

Abstract
p21-Activated serine-threonine kinase (PAK1) is implicated in breast cancer. We have shown previously that PAK1 is tyrosyl phosphorylated by prolactin (PRL)-activated Janus tyrosine kinase (JAK2). Although a role for both PRL and PAK1 in breast cancer is widely acknowledged, the mechanism remains poorly understood. In the present study, PRL-activated PAK1 stimulates the invasion of TMX2–28 human breast cancer cells through Matrigel. Three-dimensional (3D) collagen IV stimulates the secretion of the matrix proteases, metalloproteinase (MMP)-1 and -3 that is further enhanced by the PRL-dependent tyrosyl phosphorylation of PAK1. 3D collagen IV also stimulates the expression and secretion of MMP-2, but in contrast to MMP-1 and -3, PRL/PAK1 signaling down-regulates MMP-2 expression and secretion. In contrast, MMP-9 expression and secretion are stimulated by 3D collagen I, not collagen IV, and are not affected by PRL but are down-regulated by PAK1. MMP-1 and -3 are required and MMP-2 contributes to PRL-dependent invasion. ERK1/2 signaling appears to be required for the enhanced expression and secretion of MMP-1 and -3 and enhanced PRL-dependent invasion. p38 MAPK and c-Jun N-terminal kinase 1/2 pathways participate in production of MMP-1 and -3 as well as in PRL/PAK1-dependent cell invasion. Together, these data illustrate the complex interaction between the substratum and PRL/PAK1 signaling in human breast cancer cells and suggest a pivotal role for PRL-dependent PAK1 tyrosyl phosphorylation in MMP secretion.
Breast cancer is one of the most common malignancies affecting women. Colonization of distant tissues by tumor cells represents the most dangerous attribute of cancer. One hallmark of invasive breast cancer cells is increased expression and activity of matrix metalloproteinases (MMPs) that contribute to invasive potential. MMPs are a family of Zn2+-dependent enzymes composing 23 members. MMP-1 (collagenase 1) specifically degrades collagen I, a major component of the extracellular matrix (ECM) and other fibrillar collagens. MMP-1 is critical for the ECM remodeling. In clinical studies, increased MMP-1 expression is associated with advanced stages of breast cancer and may be a predictive marker for invasive disease (1). MMP-3, or stromelysin 1, degrades a variety of ECM substrates, including collagens. MMP-3 is up-regulated in breast tumors and contributes to cancer development. Indeed, mice overexpressing MMP-3 show excessive side-branching and eventual formation of mammary tumors (2). MMP-2 and MMP-9 are both type IV collagenases that contribute to tumor invasion in vitro because of their ability to break down basement membrane, degrading collagen IV in particular. Elevated circulating MMP-9 levels are apparent in breast cancers and MMP-2 and/or MMP-9 release is associated with tumor invasion and metastasis (reviewed in Refs. 3 and 4). The expression of MMPs is regulated at the transcriptional and posttranscriptional levels (including the stability of mRNA and protein, as well as the release and activation of protein) by hormones, growth factors, and cytokines. Despite efforts to discover the cellular pathways regulating MMPs, little is known as to how different cytokines cooperate with cytoskeletal proteins to regulate MMPs expression.
Cells adhere to the ECM throughout most of their lifetime. The molecular composition of the ECM, specific association of multiple growth factors/cytokines with the matrix, and “dimensionality” play major roles in the response of cells to their local matrix microenviroment (5). Three-dimensional (3D) matrix is a critical component of mammary tissue development not only under physiologic but also in pathophysiologic conditions. In vivo, women with dense mammary tissue, associated with an increasing amount of collagen in the stroma, are at 4–6 times greater risk of breast cancer and have a poor prognosis (6, 7). In vitro, increasing 3D matrix tension affects mammary cell morphogenesis and physiologic functions. Furthermore, reciprocal interactions between mammary epithelial cells, ECM, and ECM-remodeling enzymes are critical for development and differentiation during mammary gland development. Loss of this interaction leads to tumor progression (reviewed in Ref. 8).
Prolactin (PRL), a hormone of the GH/cytokine family, exerts both the endocrine and autocrine/paracrine effects and functions in both reproduction and as a cytokine exerting profound effects on a wide range of tissues, with more than 300 effects described in vertebrates (for review see Refs. 9 and 10). PRL binding to its receptor activates tyrosine kinase JAK2, PRL receptor phosphorylation, and the phosphorylation of signal transducer and activator of transcription (STAT)5a and 5b, STAT3, and STAT1. PRL also activates MAPKs, including ERK1/2, ERK5, p38 and c-Jun N-terminal kinase (JNK)1/2, protein kinase C, and PI-3 kinase (11). There is now clear evidence that PRL is involved in breast cancer (for review see Refs. 12 and 13). PRL increases motility of breast cancer cells (14). These data, combined with animal studies reporting either increased metastases with PRL administration (15) or prevention of neoplasia progression into invasive carcinoma in PRL receptor deficient mice (16), suggest that PRL is involved in the development of metastasis and tumor progression although the exact mechanism remains to be clarified. In contrast, PRL was reported to act as a suppressor of breast cancer cell invasion in vitro (17, 18), suggesting that the role of PRL in breast cancer must be explored further.
p21-Activated serine-threonine kinase (PAK1) is one of the targets of PRL-activated JAK2 (19) and has been implicated in breast cancer progression (20). PAK1 is overexpressed or up-regulated in some breast cancers. Overexpression of PAK1 was observed in 34 of 60 breast tumor specimens (21), and expression of PAK1 in human breast tumors correlates with tumor grade (22). Highly proliferating human breast cancer cell lines and tumor tissues express hyperactive PAK1 (23). Of particular interest, PAK1 plays a critical role in premalignant progression of MCF10 series of human breast epithelial cell lines grown in 3D reconstituted basal membrane overlay cultures (24). Activated PAK1 increases invasion of breast cancer cells, and expression of a kinase-dead PAK1 mutant in highly invasive breast cancer lines leads to reduced invasiveness (25). Conversely, hyperactivation of the PAK1 pathway in the noninvasive breast cancer cell line MCF-7 promotes cell migration and anchorage-independent growth (26). PAK1 phosphorylates several transcription factors, among them the C-terminal-binding protein I and snail homologue 1, both of which are important for epithelial-mesenchymal transition (27, 28). Another possible mechanism of PAK1-mediated malignant transformation is the enhancement of PAK1-regulated cell motility because PAK1 kinase activity participates in directional motility and PAK1 directly phosphorylates cytoskeletal proteins (20). We have shown that PAK1 is a JAK2 substrate and that PRL-activated JAK2 phosphorylates PAK1 in vivo on tyrosines 153, 201, and 285 (19). We have recently demonstrated that the PAK1 substrate filamin A (FLNa) plays a role in PRL-dependent cell motility (29, 30). Thus, PAK1 has become a focal point in the investigation into the mechanism and onset of human breast cancer.
Here, we extended our findings on the role of tyrosyl-phosphorylated PAK1 in breast cancer cell invasion. We used human breast cancer TMX2–28 cells, a highly invasive variant of the MCF-7 cell line. We have established TMX2–28 cell lines that stably overexpress PAK1 WT or a PAK1 Y3F mutant in which the 3 JAK2 phosphorylation sites (Tyr(s) 153, 201, and 285) were mutated to phenylalanine. We have demonstrated that PAK1 phosphorylation is required for maximal PRL-dependent invasion of TMX2–28 cells through Matrigel and that 3D collagen IV acts synergistically with PRL-activated PAK1 to induce the expression of MMP-1 and MMP-3. In contrast, PRL-activated PAK1 decreases the 3D collagen IV induction of MMP-2. MMP-1 and -3 are required and MMP-2 contributes to PRL-dependent invasion. Taken together, these data suggest that PRL can exert differential effects on the expression of different MMPs, depending on the extracellular environment.
Results
Characterization of TMX2–28 cell lines expressing PAK1 WT and PAK1 Y3F mutant
We have shown previously that the JAK2 phosphorylation of PAK1 tyrosines 153, 201, and 285 promotes PRL-dependent changes in the actin cytoskeleton and cell motility (19, 29). To assess the significance of tyrosyl-phosphorylated PAK1 (pTyr-PAK1) in breast cancer cell invasion, we used TMX2–28 cells, a variant of the MCF-7 cell line (31). These cells are PRL responsive and highly invasive compared with poorly invasive MCF-7 cells (32). We have established TMX2–28 cell lines that stably express green fluorescent protein (GFP) alone (as vector control) or with either myc-tagged PAK1 WT or a PAK1 Y3F mutant in which the 3 JAK2 phosphorylation sites (Tyr[s]) 153, 201, and 285) were mutated to phenylalanine. These retroviral constructs include internal ribosome entry site (IRES) elements that allow the transcription of a single bicistronic mRNA of myc-PAK1-IRES2-EGFP, and so produce PAK1 with N-terminal myc tag together with EGFP as a reporter for expression of PAK1 (Figure 1A).
Figure 1.

Characterization of TMX2–28 Cell Lines Stably Expressing GFP, or GFP with myc-Tagged PAK1 WT and PAK1 Y3F. A, Parental TMX2–28 cells (lane 1), TMX2–28 cells stably expressing either GFP (lane 2), myc-PAK1 WT (lane 3), or myc-PAK1 Y3F (lane 4) were lysed, and proteins were resolved by SDS-PAGE. Overexpressed proteins were visualized by immunoblotting with anti-myc Ab (upper panel). The same polyvinylidene difluoride (PVDF) membrane was stripped and reblotted with anti-PAK1 Ab to visualize endogenous (lanes 1 and 2) and overexpressed PAK1 (lanes 3 and 4) (upper middle panel) and with anti-GFP Ab (low middle panel). Immunoblotting with antiactin Ab was used as a loading control (bottom panel). B, myc-PAK1 was IP'd with anti-myc Ab from TMX2–28 PAK1 WT (lanes 1–3) or TMX2–28 PAK1 Y3F cells (lanes 4–6) transfected with vector (lanes 1 and 4), constitutive active Rac1 V12 (lanes 2 and 5), or constitutive active Cdc42 L61 (lanes 3 and 6) and immunoblotted with the indicated antibodies. The migrations of proteins are indicated. C, TMX2–28 PAK1 WT (left panel) and TMX2–28 PAK1 Y3F cells (right panel) were deprived of serum overnight and treated with or without 30 ng/mL HRG for 15 minutes. Myc-PAK1 was IP'd with anti-myc Ab and subjected to an in vitro kinase assay with H4 histone as a substrate and probed with anti-PAK1 Ab. Relative PAK1 kinase activity was then normalized by the amount of IP'd PAK1 for each lane. The numbers at the bottom of upper and middle panels give the relative fold increase of 32P incorporation into H4 histone (upper panel) and into PAK1 protein (middle panel). The migrations of proteins are indicated. All blots are representative of at least 3 experiments. D, TMX2–28 PAK1 WT and TMX2–28 PAK1 Y3F cells were deprived of serum overnight and treated with or without 200 ng/mL PRL for 20 minutes. Myc-PAK1 was IP'd with anti-myc Ab and subjected to the in vitro kinase assay as described in panel C. Relative PAK1 kinase activity was then normalized by the amount of IP'd PAK1 for each lane and plotted (right panel). The migrations of proteins are indicated. Bars represent mean ± S.E. *, P < .05 compared with the same cells untreated with PRL (n = 3). All blots are representative of at least 3 experiments.
