

Abstract
Chronic pain affects approximately one in five adults, resulting in a greatly reduced quality of life and a higher risk of developing co-morbidities such as depression. Available treatments often provide inadequate pain relief, but it is hoped that through deeper understanding of the molecular mechanisms underlying chronic pain states we can discover new and improved therapies. Although genetic research has flourished over the past decade and has identified many key genes in pain processing, the budding field of epigenetics promises to provide new insights and a more dynamic view of pain regulation. This review gives an overview of basic mechanisms and current therapies to treat pain, and discusses the clinical and preclinical evidence for the contribution of genetic and epigenetic factors, with a focus on how this knowledge can affect drug development.
The cost of chronic pain
The impact of chronic pain is staggering. Affecting approximately one in five adults, chronic pain is associated with a significantly reduced quality of life and a higher risk of depression and other mental health disorders [1,2]. The economic costs of chronic pain reflect this: for example, in the UK, back pain alone is responsible for an estimated £5 billion of public funds each year [3]. Critically, current therapies to treat pain often fall short of patient expectations. In a recent survey, 40% of sufferers reported inadequate pain control [1]. The need for improved treatment options is clear.
Pain is still primarily treated with non-steroidal anti-inflammatory agents (NSAIDs), paracetamol and weak opioids, all of which have their shortcomings. NSAIDs and opioids have less than ideal side-effect profiles, inducing stomach ulceration and blood thinning in the case of NSAIDs and sedation, constipation and possible dependency in the case of opioids. Paracetamol reaches toxicity at relatively low doses, compromising its maximal analgesic effect. Within the pharmaceutical industry, the approach to find novel analgesics has primarily relied on our understanding of how current medication works and attempting to improve delivery to reduce side effects. But ultimately these drugs all function through the same mechanisms and do not provide greatly improved analgesia to patients.
It has not been easy to develop novel and effective classes of analgesic drugs - there have been almost no new registrations in the past 15 years. There has been much discussion about the reasons for past failures and this has stimulated an interest in exploring novel mechanisms, such as epigenetics [4]. One exception has been the recent use of biologics, drugs that are designed to mimic or block products made by the immune system. This approach was taken from the immunology field, where it was discovered that anti-tumor-necrosis factor alpha (TNFα) therapy can have rapid analgesic effects [5]. One study, which delivered the anti-TNFα drug etancercept perispinally, observed pain relief in patients within 20 minutes of application, probably before disease modification can have occurred [6]. Since then, the use of biologics to target known pain mediators has resulted in some of the most dramatic examples of analgesic drug efficacy in recent history. Tanezumab, an antibody directed against nerve growth factor, was found to radically reduce pain in a population of osteoarthritis patients [7]. Although initially the US Food and Drug Administration (FDA) halted trials because of the perceived increase in adverse events in the treatment group, this hold has been lifted and new trials will be permitted [8].
This example indicates that new approaches, based firmly on both preclinical and patient data, may give rise to greatly improved analgesics. There are several biological mechanisms that maintain chronic pain at the cellular level [9] and that may serve as potential targets (Box 1). Increasingly, genetic and epigenetic factors are being identified and implicated in these mechanisms. This review discusses what is known about these factors and how they might be harnessed for effective therapy.
Pain genetics
There is good evidence from twin [10-13] and population-based studies [14] that genetic risk factors can explain some of the individual differences in pain perception and the etiology of chronic pain conditions. For instance, heritability estimates range from 0.3 to 0.6 for chronic lower back pain and seem to be higher the more severe the condition [15,16]. Research has been focused on uncovering the genes responsible for these associations, in the hope that knowing their identity might not only lead to a deeper mechanistic understanding of chronic pain, but also to new therapeutic approaches. As in other fields, two main strategies have been adopted: one is to study rare familial pain conditions with Mendelian inheritance patterns, the other to use either candidate-gene or genome-wide association studies (GWASs) to identify polymorphisms that segregate with complex pain disorders (see [17] for a review).
Families with abnormal pain processing, in particular congenital insensitivity or indifference to pain, are very rare, probably because of the crucial importance of this sensation for survival. The condition most often co-occurs with neuropathy, falling under the umbrella term of 'hereditary and sensory autonomic neuropathy' (types 1 to 5). Point mutations have been identified in various genes as the underlying cause of different hereditary and sensory autonomic neuropathy types [18], most notably the gene encoding the TrkA receptor. Loss-of-function mutations in this gene result in a marked absence of small diameter sensory neurons [19]. Recently, a small number of families have been identified that present with insensitivity to pain without concomitant cell loss. Apart from an inability to experience pain and an impaired sense of smell, these individuals are ostensibly normal [20-22]. Mutations in the gene for the sodium channel Nav1.7 (SCN9A) were found to be responsible, supporting previous preclinical data from a transgenic knockout mouse that indicated this channel's critical role in normal nociceptive processing [23,24]. Sequence abnormalities in SCN9A can also result in the opposite phenotype of increased pain, with affected family members suffering from erythromelalgia (characterized by severe burning pain in the extremities commonly triggered by heat, pressure, exertion or stress [25]) [26,27] or paroxysmal extreme pain [28], depending on the location of the mutation (Figure 1).
Figure 1.

How polymorphisms can confer risk to pain. Single nucleotide polymorphisms (SNPs) can confer increased risk to pain by causing missense mutations that alter protein function. One of the most dramatic examples of this phenomenon is SNPs in the voltage-gated sodium channel Nav1.7. In this case, a SNP causing a change from an isoleucine to threonine residue in the loop domain leads to loss of channel inactivation, which is responsible for inherited paroxysmal pain disorder [27]. (a) Structure of Nav1.7. Arrow indicates the mutation in the loop domain. (b) Human embryonic kidney (HEK) cells transfected with wild-type Nav1.7 show normal channel inactivation. (c) HEK cells transfected with mutant Nav1.7 are unable to inactivate. Adapted with permission from [27].
Despite few families suffering from these conditions, the genes identified by studying them have given rise to promising new therapies. Several Trk kinase inhibitors are being developed [29], also on the basis of extensive preclinical work showing that neurotrophins (which areTrkA ligands) can act as potent pain mediators [30]. However, perhaps the most promising target to derive from genetic studies is Nav1.7. Historically, the development of selective blockers for sodium channels has proven difficult because of the high structural homology between isoforms, many of which have important roles in the heart and central nervous system [31]. Improved drug design has led to the development of new compounds that seem to have greater selectivity [32-36], and currently there are at least three phase II clinical trials underway to test their efficacy against pain of diverse etiologies [37-39]. Recently, Xenon Pharmaceuticals published results from a pilot study conducted in a small number of erythromelalgia patients with confirmed SCN9A mutations [40]. After 2 days of treatment with an orally administered Nav1.7 antagonist, the researchers induced pain in patients by warming of the skin or exercise. Treatment increased the time to reach maximal pain and significantly reduced pain after induction. Although preliminary, these results indicate that this may be an effective treatment when Nav1.7 is implicated in the pain pathophysiology [39].
Contrary to data derived from familial pain syndromes, results from genetic association studies are more applicable to the general population and, in the case of GWASs, should be able to give rise to the discovery of completely new targets. Many putative 'pain genes' have indeed been genetically linked to various chronic pain conditions [17,18,41], but study results have proven difficult to replicate and consequently are yet to have real impact on treatment approaches. Of a wide range of candidates, three have received particular attention from researchers and can be used to illustrate the contradictory nature of the findings in the field: GCH1, which encodes GTP cyclohydrolase; COMT, an enzyme that eliminates catecholamines; and OPRM1, the μ-opioid receptor gene. A GCH1 haplotype has been associated with reduced pain ratings in healthy volunteers and patients suffering from persistent leg pain [42,43]. However, the same association or indeed the same haplotype could not be identified in a larger cohort [44] or a different ethnic population of patients with HIV-associated neuropathy [45]. Similarly, conflicting evidence has emerged from the study of COMT. The first pain-associated single nucleotide polymorphism (SNP) was reported in 2003 [46] and has since been confirmed in various patient groups, healthy individuals [47,48] and animal models [49]. However, the importance of the original SNP (Val158Met) has been disputed [50], and the link between the COMT variants and increased pain perception does not replicate across populations. Most recently, a large study of more than 7,000 individuals revealed no association between chronic body pain and COMT SNPs [51]. Instead, the authors [51] found a haplotype within the ADRB2 gene (encoding the beta-2 adrenergic receptor), which predicted both pain severity and duration after controlling for confounding factors such as gender, social class and body mass index. Finally, significant linkage between pain and polymorphisms in the OPRM1 gene has been reported for more than a decade [52,53]. But, yet again, the connection failed to hold up in a larger dataset collated by meta-analysis [54].
