

Abstract
The scope and eligibility of patents for genetic sequences have been debated for decades, but a critical case regarding gene patents (Association of Molecular Pathologists v. Myriad Genetics) is now reaching the US Supreme Court. Recent court rulings have supported the assertion that such patents can provide intellectual property rights on sequences as small as 15 nucleotides (15mers), but an analysis of all current US patent claims and the human genome presented here shows that 15mer sequences from all human genes match at least one other gene. The average gene matches 364 other genes as 15mers; the breast-cancer-associated gene BRCA1 has 15mers matching at least 689 other genes. Longer sequences (1,000 bp) still showed extensive cross-gene matches. Furthermore, 15mer-length claims from bovine and other animal patents could also claim as much as 84% of the genes in the human genome. In addition, when we expanded our analysis to full-length patent claims on DNA from all US patents to date, we found that 41% of the genes in the human genome have been claimed. Thus, current patents for both short and long nucleotide sequences are extraordinarily non-specific and create an uncertain, problematic liability for genomic medicine, especially in regard to targeted re-sequencing and other sequence diagnostic assays.
Introduction
Gene patents are a class of intellectual property that give the patentee rights to the specific sequences in the claims of a patent, providing the exclusive right to make, use, sell, and import a molecule consisting of a claimed sequence. In 2001, the US patent office issued formal guidelines on what is acceptable patent material in the human genome. It stated that DNA is eligible if it is 'isolated from its natural state and processed through purifying steps that separate the gene from other molecules naturally associated with it.' These guidelines specified that any gene or sequence patent also needs to show 'specific, credible, and substantial utility' [1]. To date, there are over 40,000 patents on DNA molecules [2,3], including those on the breast and ovarian cancer genes BRCA1 and BRCA2 [4], indicating that patents on DNA are a widespread and significant class of intellectual property that have increased consistently since the 1980s (Figure 1).
Figure 1.

Increasing number of DNA-based patents. We searched for any US patent that has DNA sequences present in the claims since 1983 and observed a continual increase of DNA patents, with the greatest increase around the time of the completion of the human genome draft sequence in 2001.
Some DNA patents are for a very specific series of nucleotides (such as 5'-ATGCGACGGATCGATC-3') or an exact chemical structure (such as a DNA molecule modified with a fluorescent probe), but diagnostic DNA-based patents have broader claims [5]. These patents are used to find mutations in various disease-related genes, and the specified DNA sequence as well as any other similar sequence are often covered within the patent claim. This is because there are many (at least [(2N) - 1]) possible combinations of mutations for a gene [6]. Diagnostic gene patents are therefore written to find any known or unknown variation of a gene. For example, in the Association of Molecular Pathologists (AMP) v. Myriad case, the broadest intellectual property rights on BRCA sequences come from several related claims in patent 5,747,282:
Claim #1. An isolated DNA coding for a BRCA1 polypeptide, said polypeptide having the amino acid sequence set forth in SEQ ID NO:2 (the BRCA1 cDNA).
Claim #2. The isolated DNA of claim 1, wherein said DNA has the nucleotide sequence set forth in SEQ ID NO:1 (the BRCA1 gene).
Claim #5. An isolated DNA having at least 15 nucleotides of the DNA of claim 1
Claim #6. An isolated DNA having at least 15 nucleotides of the DNA of claim 2.
In 2010, in the first ruling on AMP v. Myriad, a US District Court stated that all of these patent claims were invalid and that isolated DNA is 'not patentable subject matter'. Then, in 2011, the US Court of Appeals for the Federal Circuit overturned this ruling (2 to 1 decision) and stated that an isolated DNA molecule is 'markedly different' from native genomic DNA and that fragments of the BRCA genes can be patented. After a re-hearing in light of another case (Mayo v. Prometheus), the same decision was issued by the Federal Circuit in August 2012. Recently, the Supreme Court decided to hear arguments in the case, opening a re-examination of the lower courts' decisions and rationale on these gene patents and their claims.
Notably, the Federal Circuit's decision declared that even a short, isolated DNA molecule such as 'ACGT' is different from the 'NNNNN-ACGT-NNNNN' present within a chromosome (AMP v. Myriad, Federal Circuit 2012), because it will not be connected to a sugar via a phosphodiester bond and will have a hydroxyl group instead of a bond to a phosphate (Figure 2). Thus, even a 15 nucleotide fragment of DNA in Claim #6 from Patent '282 is claimed to be 'markedly different'. However, we observe that the Court's ruling is overly broad for at least three reasons. First, it relies on the sequences having chemical features and side-chains that are not actually present in the patents (Figure 2): the claims are for a linear series of nucleotides, not a specific chemical structure. Second, if allowed to be so broad, these claims could also create a monopoly on all epigenetic and chemical variations of these sequences. Third, and perhaps most importantly, the non-specificity of 15mer sequences creates unclear infringement liability that has been even noted by the Court. Specifically, Judge Bryson declared that claim 6 'is so broad that it includes products of nature (the BRCA1 exons) and portions of other genes. ... The other claim to a short segment of DNA, claim 5 of the '282 patent, is breathtakingly broad' (AMP v. Myriad, Federal Circuit 2012). To date, however, there has not been a genome-wide analysis of the uniqueness of 15mer sequences in patented genes.
Figure 2.