To determine whether PAK1 Y3F protein was functional and activated by active Rac1 and Cdc42, myc-tagged PAK1 WT and PAK1 Y3F were immunoprecipitated (IP'd) from cell lines transiently transfected with constitutively active Rac1 V12 or constitutively active Cdc42 L61. The activation states of the IP'd PAK1 WT and PAK1 Y3F were assessed by blotting with anti-phospho-T423-PAK1 Ab (phosphorylated threonine 423 is a marker of PAK1 activation). Both PAK1 WT and PAK1 Y3F were similarly activated by either Rac1 V12 or by Cdc42 L61 (Figure 1B). Heregulin (HRG), a ligand for HER3 (human epidermal growth factor receptor-3) and HER4 (human epidermal growth factor receptor-4), activates PAK1 in MCF-7 cells (33). To confirm that PAK1 Y3F retains its kinase activity, myc-tagged PAK1 WT and PAK1 Y3F were IP'd from the TMX2–28 PAK1 WT and TMX2–28 PAK1 Y3F clones treated with HRG (30 ng/mL, 15 min), and their kinase activities were measured in an in vitro kinase assay with H4 histone as a substrate. As shown in Figure 1C, HRG-induced PAK1 kinase activity and autophosphorylation were similar in the cells expressing PAK1 WT and PAK1 Y3F.
To determine whether PRL treatment altered PAK1 kinase activity, TMX2–28 clones were treated without or with PRL, and PAK1 kinase activity was measured using an in vitro kinase assay. PRL treatment more than doubled PAK1 WT kinase activity, compared with untreated control (Figure 1D). PRL can activate both PAK1 WT and PAK1 Y3F through Rac1 (PRL activates Rac1 (34)). However, in the presence of PRL, the kinase activity of PAK1 WT was significantly stronger than PAK1 Y3F (Figure 1D). These data demonstrate that PAK1 WT and PAK1 Y3F have similar kinase activities in response to active Rac1/Cdc42 and HRG. PRL stimulates both PAK1 WT and PAK1 Y3F probably through Rac1/Cd42 and further increases PAK1 WT kinase activity, suggesting that tyrosines 153, 201, and 285 are involved in regulating this enhanced activity.
PRL-activated PAK1 stimulates cell invasion
Because PAK1 activation is closely linked with the invasiveness of MCF-7 cells (poorly invasive), as well as MDA-MB435 and MDA-MB231 cells (highly invasive) (25, 33), we asked whether the PRL-dependent tyrosyl phosphorylation of PAK1 is also required for the maintenance of the invasive phenotype in breast cancer cells. The effect of pTyr-PAK1 WT on PRL-induced invasion was demonstrated by evaluating migration through Matrigel (Figure 2). Overexpression of PAK1 WT accelerated migration in response to PRL (78 invaded cells) as compared with cells overexpressing GFP (51 invaded cells), whereas overexpression of PAK1 Y3F significantly inhibited cell invasion (35 invaded cells) (Figure 2A). These data suggest that PAK1 Y3F inhibits cell invasion and implicates tyrosyl phosphorylation in the potentiating effect of PAK1 on cell invasiveness.
Figure 2.

Maximal Invasion of TMX2–28 Cells in Response to PRL Requires Tyrosyl Phosphorylation of PAK1. A, TMX2–28 cells stably overexpressing GFP, PAK1 WT, or PAK1 Y3F were serum deprived, and equal amounts of cells were loaded into the upper part of the Boyden chamber coated with the Matrigel. The number of cells that migrated to the lower surface of the chamber toward PRL (100 ng/mL) (black bar) or buffer control (white bar) after 48 hours were counted in 5 random fields and plotted. B, TMX2–28 cells were transfected with control or PAK1 siRNA and assessed for invasion as in panel A. Silencing efficiency was judged by immunoblotting with anti-PAK1 Ab 48 and 72 hours after transfection. The expression levels of actin were used as an internal control. Bars represent mean ± SE. *, P < .05 compared with untreated cells. Each experiment was repeated three times. Scale bar, 300 μm.
To directly establish the significance of PAK1 signaling on PRL-dependent cell invasion, we next examined the effect of knockdown of endogenous PAK1 by PAK1-specific small interfering RNA (siRNA). We found that PAK1-depleted cells maintained basal invasion (white bars in plots in Figure 2B) but demonstrated ablation of PRL-induced migration (black bars in plots in Figure 2B).
Tyrosyl-phosphorylated PAK1 regulates the production of invasive-relevant MMPs in response to PRL
An important step in the invasion process is the destruction of ECM by MMPs. To test whether altered secretion of MMPs by PRL-activated PAK1 regulates the invasion of TMX2–28 cells, we assessed the secretion of MMP-1, MMP-2, MMP-3, and MMP-9, because these MMPs play an important role in breast cancer invasion and metastasis (35, 36). Both collagen I or IV induced MMP transcription and/or secretion, and therefore we plated cells onto dishes covered with either collagen I or IV. Because a role of the 3D collagen lattice in the transcriptional control and/or activation of different MMPs has been demonstrated previously (eg Refs. 37–40), we also embedded the cells in either collagen I or IV, after which we treated them with or without PRL and measured MMP secretion (Figure 3).
Figure 3.


Collagens and PRL-Dependent Tyrosyl Phosphorylation of PAK1 Regulate Secretion of Invasive-Relevant MMPs. TMX2–28 cells stably overexpressing GFP, PAK1 WT, or PAK1 Y3F were plated on plastic, on plastic covered with collagen I, embedded in collagen I, on plastic covered with collagen IV, or embedded in collagen IV. After 24 hours of serum deprivation, the cells were treated with (black bars) or without (white bars) 100 ng/mL PRL for 48 hours, and the conditioned medium was assessed for protein level of secreted MMP-1 (A), MMP-3 (B), MMP-2 (C), and MMP-9 (D) by ELISA. E, TMX2–28 cells were transfected with control or PAK1 siRNA, stimulated (black bars) or not (white bars) with PRL, and assessed for secretion of MMPs as in panels A–D. Bars represent mean ± SE. *, P < .05 compared with the same cells untreated with PRL. #, P < .05 compared with cells expressing GFP with the same treatment. Each experiment was repeated 3 times.
In TMX2–28 lines plated on plastic, collagen I or IV, a moderate increase in MMP-1 release was observed in PAK1 WT cells treated with PRL, but plating cells on either type of collagen had no effect (Figure 3A). In contrast, cells overexpressing PAK1 WT and embedded in collagen IV exhibited dramatically induced MMP-1 secretion (white bar for WT cells embedded in collagen IV in Figure 3A); treatment of these cells with PRL further stimulated MMP-1 secretion (black bar). 3D collagen IV did not affect MMP-1 secretion in the cells overexpressing GFP and PAK1 Y3F regardless of PRL treatment, suggesting that tyrosines 153, 201, and 285 of PAK1 are required for the effect of collagen IV on MMP-1 secretion. Interestingly, cells embedded in collagen I also exhibited enhanced MMP-1 secretion (Figure 3A), although not as strong as 3D collagen IV (2-fold increase for WT cells treated with PRL as compared with GFP cells treated with PRL for 3D collagen I vs 15-fold increase for the WT cells embedded in collagen IV). Collectively, TMX2–28 cells overexpressing PAK1 WT, treated with PRL, and embedded in collagen IV exhibited the strongest effect on MMP-1 release as compared with control GFP and PAK1 Y3F cells or PAK1 WT cells grown on the surface of collagen IV.
MMP-3 regulation by PRL-activated PAK1 and the different substrates was similar to that observed for MMP-1. 3D collagen IV significantly increased MMP-3 secretion, especially when combined with PRL treatment (Figure 3B). This effect was again pTyr-PAK1 PRL-dependent dependent because PAK1 Y3F cells failed to induce MMP-3 secretion in response to 3D collagen IV with or without PRL (Figure 3B).
We next assessed MMP-2 secretion in TMX2–28 cell clones plated on either plastic, collagen I, collagen IV, or embedded in collagen I or IV. Embedding cells in 3D collagen IV induced MMP-2 secretion relative to the other substrates (Figure 3C). Without PRL treatment, this effect was PAK1-independent (compare white bars for GFP, PAK1 WT, and PAK1 Y3F cells for each type of substrate). However, after PRL treatment, control GFP cells and cells overexpressing PAK1 WT embedded in collagen IV secreted significantly less MMP-2 than without PRL (compare black and white bars for GFP and WT cells embedded in collagen IV in Figure 3C). In contrast, PRL had no effect on MMP-2 secretion by PAK1 Y3F cells, confirming the dominant-negative role of this mutant in the regulation of PRL-PAK1-dependent MMP-2 secretion.
We assessed secretion of MMP-9 under the conditions described above. In contrast to MMPs 1, 2, and 3, secretion of MMP-9 was PRL-independent (compare white and black bars in Figure 3D) and negatively regulated by PAK1. WT PAK1 and PAK1 Y3F both inhibited MMP-9 secretion. Neither 2D nor 3D collagen IV affected MMP-9 secretion, whereas 3D collagen I dramatically induced secretion (3-fold increase in GFP cells embedded in collagen I as compared with GFP cells plated on plastic), which is in contrast to MMPs 1, 2, and 3.
To assess the role of PAK1 in PRL-induced MMP secretion, we knocked down PAK1 in TMX2–28 cells and evaluated secretions of MMP-1, -2, -3, and -9. We found that PAK1-depleted cells exhibited decreased basal and PRL-induced secretion of MMP-1, -2, and -3 (Figure 3E). Interestingly, secretion of MMP-9 was stimulated by the absence of PAK1, which is in agreement with our data on the inhibitory effect of PAK1 on MMP-9 production (Figure 3D).
Our data suggest that 3D collagen IV strongly stimulates the secretion of MMP-1 and -3 via a PAK1-dependent pathway and that PRL treatment augments this effect via the PAK1 tyrosyl phosphorylation. 3D collagen IV also stimulates MMP-2 secretion that was inhibited by p(Tyr)-PAK1. In contrast, MMP-9 secretion is PRL-independent and stimulated by 3D collagen I (but not IV), and inhibited by PAK1.
Tyrosyl-phosphorylated PAK1 regulates the transcription of invasive-relevant MMPs in response to PRL
MMP expression is regulated mainly at the transcriptional level. To assess MMP-1, -2, -3, and -9 transcription, TMX2–28 cell clones were plated on either plastic, collagen I, or collagen IV, or embedded in collagen I or IV, treated with or without PRL, and mRNA levels were assessed by real-time RT-PCR (Figure 4A-D). In agreement with our ELISA data, expression of MMP-1 and MMP-3 was up-regulated by embedding TMX2–28 PAK1 WT cells in 3D collagen IV matrix and by treatment of the cells with PRL. Although 3D collagen IV also stimulated MMP-2 mRNA expression (not shown in Figure 4C because all data were separately normalized to untreated control cells for each substrate), PRL treatment significantly down-regulated MMP-2 mRNA in GFP and PAK1 WT-expressing cells but not in PAK1 Y3F-expressing cells. PRL failed to affect MMP-9 mRNA under any plating conditions tested, and both PAK1 WT and PAK1 Y3F strongly attenuated MMP-9 mRNA expression. Overall, MMPs 1, 2, 3, and 9 secretion levels correlated with mRNA levels, suggesting coordinate MMP mRNA and proteins regulation by matrix type, PRL treatment, and PAK1 activity.
Figure 4.

Collagens and PRL-Dependent Tyrosyl Phosphorylation of PAK1 Regulate MMP 1–3 and -9 Transcription. TMX2–28 cells stably overexpressing GFP, PAK1 WT, or PAK1 Y3F were plated and treated as in Figure 3. The mRNA level for MMP-1 (A), MMP-3 (B), MMP-2 (C), and MMP-9 (D) was analyzed by real-time RT-PCR. Bars represent mean ± SE. *, P < .05 compared with the same cells untreated with PRL. #, P < .05 compared with cells expressing GFP with the same treatment. Each experiment was repeated 3 times.