There are many possible reasons for the lack of consensus in this area. Studies may not be examining cohorts that are uniform enough given the heterogeneity of chronic pain conditions: different biological mechanisms can give rise to similar symptoms and different symptoms can be caused by the same mechanism. Attempts to conduct more rigorous phenotyping and a move towards a mechanism-based classification of pain syndromes might help resolve this confounding factor [55]. Another issue that has also been observed in other fields is that genetic association data tend to be very population specific, partly because the tagging SNPs are often in different linkage disequilibrium blocks in different ethnic cohorts (for a review see [56]).
It also helps to remember that even 'genome-wide' association studies - currently still a rarity in the pain field - classically capture only common variants with a minor allelic frequency of ≥5%. Most variants identified have disappointingly small effect sizes, and thus explain only a fraction of the genetic contribution to a particular trait or disease [57]. More success might be achieved looking at structural variation, such as changes in copy number [58], or indeed highly penetrant rare variants with a minor allelic frequency of 0.5% or less [59]. Two recent publications used exon sequencing in large cohorts (n = 2,440, n = 14,000) and both reported the overwhelming majority of variants in European and African populations (about 90%) to be rare [60,61]. Although a first attempt at correlating rare variants with pain sensitivity in a healthy twin cohort did not reveal a single strong candidate, the authors [13] did identify a cluster of 30 genes within the angiotensin II pathway that segregated with thermal pain perception [13].
A final shortcoming of human genetic studies in pain research is that they have not taken into account potential gene-gene or gene-environment interactions. In mice, gene-environment interactions have repeatedly been demonstrated for nociceptive measures [62-64] and are very likely to exist in humans. The twin studies mentioned above [10-13] suggest that environmentally induced changes throughout an organism's lifetime account for at least half of the variance in pain perception and prevalence. Moreover, the environment can have a persistent impact, with childhood experiences affecting later pain outcomes [65,66]. Uncovering the mechanisms that allow such changes to occur is another avenue that might allow the discovery of new treatments. One possibility is that the environment exerts long-lasting influence on the genome through epigenetics; that is, stable or heritable changes in gene function that are not caused by changes in the DNA sequence itself. This is discussed in the following sections.
Pain epigenetics
The emerging field of epigenetics is an exciting new hope for pain research. Examples of epigenetic mechanisms include DNA methylation and post-translational histone modifications, both of which have consequences for gene expression and cell identity. Historically, research in this area has focused on dividing cells and mitotic heritability, but in recent years there has been much interest in studying epigenetic processes within the postmitotic environment of the nervous system.
Within the study of pain, it has become clear that widespread transcriptional dysregulation occurs throughout the pain neuraxis [67], a process that is thought to contribute to the hypersensitized state of the system. Whether these changes invoke epigenetic marks that allow their persistence is a question that is only beginning to be answered [68]. In the following sections, we discuss the early evidence for epigenetic modulation in pain, as well as the therapeutic potential of targeting relevant epigenetic mechanisms (summarized in Figure 2).
Figure 2.
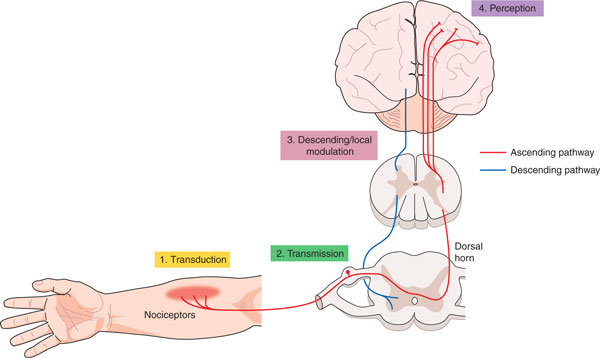
Evidence for epigenetic modulation in pain. Evidence has been obtained for such modulation at four different levels, numbered here in order from peripheral to central. 1, Pain-associated hyperacetylation of MIP2 and CXCR5 in the nerve after partial sciatic nerve ligation (PSL) [86] (shown in yellow). 2, Decreased expression of MeCP2 target genes after CFA [91]; miRNA expression changes [104,106]; intrathecal HDAC inhibitor treatment reduces acute pain after CFA [83] (shown in green). 3, GAD2 hypoacetylation after CFA leads to loss of descending inhibition [84] (shown in pink). 4, Carrageenan-associated miRNA dysregulation in the prefrontal cortex [105] (shown in purple).
Histone acetylation
The repeating subunit of chromatin is the nucleosome, composed of 147 bp of DNA wrapped around a core histone octamer. Histones are small, basic proteins whose tails are capable of being post-translationally modified in many ways, including acetylation, methylation and phosphorylation, all of which are thought to alter chromatin dynamics and gene expression in various ways [69]. Within the pain field there has been a growing interest in histone acetylation in particular, probably as a result of the availability of pharmacological inhibitors against effector proteins (discussed below), which have shown efficacy in processes that contribute to pain, such as inflammation (recently reviewed by Shakespear et al. [70]) and long-term potentiation [71-73]. Acetylation of histones occurs at lysine residues and is carried out by the histone acetyltransferase (HAT) enzyme family. This mark is thought to activate transcription directly through neutralization of the charged lysine, allowing DNA to be less tightly associated with the nucleosome, and also through recruitment of bromodomain proteins that can 'read' the mark and recruit transcriptional machinery. Acetylation is a dynamic process: deacetylation, associated with chromatin compaction and transcriptional repression, is carried out by the histone deacetylase family of enzymes (HDACs) [74].
Several compounds have been generated to block HDAC activity, which are generically called HDAC inhibitors. These drugs have varying degrees of specificity for the different classes of HDACs, although recent evidence suggests that the majority of them primarily function through targeting Class I HDACs [75,76], which may call for careful interpretation of study results claiming a role of a particular class of HDACs solely based on pharmacology. A further complication is that HDACs are not selective for histones and can also deacetylate targets in the cytoplasm, thereby exerting their effect through non-epigenetic mechanisms. As yet, it remains unclear whether it is more important to block particular HDACs than others in the treatment of pain. To explore this further, the development of isoform-specific HDACs or the use of transgenic animals will be required. In the past few years a limited number of isoform-specific HDAC inhibitors have been described (for example, compounds specific for HDAC3 [77] and HDAC6 [78]), but so far no Class IIa-specific inhibitors have been developed; these could be an interesting area for drug discovery, as Class IIa HDACs have a more restricted expression pattern than Class I HDACs, potentially reducing off-target effects. They also have been described to have important roles in learning and memory [79] and addiction [80,81], which share common neurobiological substrates and mechanisms with pain.
