/978-3-7091-0353-1_42. [DOI] [PubMed] [Google Scholar]
- Busija DW, Leffler CW, Pourcyrous M. Hyperthermia increases cerebral metabolic rate and blood flow in neonatal pigs. Am J Physiol. 1988;255:H343–H346. doi: 10.1152/ajpheart.1988.255.2.H343. [DOI] [PubMed] [Google Scholar]
- Oddo M, Frangos S, Milby A, Chen I, Maloney-Wilensky E, Murtrie EM, Stiefel M, Kofke WA, Le Roux PD, Levine JM. Induced normothermia attenuates cerebral metabolic distress in patients with aneurysmal subarachnoid hemorrhage and refractory fever. Stroke. 2009;40:1913–1916. doi: 10.1161/STROKEAHA.108.534115. [DOI] [PubMed] [Google Scholar]
- Rossi S, Zanier ER, Mauri I, Columbo A, Stocchetti N. Brain temperature, body core temperature, and intracranial pressure in acute cerebral damage. J Neurol Neurosurg Psychiatry. 2001;71:448–454. doi: 10.1136/jnnp.71.4.448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackowiak PA. Concepts of fever. Arch Intern Med. 1998;158:1870–1881. doi: 10.1001/archinte.158.17.1870. [DOI] [PubMed] [Google Scholar]
- Scaravilli V, Tinchero G, Citerio G. Fever management in SAH. Neurocritical Care. 2011;15:287–294. doi: 10.1007/s12028-011-9588-6. [DOI] [PubMed] [Google Scholar]
- Cormio M, Citerio G, Spear S, Fumagalli R, Pesenti A. Control of fever by continuous, low-dose diclofenac sodium infusion in acute cerebral damage patients. Intensive Care Med. 2000;26:552–557. doi: 10.1007/s001340051203. [DOI] [PubMed] [Google Scholar]
- Zandstra DF, Stoutenbeek CP, Alexander JP. Antipyretic therapy with diclofenac sodium: observations on effect and serious side effects in critically ill patients. Intensive Care Med. 1983;9:21–23. doi: 10.1007/BF01693701. [DOI] [PubMed] [Google Scholar]
- Cormio M, Citerio G. Continuous low dose diclofenac sodium infusion to control fever in neurosurgical critical care. Neurocrit Care. 2007;6:82–89. doi: 10.1007/s12028-007-0002-3. [DOI] [PubMed] [Google Scholar]
- Raabe A, Beck J, Keller M, Vatter H, Zimmermann M, Seifert V. Relative importance of hypertension compared with hypervolemia for increasing cerebral oxygenation in patients with cerebral vasospasm after subarachnoid hemorrhage. J Neurosurg. 2005;103:974–981. doi: 10.3171/jns.2005.103.6.0974. [DOI] [PubMed] [Google Scholar]
- Schmidt JM, Ko SB, Helbok R, Kurtz P, Stuart RM, Presciutti M, Fernandez L, Lee K, Badjatia N, Connolly ES, Claassen J, Mayer SA. Cerebral perfusion pressure thresholds for brain tissue hypoxia and metabolic crisis after poor-grade subarachnoid hemorrhage. Stroke. 2011;42:1351–1356. doi: 10.1161/STROKEAHA.110.596874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramakrishna R, Stiefel M, Udoetuk J, Spiotta A, Levine JM, Kofke WA, Zager E, Yang W, Leroux P. Brain oxygen tension and outcome in patients with aneurysmal subarachnoid hemorrhage. J Neurosurg. 2008;109:1075–1082. doi: 10.3171/JNS.2008.109.12.1075. [DOI] [PubMed] [Google Scholar]
- Sarrafzadeh A, Haux D, Kuchler I, Lanksch WR, Unterberg AW. Poor-grade aneurysmal subarachnoid hemorrhage: relationship of cerebral metabolism to outcome. J Neurosurg. 2004;100:400–406. doi: 10.3171/jns.2004.100.3.0400. [DOI] [PubMed] [Google Scholar]
- Oddo M, Levine JM, Kumar M, Iglesias K, Frangos S, Maloney-Wilensky E, Le Roux PD. Anemia and brain oxygen after severe traumatic brain injury. Intensive Care Med. 2012;38:1497–1504. doi: 10.1007/s00134-012-2593-1. [DOI] [PubMed] [Google Scholar]
- Connolly ES Jr, Rabinstein AA, Carhuapoma JR, Derdeyn CP, Dion J, Higashida RT, Hoh BL, Kirkness CJ, Naidech AM, Ogilvy CS, Patel AB, Thompson BG, Vespa P. Guidelines for the management of aneurysmal subarachnoid hemorrhage: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2012;43:1711–1737. doi: 10.1161/STR.0b013e3182587839. [DOI] [PubMed] [Google Scholar]