Role of MAPKs and nuclear factor-κB (NFκB) pathways in MMPs 1, 2, 3, and 9 secretion
Data from the literature suggest that MMP transcription is regulated through multiple signaling pathways, including MAPK ERK1/2, p38 and JNK1/2, and NFκB signaling (3). Because PAK1 can modulate each of these pathways (20), and because PRL also stimulates MMP transcription (this study), we sought to investigate the involvement of these signaling molecules in the PRL-PAK1 regulation of MMP secretion.
To investigate the ability of PRL to activate MAPKs in TMX2–28 clones, the cells were treated over a time course with PRL, and cell lysates were analyzed for MAPK phosphorylation. PRL induced ERK1/2 phosphorylation faster in PAK1 WT and PAK1 Y3F clones as compared with the control cells, although the level of pERK1/2 was similar in all three clones by 30 minutes of PRL treatment (Figure 5A). The activation of JNK1/2 was apparent at 30 minutes and remained elevated for at least 140 minutes. Both PAK1 WT and PAK1 Y3F demonstrated significantly stronger activation of JNK1 and JNK2 as compared with the control GFP cells (Figure 5A). Surprisingly, phosphorylation of p38 was strongly enhanced in PAK1 Y3F cells as compared with the PAK1 WT and GFP cells (Figure 5A). These data suggest that both PAK1 and PAK1 Y3F stimulate activation of JNK1/2 and enhanced early activation of ERK 1/2 in response to PRL. In contrast, phosphorylation of one or more of these 3 tyrosines on PAK1 inhibits p38 activation, suggesting that PAK1-dependent regulation of PRL response is a combination of positive phospho-tyrosyl-independent effect of PAK1 on JNK1/2 and ERK1/2 activation and negative phospho-tyrosyl-dependent effect of PAK1 on p38 activation.
Figure 5.

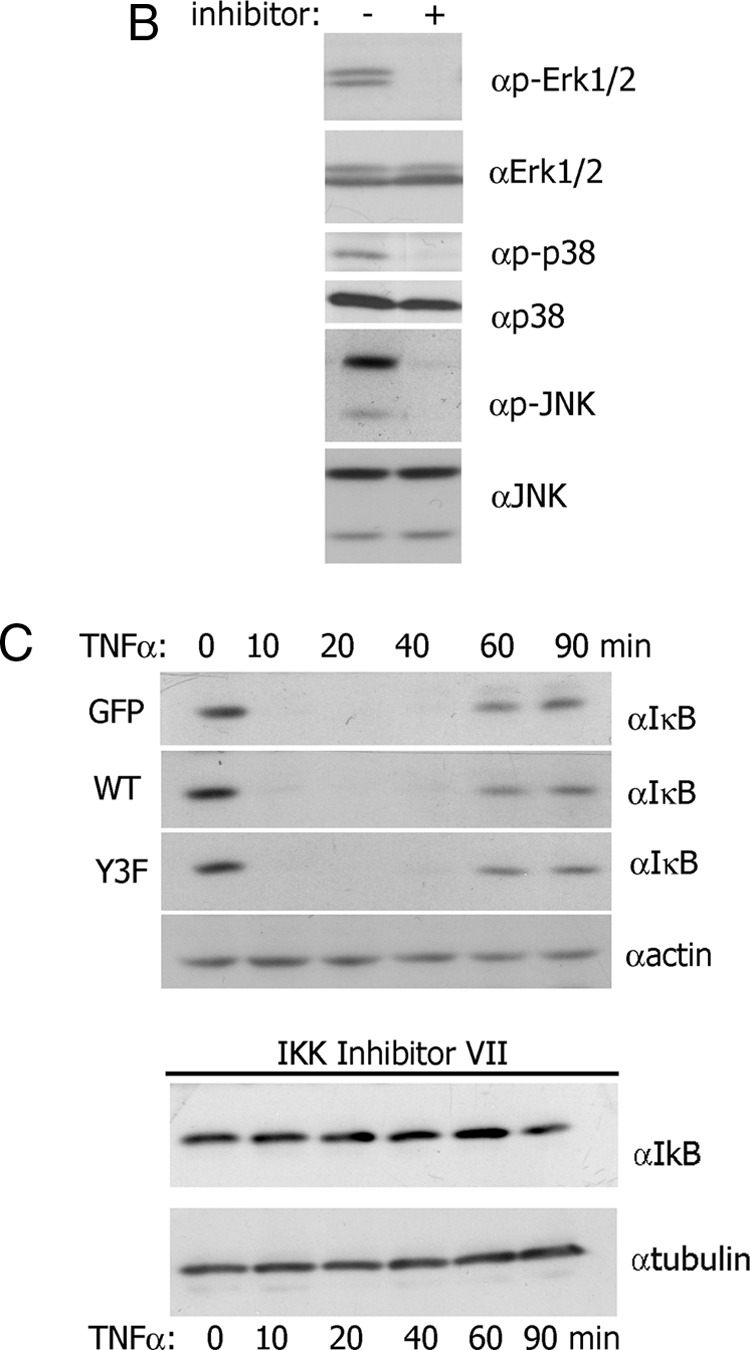
Selective Inhibitors Inhibit PRL-Activated Multiple MAPKs. A, TMX2–28 cells stably overexpressing GFP, PAK1 WT, or PAK1 Y3F were serum deprived for 72 hours and treated with 100 ng/mL PRL for the time indicated. Cells were lysed, and proteins were resolved by SDS-PAGE and analyzed by immunoblotting with the indicated antibodies. The graphs represent densiometric analysis of the band obtained for phosphorylated MAPKs normalized with total MAPKs for each lane. *, P < .05 compared with cells expressing GFP with the same treatment. Each experiment was repeated 3 times. B, TMX2–28 cells were serum deprived for 72 hours and treated with 100 ng/mL PRL and the following inhibitors: ERK 1/2 inhibitor UO 126 (10 μM, 1 h), p38 inhibitor SB 203580 (10 μM, 6 h), and JNK1/2 inhibitor SP 600125 (30 μM, 1 h). Cells were lysed, and proteins were resolved by SDS-PAGE and analyzed by immunoblotting with the indicated antibodies. C, TMX2–28 cells stably overexpressing GFP, PAK1 WT, or PAK1 Y3F were treated with TNF-α (10 ng/mL) for the time indicated. Cells were lysed, and proteins were resolved by SDS-PAGE and analyzed by immunoblotting with anti-IkB Ab. The bottom immunoblots: The TMX2–28 cells were preincubated with 5 μM inhibitor of IKK VII overnight and treated with TNF-α for the time indicated. The expression levels of γ-tubulin (αtubulin) or actin were used as an internal control.
To investigate the role of PRL-activated MAPKs in MMP secretion, we used selective MAPK inhibitors. Because PD98059 only moderately inhibited PRL-induced ERK1/2 phosphorylation (41), we used UO 126. UO 126 (10 μM) blocked the PRL-induced ERK1/2 phosphorylation in 1 hour (Figure 5B). SB 203580 (10 μM), a p38-selective inhibitor, blocked PRL-dependent activation of p38 in 6 hours whereas SP 600125 (30 μM), a JNK1/2-selective inhibitor, inhibited activation of JNK1/2 in 1 hour (Figure 5B). It has been shown that ERK5 plays a role in cell invasion (42). Although UO 126 blocks EGF-stimulated phosphorylation of ERK5 in HeLa cells (43), it does not affect ERK5 phosphorylation in response to PRL (41). In agreement with the latest, we have shown that PRL activated ERK5 over a time course (Supplemental Figure 1A; published on The Endocrine Society's Journals Online web site at http://mend.endojournals.org) and UO 126 (10 μM, 1 h) blocked the PRL-induced ERK1/2 phosphorylation, without affecting ERK5 phosphorylation (Supplemental Figure 1B). We therefore used UO 126 to further investigate a role of PRL-dependent activation of ERK1/2, but not ERK5, in the cell invasion. To assess NFκB signaling, TMX2–28 clones treated with TNF-α were analyzed for IkB-α degradation with and without IkB kinase (IKK) inhibitor VII (Figure 5C). TNF-α induced IkB-α degradation in 10 minutes, with IkB-α resynthesis in 60 minutes in all 3 clonal lines. However, preincubation with IKK inhibitor VII overnight abolished IkB-α degradation (Figure 5C). Because the MMP ELISAs required 48 hours of PRL treatment, we tested all inhibitors for 48 hours for impact on cell viability. SB203580 (10 μM) and UO126 (10 μM) did not affect viability over 48 hours whereas SP600125 (30 μM) and IKK inhibitor VII (5 μM) were slightly toxic to the cells (data not shown). For ELISA, cells were preincubated with 30 μM SP600125 for 6 hours or 5 μM IKK inhibitor VII overnight and then incubated with or without PRL for 48 hours in the presence of 15 μM SP600125 or 0.5 μM IKK inhibitor VII (neither concentration was toxic).
These inhibitors were used to determine whether PRL-dependent PAK1 phosphorylation regulates MMP secretion through ERK1/2, p38, JNK1/2, and NFκB. As shown in Figure 6, the ERK1/2 inhibitor UO126 inhibited secretion of MMP-1 and -3 in cells expressing PAK1 WT and embedded in collagen IV (compare bars for UO126 treatment with bars for control cells in Figure 6, A and B). Inhibition of p38 led to full inhibition of MMP-3 secretion (Figure 6B) and substantial diminishment of MMP-1 production (Figure 6A). MMP-1 and -3 secretion was blocked only partly by SP600125 (inhibition of JNK1/2). Inhibition of NFκB significantly inhibited both MMP-1 and -3 secretion (Figure 6, A and B).
Figure 6.