Although there are many caveats to keep in mind when interpreting the literature, there is growing evidence to suggest that interfering with HDAC activity can attenuate pain-related behavior in several preclinical inflammatory pain models [82-84]. As HDAC inhibitors have been shown to have anti-inflammatory properties in other models [70] this is perhaps unsurprising. The first group to show efficacy of HDAC inhibitors treatment in pain used repeated dosing with the HDAC inhibitors MS-275 or SAHA before testing animals using a model in which a noxious substance, formalin, is injected into the paw, which results in a characteristic biphasic nociceptive behavioral response. Treatment with HDAC inhibitors reduced the nociceptive phenotype in the second phase of the test, which was associated with increased acetylation of the NF-κB subunit p65/RelA and increased expression of one of its target genes, mGlu2, in the spinal dorsal horn and dorsal root ganglia. The anti-nociceptive effect of MS-275 was blocked with an mGlu2/3 receptor antagonist [82]. Another group, Bai et al. [83], has shown that intrathecal pre-treatment of the HDAC inhibitors SAHA, TSA, LAQ824, VPA and 4-PB could be anti-nociceptive in the acute stages of a different inflammatory pain model, injection of complete Freund's adjuvant (CFA), although whether the effects are mediated at the chromatin level or by enhanced acetylation of non-histone proteins is not clear [83].
More recently, Zhang et al. [84] determined that repeated injection of the HDAC inhibitor TSA into the nucleus raphe magnus (NRM) was able to attenuate CFA-induced hypersensitivity (Figure 3). The effect was attributed to the drug's ability to increase acetylation at the hypoacetylated Gad2 promoter, enhancing GAD65 expression and γ-aminobutyric acid (GABA) inhibition, and thus altering descending pain modulation [84]. Intriguingly, this group also demonstrated hypoacetylation of Gad2 in a model of neuropathic pain, indicating that this may be a common event in chronic pain states. It will be interesting to see whether HDAC inhibitor treatment in the NRM will be as effective in this and other models of chronic pain. A paper from Tran et al. [85] reports analgesic efficacy of intracerebroventricular administration of the same HDAC inhibitor, TSA, in a stress-induced visceral pain model. Whether this is mediated through the same GABAergic mechanism is not addressed, but this early evidence for the therapeutic potential of HDAC inhibitors warrants further investigation.
Figure 3.
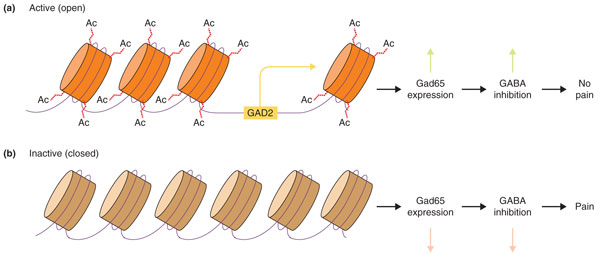
How epigenetic mechanisms can influence pain processing. (a) Under normal conditions, histone tails are acetylated at the GAD2 promoter in the nucleus raphe magnus (NRM). (b) After application of complete Freund's adjuvant (CFA), Gad65 expression is suppressed through hypoacetylation of the GAD2 promoter, leading to loss of descending inhibition from the NRM [84]. GABA, γ-aminobutyric acid.
In the neuropathic pain literature, there has only been one study that has looked directly at histone acetylation [86]. After experimentally induced peripheral nerve injury, hyperacetylation of histone H3 at lysine 9 (H3K9) was observed in the injured peripheral nerve. In this model, systemic administration of a HAT inhibitor was able to attenuate nociceptive behavior. Inhibition of p300, another HAT, has recently been reported to be analgesic in the chronic constriction injury model of neuropathic pain [87]. Further work to characterize the expression of chromatin modifying enzymes and their targets in pain-relevant anatomical substrates would help to inform future studies.
DNA methylation
DNA methylation is an epigenetic mechanism that is primarily associated with transcriptional repression. In vertebrates the majority of DNA methylation occurs at CpG dinucleotide sites, commonly found in gene promoters. DNA methylation is catalyzed by the DNA methyltransferase family of enzymes, DNMTs. Within this family are two distinct types of DNMTs: maintenance and de novo DNMTs. The canonical maintenance DNMT is Dnmt1, which recognizes and catalyzes addition of methyl groups to hemimethylated CpGs, thus allowing maintenance of methylation status across multiple cell divisions. In contrast, de novo DNMTs such as Dnmt3 can catalyze addition of methyl groups to areas regardless of current methylation state. These enzymes therefore are thought to confer a level of plasticity within this system, and indeed it has been shown that neurons can undergo rapid methylation changes in response to stimulation in vivo [88].
DNA methylation is thought to exert its repressive effects either directly by excluding DNA binding proteins from their target sites, such as in the case of binding of the transcriptional repressor CTCF at the H19 locus [89], or indirectly through the action of methyl mark readers, such as the methyl-CpG binding protein MeCP2 promoting recruitment of negative regulatory proteins [90].
Although regulation of DNA methylation has been found to be significant in other pathological states, so far only a few studies have addressed the potential role of DNA methylation in pain [91-96]. In the first of these, by Géranton et al. in 2007 [91], CFA was injected into the rat ankle joint and transcriptional changes within the spinal dorsal horn were assessed by microarray at various times after the injection. Analysis of the early transcription changes showed that several genes described as being targets of MeCP2 were highly upregulated, implying that loss of MeCP2 function is associated with early changes to central processing of pain. This group has extended their study of MeCP2 to other models, recently showing it to be downregulated in a rat neuropathic pain model, accompanied by concomitant changes to expression of HDAC1 and HDAC2, known effectors of MeCP2 action [92]. Further work to investigate causality - for example, by exogenously increasing MeCP2 expression in the dorsal horn - would provide strong evidence for this mechanism in the development of central sensitization, particularly as it has become clear since the first study [91] was published that neuronal MeCP2 binds globally across the mouse genome and does not have 'targets' as such [97].
Three studies have looked at promoter methylation in back pain [93,94] and cancer pain [95]. In these studies, pain was associated with methylation of target genes. In Viet et al. [95], inducing expression of the repressed gene attenuated pain behavior. Work from Wang et al. [96] also indicated that this strategy might be promising in a neuropathic pain model. After chronic constriction injury, this group [96] administered 5-azacytidine, a drug that works during cell division to block incorporation of methylated cytosine residues, and found that it could reduce nociceptive behavior. It will be of great interest to determine the cell types responsible for this effect.
The early preclinical data for chromatin-modifying drugs suggest that they may have some relevance for treating pain, particularly in the context of inflammation [82-84,86,96]. Many drugs have been developed against epigenetic mediators such as HDACs and HATs, and against epigenetic processes, such as maintenance of DNA methylation; a small minority of these compounds have been approved by the FDA for cancer therapy [98]. Although these compounds are available, their analgesic potential has not been systematically characterized in the clinic. Further work to determine which patient groups might benefit from this type of therapy is required.
MicroRNAs
Though not considered a classical epigenetic mechanism, the ability of microRNAs (miRNAs) to regulate gene expression has led some to consider these molecules as mediators of epigenetic change. As chronic pain is associated with aberrant gene expression [67], miRNAs may provide a unique therapeutic option whereby delivery of miRNAs may be used to return cells from a sensitized state to a baseline state through downregulation of pro-algesic transcripts. However, delivery of these molecules would be a challenge (for review, see [99]).
MicroRNAs are endogenously expressed, small 19- to 24-nucleotide RNA molecules that bind target mRNAs with imperfect complementarity, inhibiting translation and leading to downregulation of target proteins [100]. So far, various papers have been published that demonstrate the dysregulation of miRNAs in a number of preclinical pain models and tissues [101-106], indicating that this may be a fruitful area for further research, providing new targets for drug discovery. From these studies, 78 miRNAs have so far been reported to be transcriptionally altered in pain states, but only 8 miRNAs have been replicated in more than one study, probably because of the diverse models, organisms and tissues profiled. Further work to validate the function of these miRNAs would strengthen these associations with the respective pain states.