- Frontera JA, Claassen J, Schmidt JM, Wartenberg KE, Temes R, Connolly ES Jr, Mac-Donald RL, Mayer SA. Prediction of symptomatic vasospasm after subarachnoid hemorrhage: the modified Fisher scale. Neurosurgery. 2006;59:21–27. doi: 10.1227/01.neu.0000243277.86222.6c. [DOI] [PubMed] [Google Scholar]
- Broessner G, Beer R, Lackner P, Helbok R, Fischer M, Pfausler B, Rhorer J, Kuppers-Tiedt L, Schneider D, Schmutzhard E. Prophylactic, endovascularly based, long-term normothermia in ICU patients with severe cerebrovascular disease: bicenter prospective, randomized trial. Stroke. 2009;40:e657–e665. doi: 10.1161/STROKEAHA.109.557652. [DOI] [PubMed] [Google Scholar]
- Oddo M, Levine JM, Frangos S, Maloney-Wilensky E, Carrera E, Daniel RT, Levivier M, Magistretti PJ, LeRoux PD. Brain lactate metabolism in humans with subarachnoid hemorrhage. Stroke. 2012;43:1418–1421. doi: 10.1161/STROKEAHA.111.648568. [DOI] [PubMed] [Google Scholar]
- Zeger SL, Liang KY. Longitudinal data analysis for discrete and continuous outcomes. Biometrics. 1986;42:121–130. [PubMed] [Google Scholar]
- Rosenthal G, Hemphill JC, Sorani M, Martin C, Morabito D, Obrist WD, Manley GT. Brain tissue oxygen tension is more indicative of oxygen diffusion than oxygen delivery and metabolism in patients with traumatic brain injury. Crit Care Med. 2008;36:1917–1924. doi: 10.1097/CCM.0b013e3181743d77. [DOI] [PubMed] [Google Scholar]
- Jaeger M, Soehle M, Schuhmann MU, Winkler D, Meixensberger J. Correlation of continuously monitored regional cerebral blood flow and brain tissue oxygen. Acta Neurochir (Wien) 2005;147:51–56. doi: 10.1007/s00701-004-0408-z. [DOI] [PubMed] [Google Scholar]
- Jaeger M, Schuhmann MU, Soehle M, Nagel C, Meixensberger J. Continuous monitoring of cerebrovascular autoregulation after subarachnoid hemorrhage by brain tissue oxygen pressure reactivity and its relation to delayed cerebral infarction. Stroke. 2007;38:981–986. doi: 10.1161/01.STR.0000257964.65743.99. [DOI] [PubMed] [Google Scholar]
- Gan TJ. Diclofenac: an update on its mechanism of action and safety profile. Curr Med Res Opin. 2010;26:1715–1731. doi: 10.1185/03007995.2010.486301. [DOI] [PubMed] [Google Scholar]
- Attwell D, Buchan AM, Charpak S, Lauritzen M, Macvicar BA, Newman EA. Glial and neuronal control of brain blood flow. Nature. 2010;468:232–243. doi: 10.1038/nature09613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soukup J, Zauner A, Doppenberg EM, Menzel M, Gilman C, Young HF, Bullock R. The importance of brain temperature in patients after severe head injury: relationship to intracranial pressure, cerebral perfusion pressure, cerebral blood flow, and outcome. J Neurotrauma. 2002;19:559–571. doi: 10.1089/089771502753754046. [DOI] [PubMed] [Google Scholar]
- Nakamura K. Central circuitries for body temperature regulation and fever. Am J Physiol. 2011;301:R1207–1228. doi: 10.1152/ajpregu.00109.2011. [DOI] [PubMed] [Google Scholar]
- Dunser MW, Hasibeder WR. Sympathetic overstimulation during critical illness: adverse effects of adrenergic stress. J Intensive Care Med. 2009;24:293–316. doi: 10.1177/0885066609340519. [DOI] [PubMed] [Google Scholar]
- Temes RE, Tessitore E, Schmidt JM, Naidech AM, Fernandez A, Ostapkovich ND, Frontera JA, Wartenberg KE, Di Tullio MR, Badjatia N, Connolly ES, Mayer SA, Parra A. Left ventricular dysfunction and cerebral infarction from vasospasm after subarachnoid hemorrhage. Neurocritical Care. 2010;13:359–365. doi: 10.1007/s12028-010-9447-x. [DOI] [PubMed] [Google Scholar]
- American Society of Health-System Pharmacists. Following IV administration of diclo-fenac sodium in healthy adults. Drug Information. 2011;1:2114. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Flow chart of patients and interventions included in the study. This file contains a flow chart of patients and interventions included in or excluded from the study.