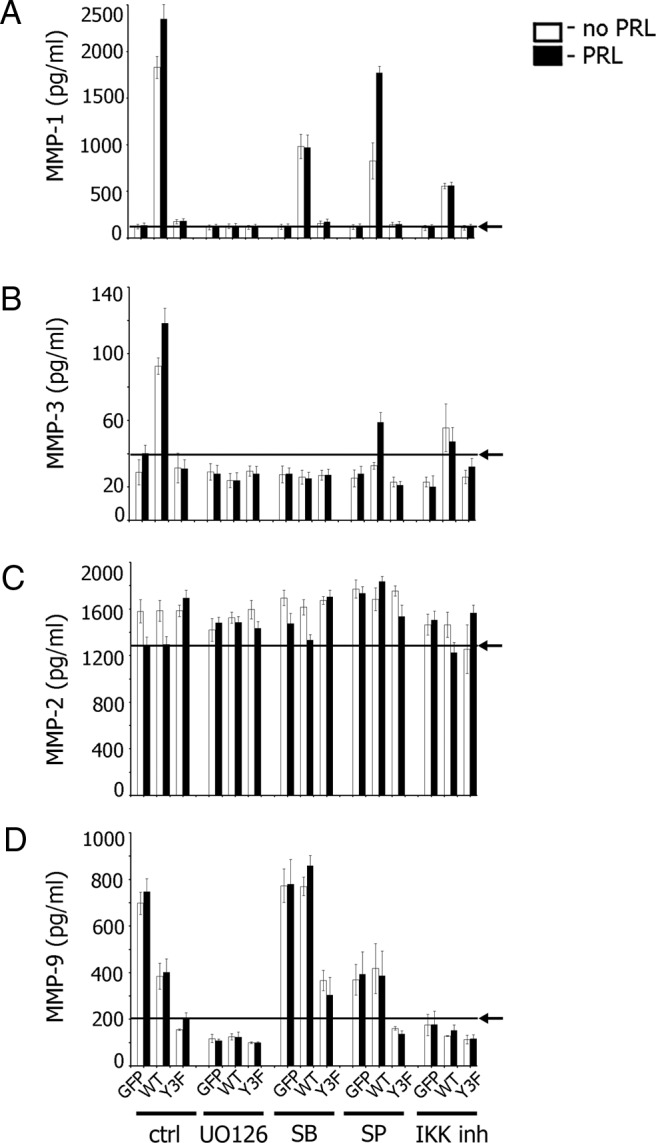
Collagens and PRL-Dependent Tyrosyl Phosphorylation of PAK1 Regulate MMP Secretions via MAPKs and NFκB. TMX2–28 cells stably overexpressing GFP, PAK1 WT, or PAK1 Y3F were embedded in collagen IV (A–C) or in collagen I (D) and treated with inhibitors. For ERK1/2 inhibition, cells were deprived for 24 hours and treated with UO 126 (10 μM) for 1 hour prior to plating with or without PRL for 48 hours in the presence of UO 126 (final concentration, 10 μM). For p38 inhibition, cells were deprived for 24 hours, treated with 10 μM SB 203580 for 6 hours prior to plating the cells with or without PRL for 48 hours in the presence of SB 203580 (final concentration, 10 μM). For JNK1/2 inhibition, cells were deprived for 24 hours and treated with inhibitor (30 μM) for 1 hour prior to plating with or without PRL for 48 hours in the presence of SP 600125 (final concentration, 15 μM). For IKK inhibition, cells were deprived for 8 hours, followed by overnight incubation with IKK inhibitor VII (5 μM). Cells were plated the next day and incubated with or without PRL for 48 hours in the presence of IKK inhibitor VII (final concentration, 0.5 μM). The conditioned medium was assessed for secreted MMP-1 (A), MMP-3 (B), MMP-2 (C), and MMP-9 (D) by ELISA. Arrows indicate the average level of MMP secretion obtained in control GFP (A–C) or PAK1 Y3F (D) expressing cells treated with PRL. Bars represent mean ± SE. Each experiment was repeated 3 times.
As before, MMP-2 secretion was slightly inhibited by PRL in control GFP and PAK1 WT cells (Figure 6C), and this inhibition was released by UO126 and SP600125, implicating ERK1/2 and JNK1/2 in MMP-2 suppression by PRL.
Secretion of MMP-9 was PRL-independent in agreement with data described above (Figure 6D). UO126 and IKK inhibitor VII inhibited MMP-9 secretion (Figure 6D). SP600125 JNK inhibitor reduced 3D collagen I-stimulated (GFP cells in Figure 6D), but not PAK1-dependent (PAK1 WT cells in Figure 6D) MMP-9 secretion. In contrast, MMP-9 secretion was stimulated by p38 inhibition in PAK1 WT and PAK1 Y3F cells (Figure 6D).
In summary, these findings suggest that activation of ERK1/2 is required for pTyr-PAK1/ 3D collagen IV-dependent regulation of MMP-1. Activated p38, JNK1/2, and NFκB are also involved to varying degrees. ERK1/2, p38, and NFκB are required for MMP-3 release, whereas activated JNK1/2 contributes to a lesser extent. ERK1/2 and JNK1/2 play a role for the pTyr-PAK1/ 3D collagen IV-dependent regulation of MMP-2. Activation of ERK1/2 and NFκB are also required for MMP-9 release, whereas p38 inhibition potentiates MMP-9 release.
Role of MMPs and MAPK pathways in cell invasion
To demonstrate a mechanistic role of MMP-1, -2, -3, and -9 as well as MAPK pathways in PRL-induced cell invasion, we first knocked down MMP-1 in TMX2–28 clones and assessed the cell invasion through Matrigel. We found that all MMP-1-depleted cell lines maintained basal invasion (white bars in plots in Figure 7A) but were deficient in PRL-induced migration (black bars in plots in Figure 7A). Next, we inhibited MMP-2, MMP-3, and MMP-9 proteins with the selective MMP inhibitors. Because the invasion assay required 48 hours, we tested all inhibitors for 48 hours for impact on cell viability. MMP-3 inhibitor I (50 μM) and MMP-9 inhibitor I (50 μM) did not affect viability over 48 hours whereas MMP-2 inhibitor I (125 μM) was slightly toxic to the cells (data not shown). For invasion assay, cells were preincubated with MMP-2 (125 μM), MMP-3 (50 μM), or MMP-9 (50 μM) inhibitors overnight and then allowed them to invade with or without PRL for 48 hours in the presence of 72.5 μM MMP-2 inhibitor I, 50 μM MMP-3, or 50 μM MMP-9 (neither concentration was toxic). MMP-3 inhibitor I abolished invasion of PAK1 WT and control GFP cells whereas invasion of PAK1 Y3F cells was the same as without the inhibitor. Inhibition of MMP-2 led to decreased invasion of PAK1 WT cells and did not have an effect on invasion of PAK1 Y3F cells. MMP-9 inhibitor I had no effect on invasion of all 3 clones (Figure 7B). These data suggest that MMP-1 is required for invasion of both PAK1 WT and PAK1 Y3F cells, whereas MMP-3 is required for, and MMP-2 contributes to, invasion of tyrosyl-phosphorylated PAK1 but not PAK1 Y3F cells.
Figure 7.
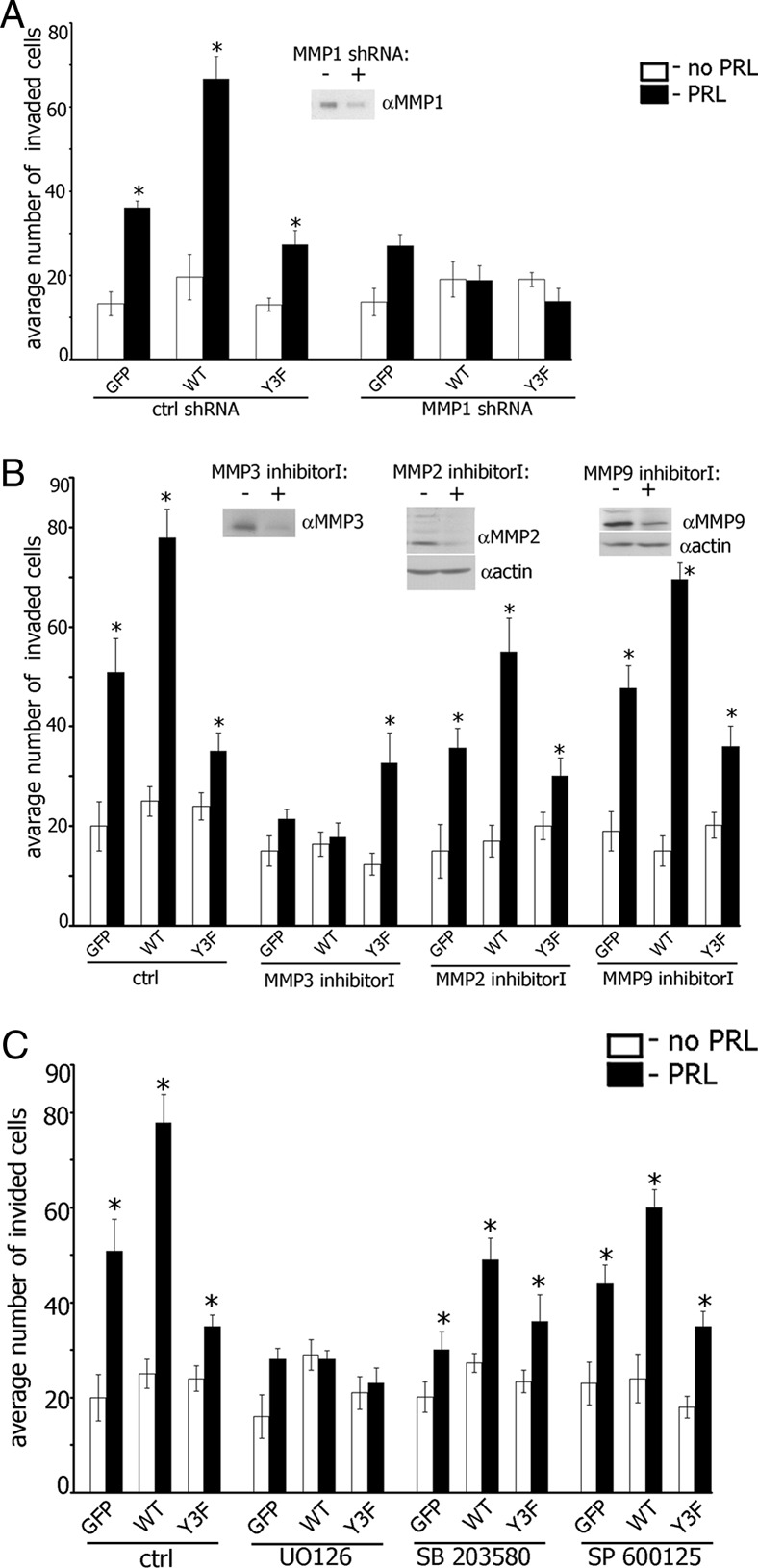
Maximal Invasion of TMX2–28 Cells in Response to PRL Requires MMPs 1, 2, 3, and 9 and MAPK Activities. A, TMX2–28 cells stably overexpressing GFP, PAK1 WT, or PAK1 Y3F were transfected with control or MMP-1 shRNA and assessed for invasion as in Figure 2. Silencing efficiency was judged by immunoblotting of conditioned medium with anti-MMP1 Ab 48 hours after transfection. B, TMX2–28 cells stably overexpressing GFP, PAK1 WT, or PAK1 Y3F were treated with either MMP-3 inhibitor I (50 μM; overnight), MMP-2 inhibitor I (125 μM; overnight) or MMP-9 inhibitor I (50 μM; overnight) and assessed for invasion as in Figure 2. The conditioned medium (for MMP-3 inhibitor I) or cell lysates (for MMP-2 inhibitor I and MMP-9 inhibitor I) were analyzed by immunoblotting with the indicated antibodies. Latent (upper band) and active (lower band) forms of MMP-2 and MMP-9 were down-regulated by MMP-2 inhibitor I and MMP-9 inhibitor I, respectively. The expression levels of actin were used as an internal control. C, TMX2–28 clones were treated with MAPK inhibitors as in Figure 6 and assessed for the invasion as in Figure 2. Bars represent mean ±/- SE. *, P < .05 compared with untreated cells. Each experiment was repeated 3 times.
To determine whether PRL-dependent PAK1 phosphorylation regulates cell invasion through ERK1/2, p38, or JNK1/2 pathways, we used selective MAPK inhibitors. As shown in Figure 7C, PRL-induced cell invasion was abolished by ERK1/2 inhibitor UO 125 in all 3 clones. Inhibition of JNK1/2 and p38 decreased invasion of GFP and PAK1 WT cells in response to PRL but not in PAK1 Y3F cells.
In summary, selective inhibition of ERK1/2 abolishes expression of MMP-1, -3, and -9 and is accompanied by inhibition of PRL-dependent cell invasion through Matrigel. MMP-1 and MMP-3 proteins are required for this invasion whereas MMP-9 has no effect. Inhibition of JNK1/2 and p38 pathways decreased production of MMP-1 and MMP-3 and also decrease cell invasion. Although inhibition of p38 pathway stimulates MMP-9 production, this stimulation does not affect cell invasion, which is in agreement with our data suggesting that MMP-9 has no effect on cell invasion.