Although the majority of work in this field has been descriptive and correlational, two recent studies have moved beyond profiling and into direct modulation of miRNAs to determine the influence of these molecules on pain states in vivo [107,108]. The first direct evidence demonstrating a role of miRNAs in pain signaling came from Zhao et al. in 2010 [107]. This group took a conditional knockout approach to delete Dicer, a critical enzyme for miRNA maturation, within the Nav1.8-positive population of sensory neurons. Dicer conditional knockouts had largely normal baseline sensory phenotypes but had a marked insensitivity to inflammatory agents both behaviorally and electrophysiologically, demonstrating the necessity of Dicer and small RNAs for normal responses to nociceptive inflammatory stimuli.
More recently, Favereaux et al. [108] used bioinformatics to identify miR-103 as a potential regulator of the voltage-gated calcium channel Cav1.2, a channel that they had previously linked to chronic-pain-associated long-term potentiation in the spinal dorsal horn [109]. Intrathecal administration of a construct that blocked miR-103 increased sensitivity to mechanical stimuli in naive rats. Conversely, in rats with a peripheral nerve injury, repeated intrathecal injection of miR-103 attenuated nociceptive behaviors. Although validation of their construct indicated that it was able to downregulate Cav1.2 and its subunits, Rattus norvegicus (rno)-miR-103 has been predicted to target 1,675 different genes [110] and it is difficult to rule out the possibility that the effect of miR-103 administration may be due to its interaction with another target or set of targets, such as Vamp1, another gene that is commonly transcriptionally deregulated in neuropathic pain models [67]. Nevertheless, the behavioral effect is promising and serves as proof of concept that modulation of specific miRNAs may be an interesting new therapeutic strategy for chronic pain.
Conclusions and future directions
In the pain field, the contribution of genetic and epigenetic mechanisms is increasingly being recognized. The study of heritable pain syndromes in humans has helped confirm preclinical evidence, identifying several genes critical for pain sensation. This includes the voltage-gated sodium channel Nav1.7, which is currently the subject of multiple independent investigations and may prove to be an important new therapeutic target. Genetic association studies have yielded data that are more contradictory regarding which genes might be involved in the generation of chronic pain conditions. Some of the confusion might be resolved through the use of more rigorously phenotyped and homogeneous populations. New strategies, such as the identification of rare variants, will give a broader perspective regarding the heritability of pain and may, in turn, give rise to novel targets for drug discovery.
The early preclinical evidence for epigenetic modifying drugs is tantalizing, indicating that they could be beneficial in treating pain of varying etiologies. However, a likely stumbling block to the use of these drugs clinically will be off-target effects resulting from the wide-ranging roles of epigenetic mediators throughout the body. Improved specificity will need to be achieved through the development of more selective compounds or tissue-targeted drug delivery. Continued exploration of the analgesic activity of epigenetic drugs in preclinical studies is warranted. This should be accompanied by descriptive studies to characterize alterations to chromatin and DNA methylation states genome-wide, in specific cell types and pain models. This work will deepen our understanding of pain processing and give insight into how chromatin-modifying drugs may exert their therapeutic benefit.
Abbreviations
CFA: complete Freund's adjuvant; DNMT: DNA methyltransferase; GABA: γ-aminobutyric acid; GWAS: genome-wide association study; HAT: histone acetyltransferase; HDAC: histone deacetylase; miRNA: microRNA; NRM: nucleus raphe magnus; NSAID: non-steroidal anti-inflammatory drug; SNP: single nucleotide polymorphism.
Competing interests
The authors declare that they have no competing interests.
Box 1
There are three main biological mechanisms that contribute to persistent pain: peripheral sensitization of primary nociceptors within the dorsal root ganglion; central sensitization of spinal interneurons; and descending modulation of the pain signal from the brainstem and higher cortical centers [9]. At all levels of processing, significant cellular and molecular changes occur, such as large alterations in the transcriptional profile of these tissues [67].
Contributor Information
Megan Crow, Email: megan.crow@kcl.ac.uk.
Franziska Denk, Email: franziska@kcl.ac.uk.
Stephen B McMahon, Email: stephen.mcmahon@kcl.ac.uk.
Acknowledgements
This work was supported by grants to MC and SBM from the Wellcome Trust.
References
- Breivik H, Collett B, Ventafridda V, Cohen R, Gallacher D. Survey of chronic pain in Europe: Prevalence, impact on daily life, and treatment. Eur J Pain. 2006;10:287–333. doi: 10.1016/j.ejpain.2005.06.009. [DOI] [PubMed] [Google Scholar]
- Gureje O. VKMSGEGR. Persistent pain and well-being: A world health organization study in primary care. JAMA. 1998;280:147–151. doi: 10.1001/jama.280.2.147. [DOI] [PubMed] [Google Scholar]
- The British Pain Society - FAQs. http://www.britishpainsociety.org/media_faq.htm
- Kissin I. The development of new analgesics over the past 50 years: a lack of real breakthrough drugs. Anesth Analg. 2010;110:780–789. doi: 10.1213/ANE.0b013e3181cde882. [DOI] [PubMed] [Google Scholar]
- Elliott MJ, Maini RN, Feldmann M, Kalden JR, Antoni C, Smolen JS, Leeb B, Breedveld FC, Macfarlane JD, Bijl JA, Woody JN. Randomised double-blind comparison of chimeric monoclonal antibody to tumour necrosis factor α (cA2) versus placebo in rheumatoid arthritis. Lancet. 1994;344:1105–1110. doi: 10.1016/S0140-6736(94)90628-9. [DOI] [PubMed] [Google Scholar]
- Tobinick E, Davoodifar S. Efficacy of etanercept delivered by perispinal administration for chronic back and/or neck disc-related pain: a study of clinical observations in 143 patients. Curr Med Res Opin. 2004;20:1075–1085. doi: 10.1185/030079903125004286. [DOI] [PubMed] [Google Scholar]
- Lane NE, Schnitzer TJ, Birbara CA, Mokhtarani M, Shelton DL, Smith MD, Brown MT. Tanezumab for the treatment of pain from osteoarthritis of the knee. N Engl J Med. 2010;363:1521–1531. doi: 10.1056/NEJMoa0901510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanezumab: Arthritis Advisory Committee Briefing Document. http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/ArthritisAdvisoryCommittee/UCM295205.pdf
- Costigan M, Scholz J, Woolf CJ. Neuropathic pain: a maladaptive response of the nervous system to damage. Annu Rev Neurosci. 2009;32:1–32. doi: 10.1146/annurev.neuro.051508.135531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norbury TA, MacGregor AJ, Urwin J, Spector TD, McMahon SB. Heritability of responses to painful stimuli in women: a classical twin study. Brain. 2007;130:3041–3049. doi: 10.1093/brain/awm233. [DOI] [PubMed] [Google Scholar]