Discussion
The data presented provide evidence that PAK1 stimulates the PRL-dependent invasion of TMX2–28 human breast tumor cells through Matrigel by up-regulating the expression and secretion of MMP-1, -2, and -3 in 3D collagen IV, which makes up 31% of Matrigel protein composition. PRL-induced PAK1 tyrosyl phosphorylation leads to a further increase in MMP-1 and MMP-3 expression and modestly inhibits MMP-2 expression. The expression and secretion of MMP-9 are dramatically increased by 3D collagen I and are not affected by PRL but inhibited by PAK1. MMP-1 and -3 are required for, and MMP-2 contributes to, PRL-dependent invasion. These data suggest that the induction of each MMP is complex and regulated differently by PRL-, PAK1-, and ECM-dependent pathways.
It is well documented that the interaction of cells with 2D substrates is significantly different than in more natural 3D environments that cells encounter in vivo (44). Cells embedded in 3D matrix have higher amounts of ligated matrix-receptors, compared with the cells grown on a thin film of matrix. Collagen receptors, such as integrins and DDRs (discoidin domain receptors), are signal transducting receptors. Integrin clustering initiates an array of signaling cascades, including activation of Rho family of small GTPases, MAPKs, and phosphatidylinositol 3-kinases (45) that can regulate MMP expression. Collagen I induces expression/secretion of MMP-1 and MMP-9 (40, 46), whereas collagen IV up-regulates MMP-2 and -9 expression (47, 48). Other collagen receptors, DDR1 and DDR2, are tyrosine kinase receptors that bind to native collagens and play integrin-independent roles in signaling, cell adhesion, matrix remodeling, and MMP secretion/activation (49, 50). Thus, embedding cells in a 3D matrix can amplify signals from ligated integrins/DDRs and lead to MMP expression. Furthermore, “cross talk” and “synergy” between signaling by ECM receptors and growth factors and cytokines have been reported that involve cooperation in the downstream signal transduction pathways.
Another possible explanation of how 3D collagen results in elevated expression of MMPs 1–3 and 9 as compared with 2D collagen relates to the physical properties of 3D matrixes (reviewed in Refs. 4 and 45). The actin cytoskeleton appears to be the major cellular system for transduction of force generated by the external network. Cytoskeletal stretching correlates with the recruitment of adhesion complex proteins and triggers signals resulting in the induction of a matrix-degrading protease (51). This may explain why 3D collagen I induces expression of only MMP-9 whereas 3D collagen IV up-regulates expression of MMP-1, -2, and -3 but not MMP-9. Collagen I is a fibril-forming collagen whereas collagen IV is a network-forming collagen. We can speculate that cells embedded in the network formed by 3D collagen IV, but not collagen I, can sense geometry and the external force generated by this network. Thus, in addition to the ligation of different receptors, physical properties of the 3D collagen IV network activate cytoskeletal-triggered signaling pathways that are distinct from those activated by 3D fibrillar collagen I, resulting in induction of distinct MMPs.
We have demonstrated that, in contrast to MMP-1 and -3, PRL modestly inhibits MMP-2 secretion and does not affect MMP-9. PRL regulation of MMP expression is complex. PRL induces activation of MAPK pathways leading to activation of transcription factor AP-1 (activating protein 1) (41, 52). In parallel, PRL activates STAT5a and STAT5b (well-known targets of PRL/JAK2) that inhibit PRL-activated AP-1 activity (53). In T47D cells PRL does not alter MMP-2 expression (53) or MMP-2 and MMP-9 activities (17), but reduction of STAT5 by siRNA or inhibition of STAT5 activity increases the PRL-dependent transcription/activity of AP-1 target genes such as MMP-2 (17, 53). In turn, overexpression of WT STAT5a inhibited MMP-2 activity (17). In agreement, we demonstrated that PRL inhibits MMP-2 secretion in control GFP and PAK1 WT-overexpressing cells, although not in PAK1 Y3F, suggesting that specific PRL-dependent inhibition of MMP-2 expression is dependent on PAK1 tyrosyl phosphorylation. This inhibitory effect of PRL was only detected in cells embedded in collagen IV, but not on plastic in agreement with published data (17, 53). In support of results published previously, we did not see an effect of PRL on MMP-9 expression (17).
PAK1 facilitates MAPK and NFκB signaling. PAK1 may coordinate signaling between Raf, MEK, and ERK1/2 by acting as a scaffold for these proteins (54). Activated PAK1 phosphorylates both Raf and MEK, enhancing their association. Following growth factor or integrin stimulation, ERK phosphorylates PAK1, resulting in a negative feedback loop (55). Activated PAK1 stimulates JNK1, whereas inhibition of PAK1 activity in breast cancer cells was associated with reduction of JNK1 activity (55). PAK1 and its upstream regulators Rac, Cdc42, and PAK-interacting exchange factor βPIX have been shown to couple to and regulate the activity of p38 MAPK (20, 55). PAK1 mediates NFκB activation by Ras, Raf-1, and Rac1, and the expression of active PAK1 stimulates NFκB on its own or via activation of upstream regulatory kinase (NFκB-inducing kinase) (20, 55). In the present study we have demonstrated that PAK1 enhances activation of JNK1/2 and accelerates activation of ERK1/2 in response to PRL. This PAK1 action depends on PRL but does not depend on 3 tyrosines (PRL has dual action on PAK1: it activates kinase activity of both PAK1 WT and PAK1 Y3F and additionally stimulates phosphorylation of 3 tyrosines on PAK1 WT by JAK2). We have assessed the effects of PRL on MAPK activation in TMX2–28 clones plated on plastic. Remarkably, we have not detected any positive effect of PRL on MMP secretion in these cells plated on plastic. However, PAK1 Y3F mutant demonstrated inhibition of MMP-3 and MMP-9 secretion as compared with control and PAK1 WT cells plated on plastic. PAK1 Y3F-dependent regulation of p38 may be responsible for this effect because PAK1 Y3F mutant stimulates activation of p38 in response to PRL, and the inhibitory effect of p38 MAPK on gene regulation has been described previously (56, 57). Further experiments to assess a synergetic effect of PRL/PAK1 and 3D collagen IV/PAK1 on MAPK's activation are needed. How can we explain that PAK1 Y3F stimulates p38 activity much stronger that PAK1 WT in response to PRL (Figure 5A)? PAK1-dependent activation of p38 is enhanced by overexpressing of βPIX (58), which is a guanine nucleotide exchange factor for Rac1 and Cdc42. The authors proposed a model of p38 activation in which βPIX might activate PAK1, MKK3/6, and p38 in two ways: by activating Rac1/Cdc42 through its guanine nucleotide exchange factor activity and by interacting directly with the PAK1-regulatory N terminus. We have preliminary data that PAK1 Y3F mutant is defective in βPIX-binding in response to PRL (A. Hammer and M. Diakonova, in preparation). Thus, overexpression of PAK1 Y3F, which cannot bind to βPIX, may instead lead to increased stimulation of Rac1/Cdc42. PAK1 Y3F can be efficiently activated by Rac1/Cdc42 (Figure 1B), leading to p38 activation and inhibition of MMP gene regulation. We still do not know whether PAK1 Y3F stimulates p38 activity in cells grown in 3D matrix to the same extent as in cells plated on plastic. We are currently investigating the role of PAK1 tyrosyl phosphorylation in p38 activation in cells embedded in 3D collagens.
In the present study we demonstrated that in cells embedded in 3D matrix, activation of ERK1/2 is required for PAK1-dependent regulation of MMPs 1, 3, and 9 secretion and is important for the secretion of MMP-2. p38 MAPK pathway is required for MMP-3 secretion and important for MMPs 1 and 9 pathways whereas activity of JNK1/2 is important for MMPs 1–3. It is known that distinct MAPK pathways (ERK1/2, p38, and JNK1/2) converge at the transcription factor AP-1, and the AP-1 binding site is present in the regulatory region of MMP-1, -3, and -9 genes but not the MMP-2 gene. An NFκB-binding site has been identified within MMPs 1, 3, and 9 gene promoters but not in the MMP-2 gene promoter, which is consistent with our data on the requirement of NFκB activation for MMP-3 and -9 but not for MMP-2 secretion. Additionally, activation of ERK1/2 plays a major role in MMP-1 and -3 expression, whereas the activation of p38 had little effect on the transcriptional activity of the MMP-1 gene, in agreement with our observation (59). MMP-9 promoter activity is regulated by ERK (60), whereas MMP-3 is regulated by p38 (for example, Refs. 3 and 61). NFκB activation is required for the expression of MMP-3 and -9 (62, 63), in agreement with our data. Unexpectedly, the inhibition of MAPK p38 by pharmacologic inhibitor SB 203580 resulted in a marked up-regulation of MMP-9 expression in all three cell lines, with the highest increase in TMX2–28 PAK1 WT cells as compared with the untreated cells. As we mentioned previously, the inhibitory effect of p38 MAPK on gene regulation has been described previously (56, 57), and MMP-9 may be another example of a p38-supressed gene.
Some effects of PAK1 on the actin cytoskeleton, cell cycle arrest, and regulation of cyclin D1 promoter activity appear to be independent of PAK1 kinase activity but dependent on protein-protein interaction (25, 64–69). We have recently shown that PAK1 phosphorylates FLNa to a greater extent when PAK1 is tyrosyl phosphorylated by JAK2 that plays important role in the PRL-dependent cell motility (30). We suggest that tyrosyl phosphorylation of PAK1 by JAK2 creates high-affinity docking sites for binding to SH2-domain-containing proteins and alters the ability of PAK1 to find, bind, and/or phosphorylate intracellular targets, thereby amplifying the effect of PAK1 on cell function. Thus, in the presence of PRL, in addition to the kinase activity, 3 tyrosines of PAK1 become phosphorylated by JAK2 and serve as a docking site for proteins that brings an additional activity toward cell invasion. This may explain why the unphospho-Tyr-PAK1 Y3F mutant is less effective than pTyr-PAK1 WT on MMP secretion and cell invasion following PRL treatment: it probably cannot associate with other proteins that require the phosphotyrosine docking sites for normal interaction.
PRL has a minimal (if any) effect on MMP production on cells grown on plastic. 3D collagen IV itself stimulates MMP-1 and -3 production through WT but not Y3F PAK1, suggesting that 3 tyrosines participate in collagen IV-triggered PAK1 regulation. PRL-induced PAK1 tyrosyl phosphorylation leads to a further increase in MMP-1 and -3 production and maximal cell invasion. The cells expressing PAK1 Y3F are still able to invade in response to PRL although to a much lesser extent that the cells expressing PAK1 WT or control cells. 3D collagen IV also promotes MMP-2 secretion; however, this effect is PAK1-independent and modestly inhibited by PRL. We did not see an effect of PRL on MMP-9 expression whereas 3D collagen I induces secretion of this MMP and PAK1 negatively regulates it.
These studies establish an important role for pTyr-PAK1 in breast cancer progression. PRL-dependent PAK1 phosphorylation significantly increases invasion of TMX2–28 human breast cancer cells through Matrigel, and we propose a model for PRL-dependent regulation of secretion of MMPs that integrates our findings with previous studies (Figure 8). We hypothesize that contact with 3D collagen IV may be an important invasive stimulus for breast cancer cells. Mammary cells are normally surrounded by basement membrane, comprised mostly of type IV collagen. In normal cells, signals from collagen IV do not induce MMP expression. In contrast, in breast cancer cells PRL (breast cancer cells secrete extrapituitary PRL that could behave as an autocrine growth factor [70, 71]) initiates the JAK2-dependent tyrosyl phosphorylation of PAK1, increasing PAK1 signaling (both through PAK1 serine/threonine kinase activity and ability to initiate additional protein-protein interactions). Importantly, PAK1 expression is also elevated in breast cancer (23). FLNa can serve as a bridge between activated integrins/DDR and pTyr-PAK1 to integrate signals from cytokines (PRL) and the ECM (collagen IV/integrin). FLNa regulates PRL-dependent cell migration (29, 30). PAK1 phosphorylates FLNa, leading to actin-dependent changes, and FLNa, in turn, activates PAK1 in a positive feed-back loop (72). Tyrosyl-phosphorylated PAK1 may also create additional docking sites to recruit SH2 domain-containing proteins to facilitate JAK2-dependent invasion. PAK1 activates ERK1/2, p38 MAPK, and JNK1/2, each of which can activate AP-1. Genes encoding MMP-1 and -3 have an AP-1 binding site supporting the transcription of these MMPs (but not MMP-2) after induction by PAK1. Secretion of MMP-1 and -3 is required for PRL-dependent invasion. Given the complexity of these signaling cascades, it is likely that additional signaling molecules are also involved in the modulation of MMP expression.
Figure 8.

PRL-Dependent Tyrosyl Phosphorylated PAK1 and 3D Collagen IV Regulate MMP-1 and MMP-3 Production and Invasion via MAPK Pathways Schematic representation of the proposed working model. PRL activation of JAK2 leads to tyrosyl phosphorylation of PAK1 on tyrosines 153, 201, and 285, thereby increasing PAK1 activities (both the serine/threonine kinase activity and ability to create potential protein-protein interactions through these phosphorylates tyrosines) and stimulating phosphorylation of FLNa. Phosphorylated FLNa stimulates the kinase activity of PAK1 in a positive feedback. In turn, FLNa binds to β-integrin and transduces signals from surrounding matrix to inside of a cell. 3D collagen IV-induced signals, in combination with pTyr-PAK1, produce intense synergistic increases in MMP-1 and MMP-3 production via MAPK pathways. MMP-1 degrades type I collagen, which is a major component of the ECM, and MMP-3 degrades collagen IV, which is a main component of basement membrane, resulting in increased invasion of breast cancer cells in response to PRL. NFκB pathway is not shown.
Materials and Methods
Plasmids and antibodies
Construction of PAK1 WT in the pLNCX2 retroviral vector containing the IRES2-EGFP element was described previously (24). Tyrosines 153, 201, and 285 in PAK1 WT were mutated to phenylalanines using the QuikChange Site-Directed Mutagenesis kit (Stratagene, La Jolla, California) (PAK1 Y3F). Mutations were confirmed by sequencing by the University of Michigan DNA Sequencing Core. The final plasmids PAK1 WT and PAK1 Y3F with N-terminal myc-tags were expressed from retroviral constructs that include IRES elements that allow the transcription of a single bicistronic mRNA of myc-PAK1-IRES2-EGFP, and therefore produce myc-PAK1 together with EGFP as a reporter for expression of PAK1. cDNAs encoding the mutants Rac1 V12 and Cdc42 L61 were used with the permission of Dr. Hall (Memorial Sloan-Kettering Cancer Center, New York, New York). PAK1 pSUPER-GFP that targets the PAK1 mRNA and the mutated control PAK1 pSUPER-GFP were described early (73). Monoclonal anti-myc (9E10) antibody (Ab), polyclonal anti-IκB-α, and polyclonal anti-PAK1 Ab (N-20) were from Santa Cruz Biotechnology, Inc. (Santa Cruz, California). Polyclonal anti-phospho-T423 PAK1, rabbit monoclonal anti-phospho-p38 (Thr180/Tyr182), rabbit monoclonal anti-p38 MAPK, mouse monoclonal anti-phospho-SAPK/JNK (Thr183/Tyr185) (G9), rabbit monoclonal anti-SAPK/JNK (56G8), monoclonal anti-phospho-p44/42 MAPK (Thr202/Tyr204), polyclonal anti-p44/42 MAPK, polyclonal anti-phospho-ERK5 (Thr218/Tyr220), and polyclonal anti-ERK5 Abs were from Cell Signaling Technology, Inc. (Danvers, Massachusetts). Polyclonal anti-MMP-1 and polyclonal anti-MMP-3 Abs were from Triple Point Biologics, Inc. (Forest Grove, Oregon), polyclonal anti-MMP-2 (ab37150) was from Abcam (Cambridge, Massachusetts), and polyclonal anti-MMP-9 (AB13458) was from EMD Millipore (Billerica, Massachusetts). Monoclonal antiactin Ab (pan Ab-5, clone ACTN05) was from Thermo Scientific (Rockford, Illinois). Monoclonal anti-HA Ab was from Roche Applied Science (Indianapolis, Indiana). Monoclonal anti-γ-tubulin Ab was from Sigma-Aldrich (St. Louis, Missouri). Human PRL was purchased from the National Hormone and Peptide Program (Dr. Parlow, National Institute of Diabetes and Digestive and Kidney Diseases, Bethesda, Maryland). TNF-α was from Invitrogen (Carlsbad, California).
Cell cultures
TMX2–28 cells and their PAK1 clones were maintained in DMEM supplemented with 10% fetal bovine serum, 50 U/mL penicillin, 50 μg/mL streptomycin, and supplemented with 4 mM l-glutamine. To generate stable TMX2–28 clones overexpressing GFP, PAK1 WT, or PAK1 Y3F mutant, we used Phoenix cells (gift of Dr. Taylor, University of Toledo, Toledo, Ohio) for virus production. Phoenix cells were maintained at 37°C in DMEM containing 10% FBS, 50 U/mL penicillin, 50 μg/mL streptomycin and supplemented with 4 mM l-glutamine, until 90% confluent. The cells were transfected with pLNCX2-IRES2-EGFP, pLNCX2-myc-PAK1 WT-IRES2-EGFP, or pLNCX2-myc-PAK1 Y3F-IRES2-EGFP using a modification of the polyethylenimine method (74). In 48 hours the virus broth was collected, supplemented with polybrene at a final concentration of 12 μg/mL and added to the TMX2–28 cells at a ratio of 1:3 (viral broth to fresh medium) and cultured at 37°C. The next day, the medium was removed and fresh complete medium was added to the cells. Clonal cell lines were isolated by dilution and expanded, and at least 6 clonal lines were examined for transgene expression by anti-myc immunoblot.
Immunoblotting
The cells were rinsed 3 times with 10 mM sodium phosphate, pH 7.4, 150 mM NaCl, 1 mM Na orthovanadate. Cells were then solubilized in lysis buffer (50 mM Tris, pH 7.5, 0.1% Triton X-100, 150 mM NaCl, 2 mM EGTA, 1 mM Na orthovanadate, 1 mM phenylmethylsulfonyl fluoride, 10 μg/mL aprotinin, 10 μg/mL leupeptin) and centrifuged at 14 000 × g for 10 minutes at 4°C. The supernatant (cell lysate) was boiled for 5 minutes in a SDS-PAGE sample buffer (250 mM Tris-HCl, pH 6.8, 10% sodium dodecyl sulfate, 10% β-mercaptoethanol, 40% glycerol, 0.01% bromphenol blue). The solubilized proteins were separated by SDS-PAGE followed by immunoblotting with the indicated antibodies and visualization with the enhanced chemiluminescence detection system.
In vitro kinase assay
To assess PAK1 WT and PAK1 Y3F in vitro kinase activity, myc-tagged WT or mutated PAK1 were IP'd with anti-myc Ab from TMX2–28 cell lines treated with or without HRG (30 ng/mL, 15 min), or with or without PRL (200 ng/mL, 20 min) and subjected to an in vitro kinase assay in the presence of 10 μCi of [γ-32P]ATP (MP Biomedicals, Irvine, California), and 5 μg of histone H4 (substrate of PAK1; New England Biolabs, Beverly, Massachusetts). 32P incorporation into histone H4, an indicator of phosphorylation, was assessed by autoradiography. PAK1 bands were quantified using Multi-Analyst (Bio-Rad Laboratories, Hercules, California) software after blotting the same nitrocellulose with antimyc, and relative PAK1 kinase activity was normalized with the amount of IP'd PAK1.
Gene silencing
For synthesis of PAK1 siRNA in vivo, TMX2–28 cells were transiently transfected with cDNA encoding PAK1 pSUPER-GFP that targets the PAK1 mRNA or control pSUPER-GFP that produces a siRNA that is 2 bp different from the PAK1 siRNA using Nucleofector kit V (Amaxa Biosystems, Gaithersburg, Maryland) according to the manufacturer's protocol. MMP-1 was knocked down by using short hairpin RNA (shRNA) expression construct as described previously (75, 76). Briefly, MMP-1 shRNA expression vector was cotransfected with a pVSV-G vector into the GPG29 amphotropic packaging cell line using a modification of the polyethylenimine method (74). In 48 hours the virus broth was collected and added to the TMX2–28 clones. MMP-1 knockdown was confirmed by Western blot analysis with anti-MMP-1 in 48 hours after infection.
Cell invasion assay
Equal numbers of deprived TMX2–28 cells (5 × 105 cells) were placed in deprivation media (DM; 2% BSA) in the upper chamber of a Boyden chamber (Corning, Inc.) coated with Matrigel (BD Biosciences, Palo Alto, California). DM with or without 100 ng/mL hPRL was placed in the lower chamber. After 48 hours cells from 5 separate fields that had invaded the Matrigel were counted after fixation and staining with 4′,6-diamidino-2-phenylindole.
Preparation of collagen gels
For cells plated on collagen I (BD Biosciences) or collagen IV (Sigma), plates were coated with rat tail type I collagen or human placental collagen IV (10 μg/cm2). For embedded growth in collagens, 0.5 × 106 cells in 2 mg/mL, collagen was applied to 24-well plates and DM, with or without 100 ng/mL PRL, was added. After 48 hours conditioned medium was collected and processed for ELISA.
Treatment with inhibitors
TMX2–28 clones were treated with inhibitors before and during incubation with PRL. Cell viability was monitored by trypan blue exclusion. For treatment with IKK inhibitor VII (EMD Chemicals, Inc., Philadelphia, Pennsylvania), cells were deprived for 8 hours, followed by overnight incubation with inhibitor (5 μM). Cells were plated the next day and incubated with or without PRL for 48 hours in the presence of IKK inhibitor VII (final concentration, 0.5 μM). For treatment with SB203580 (EMD Chemicals, Inc.), cells were deprived for 24 hours, treated with 10 μM SB203580 for 6 hours prior to plating the cells with or without PRL for 48 hours in the presence of SB203580 (final concentration, 10 μM). For treatment with JNK Inhibitor II (SP600125) (EMD Chemicals, Inc.), cells were deprived for 24 hours, treated with inhibitor (30 μM) for 1 hour prior to plating with or without PRL for 48 hours in the presence of SP600125 (final concentration, 15 μM). For treatment with UO126 (Cell Signaling, Inc.), cells were deprived for 24 hours and treated with the inhibitor (10 μM) for 1 hour prior to plating with or without PRL for 48 hours in the presence of UO126 (final concentration, 10 μM). For MMP inhibition, TMX2–28 clones were treated with MMP-2 inhibitor I (125 μM; overnight) (Millipore Corp., Bedford, Massachusetts), MMP-3 inhibitor I (50 μM; overnight) (Alpha Diagnostic International, San Antonio, Texas), or MMP-9 inhibitor I (50 μM; overnight) (Millipore). Cells were allowed to invade for 48 hours in the presence of the MMP inhibitors.
ELISA
After incubation for with 48 hours with or without 100 ng/mL PRL in plain DMEM, immunoreactive MMP-1, -2, -3, and -9 in conditioned medium were quantified by ELISA (R&D Systems, Minneapolis, Minnesota). MMP-1, MMP-2, MMP-3, and MMP-9 assays recognize total (pro- and active) MMP-1, -2, -3, and -9 forms, respectively. 1-Step Ultra TMB-ELISA (Thermo Fisher Scientific, Pittsburgh, Pennsylvania; Pierce Chemical Co., Rockford, Illinois) was used for all ELISA experiments. Conditioned medium was concentrated prior to ELISA.