- Nielsen CS, Stubhaug A, Price DD, Vassend O, Czajkowski N, Harris JR. Individual differences in pain sensitivity: genetic and environmental contributions. Pain. 2008;136:21–29. doi: 10.1016/j.pain.2007.06.008. [DOI] [PubMed] [Google Scholar]
- Hartvigsen J, Nielsen J, Kyvik KO, Fejer R, Vach W, Iachine I, Leboeuf-Yde C. Heritability of spinal pain and consequences of spinal pain: a comprehensive genetic epidemiologic analysis using a population-based sample of 15,328 twins ages 20-71 years. Arthritis Rheum. 2009;61:1343–1351. doi: 10.1002/art.24607. [DOI] [PubMed] [Google Scholar]
- Williams FM, Scollen S, Cao D, Memari Y, Hyde CL, Zhang B, Sidders B, Ziemek D, Shi Y, Harris J, Harrow I, Dougherty B, Malarstig A, McEwen R, Stephens JC, Patel K, Menni C, Shin SY, Hodgkiss D, Surdulescu G, He W, Jin X, McMahon SB, Soranzo N, John S, Wang J, Spector TD. Genes contributing to pain sensitivity in the normal population: an exome sequencing study. PLoS Genet. 2012;8:e1003095. doi: 10.1371/journal.pgen.1003095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hocking LJ, Morris AD, Dominiczak AF, Porteous DJ, Smith BH. Heritability of chronic pain in 2195 extended families. Eur J Pain. 2012;16:1053–1063. doi: 10.1002/j.1532-2149.2011.00095.x. [DOI] [PubMed] [Google Scholar]
- Livshits G, Popham M, Malkin I, Sambrook PN, Macgregor AJ, Spector T, Williams FM. Lumbar disc degeneration and genetic factors are the main risk factors for low back pain in women: the UK Twin Spine Study. Ann Rheum Dis. 2011;70:1740–1745. doi: 10.1136/ard.2010.137836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacGregor AJ, Andrew T, Sambrook PN, Spector TD. Structural, psychological, and genetic influences on low back and neck pain: a study of adult female twins. Arthritis Rheum. 2004;51:160–167. doi: 10.1002/art.20236. [DOI] [PubMed] [Google Scholar]
- Mogil JS. Pain genetics: past, present and future. Trends Genet. 2012;28:258–266. doi: 10.1016/j.tig.2012.02.004. [DOI] [PubMed] [Google Scholar]
- Foulkes T, Wood JN. Pain genes. PLoS Genet. 2008;4:e1000086. doi: 10.1371/journal.pgen.1000086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Indo Y, Tsuruta M, Hayashida Y, Karim MA, Ohta K, Kawano T, Mitsubuchi H, Tonoki H, Awaya Y, Matsuda I. Mutations in the TRKA/NGF receptor gene in patients with congenital insensitivity to pain with anhidrosis. Nat Genet. 1996;13:485–488. doi: 10.1038/ng0896-485. [DOI] [PubMed] [Google Scholar]
- Cox JJ, Reimann F, Nicholas AK, Thornton G, Roberts E, Springell K, Karbani G, Jafri H, Mannan J, Raashid Y, Al-Gazali L, Hamamy H, Valente EM, Gorman S, Williams R, McHale DP, Wood JN, Gribble FM, Woods CG. An SCN9A channelopathy causes congenital inability to experience pain. Nature. 2006;444:894–898. doi: 10.1038/nature05413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss J, Pyrski M, Jacobi E, Bufe B, Willnecker V, Schick B, Zizzari P, Gossage SJ, Greer CA, Leinders-Zufall T, Woods CG, Wood JN, Zufall F. Loss-of-function mutations in sodium channel Nav1.7 cause anosmia. Nature. 2011;472:186–190. doi: 10.1038/nature09975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsen KB, Nicholas AK, Woods CG, Mellgren SI, Nebuchennykh M, Aasly J. Two novel SCN9A mutations causing insensitivity to pain. Pain. 2009;143:155–158. doi: 10.1016/j.pain.2009.02.016. [DOI] [PubMed] [Google Scholar]
- Nassar MA, Stirling LC, Forlani G, Baker MD, Matthews EA, Dickenson AH, Wood JN. Nociceptor-specific gene deletion reveals a major role for Nav1.7 (PN1) in acute and inflammatory pain. Proc Natl Acad Sci USA. 2004;101:12706–12711. doi: 10.1073/pnas.0404915101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raouf R, Quick K, Wood JN. Pain as a channelopathy. J Clin Invest. 2010;120:3745–3752. doi: 10.1172/JCI43158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith LA, Allen EV. Erythermalgia (erythromelalgia) of the extremities: a syndrome characterized by Redness, heat, and pain. Am Heart J. 1938;16:175–188. doi: 10.1016/S0002-8703(38)90693-3. [DOI] [Google Scholar]
- Yang Y, Wang Y, Li S, Xu Z, Li H, Ma L, Fan J, Bu D, Liu B, Fan Z, Wu G, Jin J, Ding B, Zhu X, Shen Y. Mutations in SCN9A, encoding a sodium channel alpha subunit, in patients with primary erythermalgia. J Med Genet. 2004;41:171–174. doi: 10.1136/jmg.2003.012153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segerdahl AR, Xie J, Paterson K, Ramirez JD, Tracey I, Bennett DL. Imaging the neural correlates of neuropathic pain and pleasurable relief associated with inherited erythromelalgia in a single subject with quantitative arterial spin labelling. Pain. 2012;153:1122–1127. doi: 10.1016/j.pain.2011.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fertleman CR, Baker MD, Parker KA, Moffatt S, Elmslie FV, Abrahamsen B, Ostman J, Klugbauer N, Wood JN, Gardiner RM, Rees M. SCN9A mutations in paroxysmal extreme pain disorder: allelic variants underlie distinct channel defects and phenotypes. Neuron. 2006;52:767–774. doi: 10.1016/j.neuron.2006.10.006. [DOI] [PubMed] [Google Scholar]
- Wang T, Yu D, Lamb ML. Trk kinase inhibitors as new treatments for cancer and pain. Expert Opin Ther Pat. 2009;19:305–319. doi: 10.1517/13543770902721261. [DOI] [PubMed] [Google Scholar]
- Pezet S, McMahon SB. Neurotrophins: mediators and modulators of pain. Annu Rev Neurosci. 2006;29:507–538. doi: 10.1146/annurev.neuro.29.051605.112929. [DOI] [PubMed] [Google Scholar]
- England S. Voltage-gated sodium channels: the search for subtype-selective analgesics. Expert Opin Investig Drugs. 2008;17:1849–1864. doi: 10.1517/13543780802514559. [DOI] [PubMed] [Google Scholar]
- Chowdhury S, Chafeev M, Liu S, Sun J, Raina V, Chui R, Young W, Kwan R, Fu J, Cadieux JA. Discovery of XEN907, a spirooxindole blocker of NaV1.7 for the treatment of pain. Bioorg Med Chem Lett. 2011;21:3676–3681. doi: 10.1016/j.bmcl.2011.04.088. [DOI] [PubMed] [Google Scholar]
- Hoyt SB, London C, Ok H, Gonzalez E, Duffy JL, Abbadie C, Dean B, Felix JP, Garcia ML, Jochnowitz N, Karanam BV, Li X, Lyons KA, McGowan E, Macintyre DE, Martin WJ, Priest BT, Smith MM, Tschirret-Guth R, Warren VA, Williams BS, Kaczorowski GJ, Parsons WH. Benzazepinone Nav1.7 blockers: potential treatments for neuropathic pain. Bioorg Med Chem Lett. 2007;17:6172–6177. doi: 10.1016/j.bmcl.2007.09.032. [DOI] [PubMed] [Google Scholar]
- McGowan E, Hoyt SB, Li X, Lyons KA, Abbadie C. A peripherally acting Nav1.7 sodium channel blocker reverses hyperalgesia and allodynia on rat models of inflammatory and neuropathic pain. Anesth Analg. 2009;109:951–958. doi: 10.1213/ane.0b013e3181b01b02. [DOI] [PubMed] [Google Scholar]
- Nguyen HN, Bregman H, Buchanan JL, Du B, Feric E, Huang L, Li X, Ligutti J, Liu D, Malmberg AB, Matson DJ, McDermott JS, Patel VF, Wilenkin B, Zou A, McDonough SI, Dimauro EF. Discovery and optimization of aminopyrimidinones as potent and state-dependent Nav1.7 antagonists. Bioorg Med Chem Lett. 2012;22:1055–1060. doi: 10.1016/j.bmcl.2011.11.111. [DOI] [PubMed] [Google Scholar]
- Schmalhofer WA, Calhoun J, Burrows R, Bailey T, Kohler MG, Weinglass AB, Kaczorowski GJ, Garcia ML, Koltzenburg M, Priest BT. ProTx-II, a selective inhibitor of NaV1.7 sodium channels, blocks action potential propagation in nociceptors. Mol Pharmacol. 2008;74:1476–1484. doi: 10.1124/mol.108.047670. [DOI] [PubMed] [Google Scholar]
- A Phase 2a, Randomized, Double-Blind, Placebo-Controlled, Two-Period Crossover Study to Evaluate the Safety, Tolerability, Preliminary Efficacy, and Systemic Exposure of Topical XPF-002 in Subjects With Postherpetic Neuralgia. http://clinicaltrials.gov/ct2/show/NCT01195636?term=xen402&rank=1
- Phase 2a, Exploratory, Double-blind, Placebo-controlled Two-part Study to Evaluate the Safety, Efficacy, Tolerability and Pharmacokinetics of Topically Applied XPF-002 (XEN402 8% w/w Ointment) in Patients With Primary/Inherited Erythromelalgia. http://clinicaltrials.gov/ct2/show/NCT01486446?term=xen402&rank=2
- A Randomized, Double Blind, Cross-over Study to Evaluate the Safety and Efficacy of CNV1014802 in Subjects With Neuropathic Pain From Lumbosacral Radiculopathy. http://clinicaltrials.gov/ct2/show/NCT01561027?term=CNV1014802&rank=2
- Goldberg YP, Price N, Namdari R, Cohen CJ, Lamers MH, Winters C, Price J, Young CE, Verschoof H, Sherrington R, Pimstone SN, Hayden MR. Treatment of Na(v)1.7-mediated pain in inherited erythromelalgia using a novel sodium channel blocker. Pain. 2012;153:8085. doi: 10.1016/j.pain.2011.09.008. [DOI] [PubMed] [Google Scholar]
- Diatchenko L, Nackley AG, Tchivileva IE, Shabalina SA, Maixner W. Genetic architecture of human pain perception. Trends Genet. 2007;23:605–613. doi: 10.1016/j.tig.2007.09.004. [DOI] [PubMed] [Google Scholar]
- Tegeder I, Costigan M, Griffin RS, Abele A, Belfer I, Schmidt H, Ehnert C, Nejim J, Marian C, Scholz J, Wu T, Allchorne A, Diatchenko L, Binshtok AM, Goldman D, Adolph J, Sama S, Atlas SJ, Carlezon WA, Parsegian A, Lötsch J, Fillingim RB, Maixner W, Geisslinger G, Max MB, Woolf CJ. GTP cyclohydrolase and tetrahydrobiopterin regulate pain sensitivity and persistence. Nat Med. 2006;12:1269–1277. doi: 10.1038/nm1490. [DOI] [PubMed] [Google Scholar]
- Campbell CM, Edwards RR, Carmona C, Uhart M, Wand G, Carteret A, Kim YK, Frost J, Campbell JN. Polymorphisms in the GTP cyclohydrolase gene (GCH1) are associated with ratings of capsaicin pain. Pain. 2009;141:114–118. doi: 10.1016/j.pain.2008.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, Dionne RA. Lack of influence of GTP cyclohydrolase gene (GCH1) variations on pain sensitivity in humans. Mol Pain. 2007;3:6. doi: 10.1186/1744-8069-3-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wadley AL, Lombard Z, Cherry CL, Price P, Kamerman PR. Analysis of a previously identified 'pain protective' haplotype and individual polymorphisms in the GCH1 gene in Africans with HIV-associated sensory neuropathy: a genetic association study. J Acquir Immune Defic Syndr. 2012;60:20–23. doi: 10.1097/QAI.0b013e31824bcc17. [DOI] [PubMed] [Google Scholar]
- Zubieta JK, Heitzeg MM, Smith YR, Bueller JA, Xu K, Xu Y, Koeppe RA, Stohler CS, Goldman D. COMT val158met genotype affects mu-opioid neurotransmitter responses to a pain stressor. Science. 2003;299:1240–1243. doi: 10.1126/science.1078546. [DOI] [PubMed] [Google Scholar]
- Mukherjee N, Kidd KK, Pakstis AJ, Speed WC, Li H, Tarnok Z, Barta C, Kajuna SL, Kidd JR. The complex global pattern of genetic variation and linkage disequilibrium at catechol-O-methyltransferase. Mol Psychiatry. 2010;15:216–225. doi: 10.1038/mp.2008.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diatchenko L, Nackley AG, Slade GD, Bhalang K, Belfer I, Max MB, Goldman D, Maixner W. Catechol-O-methyltransferase gene polymorphisms are associated with multiple pain-evoking stimuli. Pain. 2006;125:216–224. doi: 10.1016/j.pain.2006.05.024. [DOI] [PubMed] [Google Scholar]
- Segall SK, Nackley AG, Diatchenko L, Lariviere WR, Lu X, Marron JS, Grabowski-Boase L, Walker JR, Slade G, Gauthier J, Bailey JS, Steffy BM, Maynard TM, Tarantino LM, Wiltshire T. Comt1 genotype and expression predicts anxiety and nociceptive sensitivity in inbred strains of mice. Genes Brain Behav. 2010;9:933–946. doi: 10.1111/j.1601-183X.2010.00633.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, Mittal DP, Iadarola MJ, Dionne RA. Genetic predictors for acute experimental cold and heat pain sensitivity in humans. J Med Genet. 2006;43:e40. doi: 10.1136/jmg.2005.036079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hocking LJ, Smith BH, Jones GT, Reid DM, Strachan DP, Macfarlane GJ. Genetic variation in the beta2-adrenergic receptor but not catecholamine-O-methyltransferase predisposes to chronic pain: results from the 1958 British Birth Cohort Study. Pain. 2010;149:143–151. doi: 10.1016/j.pain.2010.01.023. [DOI] [PubMed] [Google Scholar]
- Bond C, LaForge KS, Tian M, Melia D, Zhang S, Borg L, Gong J, Schluger J, Strong JA, Leal SM, Tischfield JA, Kreek MJ, Yu L. Single-nucleotide polymorphism in the human mu opioid receptor gene alters beta-endorphin binding and activity: possible implications for opiate addiction. Proc Natl Acad Sci USA. 1998;95:9608–9613. doi: 10.1073/pnas.95.16.9608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendel B, Hoehe MR. The human mu opioid receptor gene: 5' regulatory and intronic sequences. J Mol Med (Berl) 1998;76:525–532. doi: 10.1007/s001090050246. [DOI] [PubMed] [Google Scholar]
- Walter C, Lotsch J. Meta-analysis of the relevance of the OPRM1 118A>G genetic variant for pain treatment. Pain. 2009;146:270–275. doi: 10.1016/j.pain.2009.07.013. [DOI] [PubMed] [Google Scholar]
- Maier C, Baron R, Tölle TR, Binder A, Birbaumer N, Birklein F, Gierthmühlen J, Flor H, Geber C, Huge V, Krumova EK, Landwehrmeyer GB, Magerl W, Maihöfner C, Richter H, Rolke R, Scherens A, Schwarz A, Sommer C, Tronnier V, Uçeyler N, Valet M, Wasner G, Treede RD. Quantitative sensory testing in the German Research Network on Neuropathic Pain (DFNS): somatosensory abnormalities in 1236 patients with different neuropathic pain syndromes. Pain. 2010;150:439–450. doi: 10.1016/j.pain.2010.05.002. [DOI] [PubMed] [Google Scholar]
- Frazer KA, Murray SS, Schork NJ, Topol EJ. Human genetic variation and its contribution to complex traits. Nat Rev Genet. 2009;10:241–251. doi: 10.1038/nrg2554. [DOI] [PubMed] [Google Scholar]