Real-time RT-PCR
Total RNA was extracted using TRIZOL reagent (Invitrogen Life Technologies) according to the manufacturer's instruction. Total RNA (1 μg) was subject to random-primed first-strand cDNA synthesis. The primer sequences (human) were as follows: MMP-1 forward, 5′-ATTGGAGCAGCAAGAGGCTGGGA-3′; and reverse, 5′-TTCCAGGTATTTCTGGACTAAGT-3′ (77); MMP-2 forward, 5′-GAGAACCAAAGTCTGAAGAG-3′; and reverse, 5′-GGAGTGAGAATGCTGATTAG-3′; MMP-3 forward, 5′- GCTGCAAGGGGTGAGGACAC-3′; and reverse, 5′- GATGCCAGGAAAGGTTCTGAAGTG-3′; MMP-9 forward, 5′- GGGAGACGCCCATTTCG-3′; and reverse, 5′-CGCGCCATCTGCGTTT-3′ (40); glyceraldehyde-3-phosphate dehydrogenase forward, 5′-CGACCACTTTGTCAAGCTCA-3′; and reverse, 5′-AGGGGAGATTCAGTGTGGTG-3′.
Real-time RT-PCR was performed using IQ SYBR Green Supermix (Bio-Rad).
Representative samples from each batch of experiments were subjected to RT-PCR without reverse transcriptase enzyme to rule out DNA contamination and possible amplification of genomic DNA. Glyceraldehyde-3-phosphate dehydrogenase was used as the control (house-keeping) gene. All real-time RT-PCRs were performed in triplicate. Each experiment was repeated at least 3 times. mRNAs from breast cancer samples were used as positive controls for all MMPs. Three different breast cancer samples were purchased from the Cooperative Human Tissue Network (Ohio State University, Columbus, Ohio).
Supplementary Material
Acknowledgments
We thank Drs Mattingly and Li (Wayne State University, Detroit, Michigan) for the construction of PAK1 WT in the pLNCX2 retroviral vector containing the IRES 2-enhanced GFP element. We also thank the following: Drs Li (University of Connecticut Health Center, Connecticut) and Wu (Yale School of Medicine, New Haven, Connecticut) for sending the cDNAs encoding PAK1 pSUPER-GFP and mutated control PAK1 pSUPER-GFP; Dr Eisenmann (University of Toledo, Toledo, Ohio) for providing TMX2–28 cells; Dr Shemshedini (University of Toledo) for providing primers for RT-PCR; Dr Yeung (University of Toledo) for providing MMP-1 shRNA expression construct and GPG29 amphotropic packaging cells; and Dr Komuniecki (University of Toledo) for help with the manuscript; Dr La Motte (Lerner Research Institute, Cleveland Clinic Foundation, Cleveland, Ohio) for endotoxin testing of collagens; and Dr Leaman (University of Toledo) for help with real-time RT-PCR and very helpful discussion.
This work was supported by National Institutes of Health Grant R01 DK88127 (to M.D.).
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- 3D
- three-dimensional
- Ab
- antibody
- DDR
- discoidin domain receptor
- DM
- deprivation media
- ECM
- extracellular matrix
- EGFP
- enhanced GFP
- FLNa
- filamin A
- GFP
- green fluorescent protein
- HRG
- heregulin
- IkB-α
- inhibitor of NFκB-α
- IKK
- IkB kinase
- IP'd
- immunoprecipitated
- IRES
- internal ribosome entry site
- JAK2
- Janus tyrosine kinase
- JNK
- c-Jun N-terminal kinase
- MMP
- metalloproteinase
- NFκB
- nuclear factor-κB
- PAK1
- p21-activated serine-threonine kinase
- βPIX
- PAK-interacting exchange factor
- PRL
- prolactin
- pTyr
- tyrosyl phosphorylated
- STAT
- signal transducer and activator of transcription
- WT
- wild type.
References
- 1. Poola I, DeWitty RL, Marshalleck JJ, Bhatnagar R, Abraham J, Leffall LD. Identification of MMP-1 as a putative breast cancer predictive marker by global gene expression analysis. Nat Med. 2005;11:481–483 [DOI] [PubMed] [Google Scholar]
- 2. Sternlicht MD, Bissell MJ, Werb Z. The matrix metalloproteinase stromelysin-1 acts as a natural mammary tumor promoter. Oncogene. 2000;19:1102–1113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Overall CM, López-Otin C. Strategies for MMP inhibition in cancer: innovations for the post-trial era. Nat Rev Cancer. 2002;2:657–672 [DOI] [PubMed] [Google Scholar]
- 4. Lu P, Takai K, Weaver VM, Werb Z. Extracellular matrix degradation and remodeling in development and disease. Cold Spring Harb Perspect Biol. 2011;3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Geiger B, Yamada KM. Molecular architecture and function of matrix adhesions. Cold Spring Harb Perspect Biol. 2011;3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Boyd NF, Lockwood GA, Martin LJ, et al. Mammographic densities and breast cancer risk. Breast Dis. 1998;10:113–126 [DOI] [PubMed] [Google Scholar]
- 7. Colpaert C, Vermeulen P, Van Marck E, Dirix L. The presence of a fibrotic focus is an independent predictor of early metastasis in lymph node-negative breast cancer patients. Am J Surg Pathol. 2001;25:1557–1558 [DOI] [PubMed] [Google Scholar]
- 8. Adriance MC, Inman JL, Petersen OW, Bissell MJ. Myoepithelial cells: good fences make good neighbors. Breast Cancer Res. 2005;7:190–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ben-Jonathan N, LaPensee CR, LaPensee EW. What can we learn from rodents about prolactin in humans? Endocr Rev. 2008;29:1–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bernichtein S, Touraine P, Goffin V. New concepts in prolactin biology. J Endocrinol. 2010;206:1–11 [DOI] [PubMed] [Google Scholar]
- 11. Carver KC, Arendt LM, Schuler LA. Complex prolactin crosstalk in breast cancer: new therapeutic implications. Mol Cell Endocrinol. 2009;307:1–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Clevenger CV. Role of prolactin/prolactin receptor signaling in human breast cancer. Breast Dis. 2003;18:75–86 [DOI] [PubMed] [Google Scholar]
- 13. Tworoger SS, Hankinson SE. Prolactin and breast cancer risk. Cancer Lett. 2006;243:160–169 [DOI] [PubMed] [Google Scholar]
- 14. Maus MV, Reilly SC, Clevenger CV. Prolactin as a chemoattractant for human breast carcinoma. Endocrinology. 1999;140:5447–5450 [DOI] [PubMed] [Google Scholar]
- 15. Liby K, Neltner B, Mohamet L, Menchen L, Ben-Jonathan N. Prolactin overexpression by MDA-MB-435 human breast cancer cells accelerates tumor growth. Breast Cancer Res Treat. 2003;79:241–252 [DOI] [PubMed] [Google Scholar]
- 16. Oakes SR, Robertson FG, Kench JG, et al. Loss of mammary epithelial prolactin receptor delays tumor formation by reducing cell proliferation in low-grade preinvasive lesions. Oncogene. 2007;26:543–553 [DOI] [PubMed] [Google Scholar]
- 17. Sultan AS, Xie J, LeBaron MJ, Ealley EL, Nevalainen MT, Rui H. Stat5 promotes homotypic adhesion and inhibits invasive characteristics of human breast cancer cells. Oncogene. 2005;24:746–760 [DOI] [PubMed] [Google Scholar]
- 18. Nouhi Z, Chughtai N, Hartley S, Cocolakis E, Lebrun JJ, Ali S. Defining the role of prolactin as an invasion suppressor hormone in breast cancer cells. Cancer Res. 2006;66:1824–1832 [DOI] [PubMed] [Google Scholar]
- 19. Rider L, Shatrova A, Feener EP, Webb L, Diakonova M. JAK2 tyrosine kinase phosphorylates PAK1 and regulates PAK1 activity and functions. J Biol Chem. 2007;282:30985–30996 [DOI] [PubMed] [Google Scholar]
- 20. Molli PR, Li DQ, Murray BW, Rayala SK, Kumar R. PAK signaling in oncogenesis. Oncogene. 2009;28:2545–2555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Balasenthil S, Sahin AA, Barnes CJ, et al. p21-activated kinase-1 signaling mediates cyclin D1 expression in mammary epithelial and cancer cells. J Biol Chem. 2004;279:1422–1428 [DOI] [PubMed] [Google Scholar]
- 22. Salh B, Marotta A, Wagey R, Sayed M, Pelech S. Dysregulation of phosphatidylinositol 3-kinase and downstream effectors in human breast cancer. Int J Cancer. 2002;98:148–154 [DOI] [PubMed] [Google Scholar]
- 23. Mira JP, Benard V, Groffen J, Sanders LC, Knaus UG. Endogenous, hyperactive Rac3 controls proliferation of breast cancer cells by a p21-activated kinase-dependent pathway. Proc Natl Acad Sci USA. 2000;97:185–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Li Q, Mullins SR, Sloane BF, Mattingly RR. p21-Activated kinase 1 coordinates aberrant cell survival and pericellular proteolysis in a three-dimensional culture model for premalignant progression of human breast cancer. Neoplasia. 2008;10:314–329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Adam L, Vadlamudi R, Mandal M, Chernoff J, Kumar R. Regulation of microfilament reorganization and invasiveness of breast cancer cells by kinase dead p21-activated kinase-1. J Biol Chem. 2000;275:12041–12050 [DOI] [PubMed] [Google Scholar]
- 26. Vadlamudi RK, Adam L, Wang RA, et al. Regulatable expression of p21-activated kinase-1 promotes anchorage-independent growth and abnormal organization of mitotic spindles in human epithelial breast cancer cells. J Biol Chem. 2000;275:36238–36244 [DOI] [PubMed] [Google Scholar]
- 27. Barnes CJ, Bagheri-Yarmand R, Mandal M, et al. Suppression of epidermal growth factor receptor, mitogen-activated protein kinase, and Pak1 pathways and invasiveness of human cutaneous squamous cancer cells by the tyrosine kinase inhibitor ZD1839 (Iressa). Mol Cancer Ther. 2003;2:345–351 [PubMed] [Google Scholar]
- 28. Yang Z, Rayala S, Nguyen D, Vadlamudi RK, Chen S, Kumar R. Pak1 phosphorylation of snail, a master regulator of epithelial-to-mesenchyme transition, modulates snail's subcellular localization and functions. Cancer Res. 2005;65:3179–3184 [DOI] [PubMed] [Google Scholar]
- 29. Rider L, Diakonova M. Adapter protein SH2B1β binds filamin A to regulate prolactin-dependent cytoskeletal reorganization and cell motility. Mol Endocrinol. 2011;25:1231–1243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hammer A, Rider L, Oladimeji P, et al. Tyrosyl Phosphorylated PAK1 regulates breast cancer cell motility in response to prolactin through filamin A. Mol Endocrinol. 2013;27:455–465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Fasco MJ, Amin A, Pentecost BT, Yang Y, Gierthy JF. Phenotypic changes in MCF-7 cells during prolonged exposure to tamoxifen. Mol Cell Endocrinol. 2003;206:33–47 [DOI] [PubMed] [Google Scholar]
- 32. Gozgit JM, Pentecost BT, Marconi SA, Otis CN, Wu C, Arcaro KF. Use of an aggressive MCF-7 cell line variant, TMX2–28, to study cell invasion in breast cancer. Mol Cancer Res. 2006;4:905–913 [DOI] [PubMed] [Google Scholar]
- 33. Adam L, Vadlamudi R, Kondapaka SB, Chernoff J, Mendelsohn J, Kumar R. Heregulin regulates cytoskeletal reorganization and cell migration through the p21-activated kinase-1 via phosphatidylinositol-3 kinase. J Biol Chem. 1998;273:28238–28246 [DOI] [PubMed] [Google Scholar]