- Hardy J, Singleton A. Genomewide association studies and human disease. New Engl J Med. 2009;360:1759–1768. doi: 10.1056/NEJMra0808700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium WTCC. Genome-wide association study of CNVs in 16,000 cases of eight common diseases and 3,000 shared controls. Nature. 2010;464:713–720. doi: 10.1038/nature08979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson G. Rare and common variants: twenty arguments. Nat Rev Genet. 2011;13:135–145. doi: 10.1038/nrg3118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson MR, Wegmann D, Ehm MG, Kessner D, St Jean P, Verzilli C, Shen J, Tang Z, Bacanu SA, Fraser D, Warren L, Aponte J, Zawistowski M, Liu X, Zhang H, Zhang Y, Li J, Li Y, Li L, Woollard P, Topp S, Hall MD, Nangle K, Wang J, Abecasis G, Cardon LR, Zöllner S, Whittaker JC, Chissoe SL, Novembre J, Mooser V. An abundance of rare functional variants in 202 drug target genes sequenced in 14,002 people. Science. 2012;337:100–104. doi: 10.1126/science.1217876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tennessen JA, Bigham AW, O'Connor TD, Fu W, Kenny EE, Gravel S, McGee S, Do R, Liu X, Jun G, Kang HM, Jordan D, Leal SM, Gabriel S, Rieder MJ, Abecasis G, Altshuler D, Nickerson DA, Boerwinkle E, Sunyaev S, Bustamante CD, Bamshad MJ, Akey JM. Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science. 2012;337:64–69. doi: 10.1126/science.1219240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesler EJ, Wilson SG, Lariviere WR, Rodriguez-Zas SL, Mogil JS. Influences of laboratory environment on behavior. Nat Neurosci. 2002;5:1101–1102. doi: 10.1038/nn1102-1101. [DOI] [PubMed] [Google Scholar]
- Devor M, Gilad A, Arbilly M, Nissenbaum J, Yakir B, Raber P, Minert A, Pisante A, Darvasi A. Sex-specific variability and a 'cage effect' independently mask a neuropathic pain quantitative trait locus detected in a whole genome scan. Eur J Neurosci. 2007;26:681–688. doi: 10.1111/j.1460-9568.2007.05704.x. [DOI] [PubMed] [Google Scholar]
- Mogil JS, Sorge RE, LaCroix-Fralish ML, Smith SB, Fortin A, Sotocinal SG, Ritchie J, Austin JS, Schorscher-Petcu A, Melmed K, Czerminski J, Bittong RA, Mokris JB, Neubert JK, Campbell CM, Edwards RR, Campbell JN, Crawley JN, Lariviere WR, Wallace MR, Sternberg WF, Balaban CD, Belfer I, Fillingim RB. Pain sensitivity and vasopressin analgesia are mediated by a gene-sex-environment interaction. Nat Neurosci. 2011;14:1569–1573. doi: 10.1038/nn.2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones GT, Power C, Macfarlane GJ. Adverse events in childhood and chronic widespread pain in adult life: Results from the 1958 British Birth Cohort Study. Pain. 2009;143:92–96. doi: 10.1016/j.pain.2009.02.003. [DOI] [PubMed] [Google Scholar]
- Maneyapanda SB, Venkatasubramanian A. Relationship between significant perinatal events and migraine severity. Pediatrics. 2005;116:e555–558. doi: 10.1542/peds.2005-0454. [DOI] [PubMed] [Google Scholar]
- LaCroix-Fralish ML, Austin JS, Zheng FY, Levitin DJ, Mogil JS. Patterns of pain: meta-analysis of microarray studies of pain. Pain. 2011;152:1888–1898. doi: 10.1016/j.pain.2011.04.014. [DOI] [PubMed] [Google Scholar]
- Denk F, McMahon SB. Chronic pain: emerging evidence for the involvement of epigenetics. Neuron. 2012;73:435–444. doi: 10.1016/j.neuron.2012.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- Shakespear MR, Halili MA, Irvine KM, Fairlie DP, Sweet MJ. Histone deacetylases as regulators of inflammation and immunity. Trends Immunol. 2011;32:335–343. doi: 10.1016/j.it.2011.04.001. [DOI] [PubMed] [Google Scholar]
- Francis YI, Fà M, Ashraf H, Zhang H, Staniszewski A, Latchman DS, Arancio O. Dysregulation of histone acetylation in the APP/PS1 mouse model of Alzheimer's disease. J Alzheimers Dis. 2009;18:131–139. doi: 10.3233/JAD-2009-1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monsey MS, Ota KT, Akingbade IF, Hong ES, Schafe GE. Epigenetic alterations are critical for fear memory consolidation and synaptic plasticity in the lateral amygdala. PLoS One. 2011;6:e19958. doi: 10.1371/journal.pone.0019958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haettig J, Stefanko DP, Multani ML, Figueroa DX, McQuown SC, Wood MA. HDAC inhibition modulates hippocampus-dependent long-term memory for object location in a CBP-dependent manner. Learn Mem. 2011;18:71–79. doi: 10.1101/lm.1986911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haberland M, Montgomery RL, Olson EN. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat Rev Genet. 2009;10:32–42. doi: 10.1038/nrg2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradner JE, West N, Grachan ML, Greenberg EF, Haggarty SJ, Warnow T, Mazitschek R. Chemical phylogenetics of histone deacetylases. Nat Chem Biol. 2010;6:238–243. doi: 10.1038/nchembio.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilgore M, Miller CA, Fass DM, Hennig KM, Haggarty SJ, Sweatt JD, Rumbaugh G. Inhibitors of class 1 histone deacetylases reverse contextual memory deficits in a mouse model of Alzheimer's disease. Neuropsychopharmacology. 2010;35:870–880. doi: 10.1038/npp.2009.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rai M, Soragni E, Chou CJ, Barnes G, Jones S, Rusche JR, Gottesfeld JM, Pandolfo M. Two new pimelic diphenylamide HDAC inhibitors induce sustained frataxin upregulation in cells from Friedreich's ataxia patients and in a mouse model. PloS One. 2010;5:e8825. doi: 10.1371/journal.pone.0008825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inks ES, Josey BJ, Jesinkey SR, Chou CJ. A novel class of small molecule inhibitors of HDAC6. ACS Chem Biol. 2012;7:331–339. doi: 10.1021/cb200134p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MS, Akhtar MW, Adachi M, Mahgoub M, Bassel-Duby R, Kavalali ET, Olson EN, Monteggia LM. An essential role for histone deacetylase 4 in synaptic plasticity and memory formation. J Neurosci. 2012;32:10879–10886. doi: 10.1523/JNEUROSCI.2089-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renthal W, Maze I, Krishnan V, Covington HE, Xiao G, Kumar A, Russo SJ, Graham A, Tsankova N, Kippin TE, Kerstetter KA, Neve RL, Haggarty SJ, McKinsey TA, Bassel-Duby R, Olson EN, Nestler EJ. Histone deacetylase 5 epigenetically controls behavioral adaptations to chronic emotional stimuli. Neuron. 2007;56:517–529. doi: 10.1016/j.neuron.2007.09.032. [DOI] [PubMed] [Google Scholar]
- Wang L, Lv Z, Hu Z, Sheng J, Hui B, Sun J, Ma L. Chronic cocaine-induced H3 acetylation and transcriptional activation of CaMKIIalpha in the nucleus accumbens is critical for motivation for drug reinforcement. Neuropsychopharmacology. 2010;35:913–928. doi: 10.1038/npp.2009.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiechio S, Zammataro M, Morales ME, Busceti CL, Drago F, Gereau RWt, Copani A, Nicoletti F. Epigenetic modulation of mGlu2 receptors by histone deacetylase inhibitors in the treatment of inflammatory pain. Mol Pharmacol. 2009;75:1014–1020. doi: 10.1124/mol.108.054346. [DOI] [PubMed] [Google Scholar]
- Bai G, Wei D, Zou S, Ren K, Dubner R. Inhibition of class II histone deacetylases in the spinal cord attenuates inflammatory hyperalgesia. Mol Pain. 2010;6:51. doi: 10.1186/1744-8069-6-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Cai YQ, Zou F, Bie B, Pan ZZ. Epigenetic suppression of GAD65 expression mediates persistent pain. Nat Med. 2011;17:1448–1455. doi: 10.1038/nm.2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran L, Chaloner A, Sawalha AH, Greenwood Van-Meerveld B. Importance of epigenetic mechanisms in visceral pain induced by chronic water avoidance stress. Psychoneuroendocrinology. 2012. doi: 10.1016/j.psyneuen.2012.09.016. [DOI] [PubMed]