- 34. Miller SL, DeMaria JE, Freier DO, Riegel AM, Clevenger CV. Novel association of Vav2 and Nek3 modulates signaling through the human prolactin receptor. Mol Endocrinol. 2005;19:939–949 [DOI] [PubMed] [Google Scholar]
- 35. Folgueras AR, Pendás AM, Sánchez LM, López-Otin C. Matrix metalloproteinases in cancer: from new functions to improved inhibition strategies. Int J Dev Biol. 2004;48:411–424 [DOI] [PubMed] [Google Scholar]
- 36. Radisky ES, Radisky DC. Matrix metalloproteinase-induced epithelial-mesenchymal transition in breast cancer. J Mammary Gland Biol Neoplasia. 2010;15:201–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Haas TL, Davis SJ, Madri JA. Three-dimensional type I collagen lattices induce coordinate expression of matrix metalloproteinases MT1-MMP and MMP-2 in microvascular endothelial cells. J Biol Chem. 1998;273:3604–3610 [DOI] [PubMed] [Google Scholar]
- 38. Ellerbroek SM, Fishman DA, Kearns AS, Bafetti LM, Stack MS. Ovarian carcinoma regulation of matrix metalloproteinase-2 and membrane type 1 matrix metalloproteinase through β1 integrin. Cancer Res. 1999;59:1635–1641 [PubMed] [Google Scholar]
- 39. Xu J, Clark RA, Parks WC. p38 mitogen-activated kinase is a bidirectional regulator of human fibroblast collagenase-1 induction by three-dimensional collagen lattices. Biochem J. 2001;355:437–447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zhou L, Yan C, Gieling RG, et al. Tumor necrosis factor-α induced expression of matrix metalloproteinase-9 through p21-activated kinase-1. BMC Immunol. 2009;10:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gutzman JH, Rugowski DE, Schroeder MD, Watters JJ, Schuler LA. Multiple kinase cascades mediate prolactin signals to activating protein-1 in breast cancer cells. Mol Endocrinol. 2004;18:3064–3075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mehta PB, Jenkins BL, McCarthy L, et al. MEK5 overexpression is associated with metastatic prostate cancer, and stimulates proliferation, MMP-9 expression and invasion. Oncogene. 2003;22:1381–1389 [DOI] [PubMed] [Google Scholar]
- 43. Mody N, Leitch J, Armstrong C, Dixon J, Cohen P. Effects of MAP kinase cascade inhibitors on the MKK5/ERK5 pathway. FEBS Lett. 2001;502:21–24 [DOI] [PubMed] [Google Scholar]
- 44. Nelson CM, Bissell MJ. Of extracellular matrix, scaffolds, and signaling: tissue architecture regulates development, homeostasis, and cancer. Annu Rev Cell Dev Biol. 2006;22:287–309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Daley WP, Peters SB, Larsen M. Extracellular matrix dynamics in development and regenerative medicine. J Cell Sci. 2008;121:255–264 [DOI] [PubMed] [Google Scholar]
- 46. Langholz O, Röckel D, Mauch C, et al. Collagen and collagenase gene expression in three-dimensional collagen lattices are differentially regulated by α 1 β 1 and α 2 β 1 integrins. J Cell Biol. 1995;131:1903–1915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Cortes-Reynosa P, Robledo T, Macias-Silva M, Wu SV, Salazar EP. Src kinase regulates metalloproteinase-9 secretion induced by type IV collagen in MCF-7 human breast cancer cells. Matrix Biol. 2008;27:220–231 [DOI] [PubMed] [Google Scholar]
- 48. Castro-Sanchez L, Soto-Guzman A, Guaderrama-Diaz M, Cortes-Reynosa P, Salazar EP. Role of DDR1 in the gelatinases secretion induced by native type IV collagen in MDA-MB-231 breast cancer cells. Clin Exp Metastasis. 2011;28:463–477 [DOI] [PubMed] [Google Scholar]
- 49. Vogel W, Gish GD, Alves F, Pawson T. The discoidin domain receptor tyrosine kinases are activated by collagen. Mol Cell. 1997;1:13–23 [DOI] [PubMed] [Google Scholar]
- 50. Vogel WF, Abdulhussein R, Ford CE. Sensing extracellular matrix: an update on discoidin domain receptor function. Cell Signal. 2006;18:1108–1116 [DOI] [PubMed] [Google Scholar]
- 51. Giannone G, Sheetz MP. Substrate rigidity and force define form through tyrosine phosphatase and kinase pathways. Trends Cell Biol. 2006;16:213–223 [DOI] [PubMed] [Google Scholar]
- 52. Gutzman JH, Nikolai SE, Rugowski DE, Watters JJ, Schuler LA. Prolactin and estrogen enhance the activity of activating protein 1 in breast cancer cells: role of extracellularly regulated kinase 1/2-mediated signals to c-fos. Mol Endocrinol. 2005;19:1765–1778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Gutzman JH, Rugowski DE, Nikolai SE, Schuler LA. Stat5 activation inhibits prolactin-induced AP-1 activity: distinct prolactin-initiated signals in tumorigenesis dependent on cell context. Oncogene. 2007;26:6341–6348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Sundberg-Smith LJ, Doherty JT, Mack CP, Taylor JM. Adhesion stimulates direct PAK1/ERK2 association and leads to ERK-dependent PAK1 Thr212 phosphorylation. J Biol Chem. 2005;280:2055–2064 [DOI] [PubMed] [Google Scholar]
- 55. Kumar R, Gururaj AE, Barnes CJ. p21-activated kinases in cancer. Nat Rev Cancer. 2006;6:459–471 [DOI] [PubMed] [Google Scholar]
- 56. Engel FB, Schebesta M, Duong MT, et al. p38 MAP kinase inhibition enables proliferation of adult mammalian cardiomyocytes. Genes Dev. 2005;19:1175–1187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Zer C, Sachs G, Shin JM. Identification of genomic targets downstream of p38 mitogen-activated protein kinase pathway mediating tumor necrosis factor-α signaling. Physiol Genomics. 2007;31:343–351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Lee SH, Eom M, Lee SJ, Kim S, Park HJ, Park D. βPix-enhanced p38 activation by Cdc42/Rac/PAK/MKK3/6-mediated pathway. Implication in the regulation of membrane ruffling. J Biol Chem. 2001;276:25066–25072 [DOI] [PubMed] [Google Scholar]
- 59. Westermarck J, Li SP, Kallunki T, Han J, Kähäri VM. p38 mitogen-activated protein kinase-dependent activation of protein phosphatases 1 and 2A inhibits MEK1 and MEK2 activity and collagenase 1 (MMP-1) gene expression. Mol Cell Biol. 2001;21:2373–2383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Lakka SS, Jasti SL, Kyritsis AP, et al. Regulation of MMP-9 (type IV collagenase) production and invasiveness in gliomas by the extracellular signal-regulated kinase and jun amino-terminal kinase signaling cascades. Clin Exp Metastasis. 2000;18:245–252 [DOI] [PubMed] [Google Scholar]
- 61. Oner C, Schatz F, Kizilay G, et al. Progestin-inflammatory cytokine interactions affect matrix metalloproteinase-1 and -3 expression in term decidual cells: implications for treatment of chorioamnionitis-induced preterm delivery. J Clin Endocrinol Metab. 2008;93:252–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Bond M, Fabunmi RP, Baker AH, Newby AC. Synergistic upregulation of metalloproteinase-9 by growth factors and inflammatory cytokines: an absolute requirement for transcription factor NF-κ B. FEBS Lett. 1998;435:29–34 [DOI] [PubMed] [Google Scholar]
- 63. Bond M, Baker AH, Newby AC. Nuclear factor kappaB activity is essential for matrix metalloproteinase-1 and -3 upregulation in rabbit dermal fibroblasts. Biochem Biophys Res Commun. 1999;264:561–567 [DOI] [PubMed] [Google Scholar]
- 64. Manser E, Huang HY, Loo TH, et al. Expression of constitutively active α-PAK reveals effects of the kinase on actin and focal complexes. Mol Cell Biol. 1997;17:1129–1143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Sells MA, Knaus UG, Bagrodia S, Ambrose DM, Bokoch GM, Chernoff J. Human p21-activated kinase (Pak1) regulates actin organization in mammalian cells. Curr Biol. 1997;7:202–210 [DOI] [PubMed] [Google Scholar]
- 66. Turner CE, Brown MC, Perrotta JA, et al. Paxillin LD4 motif binds PAK and PIX through a novel 95-kD ankyrin repeat, ARF-GAP protein: A role in cytoskeletal remodeling. J Cell Biol. 1999;145:851–863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Vadlamudi RK, Kumar R. P21-activated kinases in human cancer. Cancer Metastasis Rev. 2003;22:385–393 [DOI] [PubMed] [Google Scholar]
- 68. Thullberg M, Gad A, Beeser A, Chernoff J, Strömblad S. The kinase-inhibitory domain of p21-activated kinase 1 (PAK1) inhibits cell cycle progression independent of PAK1 kinase activity. Oncogene. 2007;26:1820–1828 [DOI] [PubMed] [Google Scholar]
- 69. Tao J, Oladimeji P, Rider L, Diakonova M. PAK1-Nck Regulates Cyclin D1 Promoter Activity in Response to Prolactin. Mol Endocrinol. 2011;25:1565–1578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Clevenger CV, Chang WP, Ngo W, Pasha TL, Montone KT, Tomaszewski JE. Expression of prolactin and prolactin receptor in human breast carcinoma. Evidence for an autocrine/paracrine loop. Am J Pathol. 1995;146:695–705 [PMC free article] [PubMed] [Google Scholar]
- 71. Ginsburg E, Vonderhaar BK. Prolactin synthesis and secretion by human breast cancer cells. Cancer Res. 1995;55:2591–2595 [PubMed] [Google Scholar]
- 72. Vadlamudi RK, Li F, Adam L, et al. Filamin is essential in actin cytoskeletal assembly mediated by p21-activated kinase 1. Nat Cell Biol. 2002;4:681–690 [DOI] [PubMed] [Google Scholar]
- 73. Li Z, Hannigan M, Mo Z, et al. Directional sensing requires G β γ-mediated PAK1 and PIX α-dependent activation of Cdc42. Cell. 2003;114:215–227 [DOI] [PubMed] [Google Scholar]
- 74. Boussif O, Lezoualc'h F, Zanta MA, et al. A versatile vector for gene and oligonucleotide transfer into cells in culture and in vivo: polyethylenimine. Proc Natl Acad Sci USA. 1995;92:7297–7301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Beshir AB, Ren G, Magpusao AN, Barone LM, Yeung KC, Fenteany G. Raf kinase inhibitor protein suppresses nuclear factor-κB-dependent cancer cell invasion through negative regulation of matrix metalloproteinase expression. Cancer Lett. 2010;299:137–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Gupta GP, Nguyen DX, Chiang AC, et al. Mediators of vascular remodelling co-opted for sequential steps in lung metastasis. Nature. 2007;446:765–770 [DOI] [PubMed] [Google Scholar]
- 77. Balduyck M, Zerimech F, Gouyer V, et al. Specific expression of matrix metalloproteinases 1, 3, 9 and 13 associated with invasiveness of breast cancer cells in vitro. Clin Exp Metastasis. 2000;18:171–178 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.