- Kiguchi N, Kobayashi Y, Maeda T, Fukazawa Y, Tohya K, Kimura M, Kishioka S. Epigenetic augmentation of the macrophage inflammatory protein 2/C-X-C chemokine receptor type 2 axis through histone H3 acetylation in injured peripheral nerves elicits neuropathic pain. J Pharmacol Exp Ther. 2012;340:577–587. doi: 10.1124/jpet.111.187724. [DOI] [PubMed] [Google Scholar]
- Zhu XY, Huang CS, Li Q, Chang RM, Song ZB, Zou WY, Guo QL. p300 exerts an epigenetic role in chronic neuropathic pain through its acetyltransferase activity in rats following chronic constriction injury (CCI). Mol Pain. 2012;8:84. doi: 10.1186/1744-8069-8-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo JU, Ma DK, Mo H, Ball MP, Jang MH, Bonaguidi MA, Balazer JA, Eaves HL, Xie B, Ford E, Zhang K, Ming GL, Gao Y, Song H. Neuronal activity modifies the DNA methylation landscape in the adult brain. Nat Neurosci. 2011;14:1345–1351. doi: 10.1038/nn.2900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hark AT, Schoenherr CJ, Katz DJ, Ingram RS, Levorse JM, Tilghman SM. CTCF mediates methylation-sensitive enhancer-blocking activity at the H19/Igf2 locus. Nature. 2000;405:486–489. doi: 10.1038/35013106. [DOI] [PubMed] [Google Scholar]
- Bird A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002;16:6–21. doi: 10.1101/gad.947102. [DOI] [PubMed] [Google Scholar]
- Géranton SM, Morenilla-Palao C, Hunt SP. A role for transcriptional repressor methyl-CpG-binding protein 2 and plasticity-related gene serum- and glucocorticoid-inducible kinase 1 in the induction of inflammatory pain states. J Neurosci. 2007;27:6163–6173. doi: 10.1523/JNEUROSCI.1306-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tochiki KK, Cunningham J, Hunt SP, Geranton SM. The expression of spinal methyl-CpG-binding protein 2, DNA methyltransferases and histone deacetylases is modulated in persistent pain states. Mol Pain. 2012;8:14. doi: 10.1186/1744-8069-8-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajerian M, Alvarado S, Millecamps M, Dashwood T, Anderson KM, Haglund L, Ouellet J, Szyf M, Stone LS. DNA methylation of SPARC and chronic low back pain. Mol Pain. 2011;7:65. doi: 10.1186/1744-8069-7-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams FM, Bansal AT, van Meurs JB, Bell JT, Meulenbelt I, Suri P, Rivadeneira F, Sambrook PN, Hofman A, Bierma-Zeinstra S, Menni C, Kloppenburg M, Slagboom PE, Hunter DJ, Macgregor AJ, Uitterlinden AG, Spector TD. Novel genetic variants associated with lumbar disc degeneration in northern Europeans: a meta-analysis of 4600 subjects. Ann Rheum Dis. 2012. doi:10.1136/annrheumdis-2012-201551. [DOI] [PMC free article] [PubMed]
- Viet CT, Ye Y, Dang D, Lam DK, Achdjian S, Zhang J, Schmidt BL. Re-expression of the methylated EDNRB gene in oral squamous cell carcinoma attenuates cancer-induced pain. Pain. 2011;152:2323–2332. doi: 10.1016/j.pain.2011.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Liu C, Guo Q-L, Yan J-Q, Zhu X-Y, Huang C-S, Zou W-Y. Intrathecal 5-azacytidine inhibits global DNA methylation and methyl- CpG-binding protein 2 expression and alleviates neuropathic pain in rats following chronic constriction injury. Brain Res. 2011;1418:64–69. doi: 10.1016/j.brainres.2011.08.040. [DOI] [PubMed] [Google Scholar]
- Skene PJ, Illingworth RS, Webb S, Kerr AR, James KD, Turner DJ, Andrews R, Bird AP. Neuronal MeCP2 is expressed at near histone-octamer levels and globally alters the chromatin state. Mol Cell. 2010;37:457–468. doi: 10.1016/j.molcel.2010.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szyf M. Epigenetics, DNA methylation, and chromatin modifying drugs. Annu Rev Pharmacol Toxicol. 2009;49:243–263. doi: 10.1146/annurev-pharmtox-061008-103102. [DOI] [PubMed] [Google Scholar]
- Garzon R, Marcucci G, Croce CM. Targeting microRNAs in cancer: rationale, strategies and challenges. Nat Rev Drug Discov. 2010;9:775–789. doi: 10.1038/nrd3179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/S0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- Aldrich BT, Frakes EP, Kasuya J, Hammond DL, Kitamoto T. Changes in expression of sensory organ-specific microRNAs in rat dorsal root ganglia in association with mechanical hypersensitivity induced by spinal nerve ligation. Neuroscience. 2009;164:711–723. doi: 10.1016/j.neuroscience.2009.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai G, Ambalavanar R, Wei D, Dessem D. Downregulation of selective microRNAs in trigeminal ganglion neurons following inflammatory muscle pain. Mol Pain. 2007;3:15. doi: 10.1186/1744-8069-3-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai S, Saeki M, Yanase M, Horiuchi H, Abe M, Narita M, Kuzumaki N, Suzuki T. Change in microRNAs associated with neuronal adaptive responses in the nucleus accumbens under neuropathic pain. J Neurosci. 2011;31:15294–15299. doi: 10.1523/JNEUROSCI.0921-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusuda R, Cadetti F, Ravanelli MI, Sousa TA, Zanon S, De Lucca FL, Lucas G. Differential expression of microRNAs in mouse pain models. Mol Pain. 2011;7:17. doi: 10.1186/1744-8069-7-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poh KW, Yeo JF, Ong WY. MicroRNA changes in the mouse prefrontal cortex after inflammatory pain. Eur J Pain. 2011;15:801 e801–812. doi: 10.1016/j.ejpain.2011.02.002. [DOI] [PubMed] [Google Scholar]
- von Schack D, Agostino MJ, Murray BS, Li Y, Reddy PS, Chen J, Choe SE, Strassle BW, Li C, Bates B, Zhang L, Hu H, Kotnis S, Bingham B, Liu W, Whiteside GT, Samad TA, Kennedy JD, Ajit SK. Dynamic changes in the microRNA expression profile reveal multiple regulatory mechanisms in the spinal nerve ligation model of neuropathic pain. PLoS One. 2011;6:e17670. doi: 10.1371/journal.pone.0017670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Lee MC, Momin A, Cendan CM, Shepherd ST, Baker MD, Asante C, Bee L, Bethry A, Perkins JR, Nassar MA, Abrahamsen B, Dickenson A, Cobb BS, Merkenschlager M, Wood JN. Small RNAs control sodium channel expression, nociceptor excitability, and pain thresholds. J Neurosci. 2010;30:10860–10871. doi: 10.1523/JNEUROSCI.1980-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Favereaux A, Thoumine O, Bouali-Benazzouz R, Roques V, Papon MA, Salam SA, Drutel G, Leger C, Calas A, Nagy F, Landry M. Bidirectional integrative regulation of Cav1.2 calcium channel by microRNA miR-103: role in pain. EMBO J. 2011;30:3830–3841. doi: 10.1038/emboj.2011.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fossat P, Dobremez E, Bouali-Benazzouz R, Favereaux A, Bertrand SS, Kilk K, Leger C, Cazalets JR, Langel U, Landry M, Nagy F. Knockdown of L calcium channel subtypes: differential effects in neuropathic pain. J Neurosci. 2010;30:1073–1085. doi: 10.1523/JNEUROSCI.3145-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betel D, Wilson M, Gabow A, Marks DS, Sander C. The microRNA.org resource: targets and expression. Nucleic Acids Res. 2008;36:D149–D153. doi: 10.1093/nar/gkm995. [DOI] [PMC free article] [PubMed] [Google Scholar]