Court-proposed molecular points of distinction that allow claims on isolated DNA sequences. On the basis of two molecular changes (small circles), specifically to a single phosphate and one hydroxyl group, the Federal Circuit suggested that a new DNA fragment (right) is patentable subject matter. However, such molecular structures rarely appear in gene patents, and they are not present in the BRCA patents.
Establishing the uniqueness of gene patents in DNA and cDNA could potentially have a large impact on the interpretation of these patents [7,8]. Previous work has examined Claim #5 with respect to other cDNAs on one chromosome (Chr1), and used these data to estimate that 15 infringing 15mers might exist for any cDNA [9]. However, these studies compared the likely uniqueness of cDNAs on the basis of an average degeneracy of the genetic code, leaving open the issue of exact DNA matches in the coding regions of genes and the genome. Also, a first estimate, made in 2005, calculated that 18% of human genes were patented [10], but many new DNA patents have emerged since (Figure 1). These results have been challenged in recent work [8], which has demonstrated that some gene patents for genetic sequences do not contain the DNA fragments within the actual claims. Thus, we sought to examine the current landscape of gene patents using empirical, exact matches to known genes that were confirmed to be present in patent claims, ranging from sequences of 15 nucleotides (15mers) to the full lengths of all patented DNA fragments.
Results
We first examined the incidence with which 15mers (k = 15) from a given gene matched 15mers in other genes using the most recent Consensus Coding Sequences (CCDS) database [11] of 18,382 high-confidence genes (see Methods and data). We incrementally divided each gene into k-mers (of between 15 and 1,000 nucleotides) and used the Bowtie alignment algorithm [12] to report every instance of a k-mer from one gene that perfectly matched the sequence of another gene. Our data showed that every gene in the CCDS database had a 15mer that matched the sequence of at least one other gene (Figure 3a). The number of matching genes ranged from as few as 5 (for MTRNR2L7) or 689 (for BRCA1) to as high as 7,688 (for TTN), corresponding to 0.01%, 4%, and 42% of all genes in the human genome. These results demonstrated that short patent sequences are extremely non-specific and that a 15mer patent claim from one gene will always 'cross-match' and patent a portion of another gene as well.
Figure 3.

Total matches and average number of other genes patented plotted against k-mer size. We searched for matches of a given fragment size (k-mer) of all genes relative to all other genes. We fragmented each Consensus Coding Sequence (CCDS) gene (n = 18,382) into variable lengths (x-axis) and then (a) summed the percentage of genes that would match another CCDS gene (y-axis). (b) We then examined, for each gene, the number of other genes that would match any of its own k-mers. We plotted the distribution of these cross-gene matches (y-axis) across all CCDS genes, with a varying k-mer size (x-axis). Boxplots show the distribution of the data as the median (middle line), 75th and 25th percentiles (top and bottom of red boxes, respectively) and any data points beyond the distribution (shown as dots).
We then examined the distribution of 'cross-matches' for varying k-mers across all human genes. We found that the number of matches decreased as the k-mer size increased, showing an inversely proportional relationship of sequence uniqueness and k-mer size (Figure 3b). Notably, even 1,000 nucleotide fragments from known genes could still match 117 other genes, showing that long gene fragments can still show substantial non-specificity. We then used the same alignment criteria to examine the uniqueness of the entire human genome (beyond just coding regions), and we found that 99.999% of 15mers in the human genome are repeated at least twice (see Methods and data). These data confirm the findings of previous studies that showed little sequence specificity for small k-mers in the human genome [13-15], but our data show for the first time that this global non-specificity of 15mers and longer k-mers impacts all gene patents, including those on BRCA1 and those claiming non-coding areas of the genome.
We next examined claimed sequences from existing gene patents, which spanned a wide range of sizes (Figure 4). We used sequences from published patent claims in the Cambia patent database (see Methods and data) and aligned them to the human CCDS gene set using the Basic Local Alignment Search Tool (BLAST). A previous analysis of patented genes carried out in 2005 estimated that 18% of known genes in the human genome were patented [10], but a recent study suggested that this estimate could be inflated as some sequences are not found in the patents' claims [8]. When we used the same criteria (150-nucleotide match, e-value = 0) to search the most recent Cambia database, ensuring that the sequences were actually present in the patents' claims (nt-inClaims.fsa, see Methods and data), we found that 21% (3,945/18,382) of human genes are currently claimed when these stringent parameters are applied. When we repeated this analysis with more commonly used BLAST parameters (e-value <0.05), we found that claimed sequences matched 9,361 (41%) of human genes (Additional file 1). Both results, using relatively stringent criteria, indicate an increase in patented genes since 2005 and show that current gene patents cover almost half of all known genes.
Figure 4.

Size distribution of gene patents. We examined the total number of base pairs (bp) included in the claims of each US Patent. The mean amount of sequence claimed is 1,781 bp with the median of 21,950 bp. This disparity indicates that there are many patents that claim a large amount of sequence.
Additionally, when we took existing gene patents and matched their 15mers to known genes, we found that 100% of known genes have at least one 15mer claimed in a known patent. Current gene patents were observed to match each gene many times, with 1,295 matches to other genes on average (standard deviation 1,208). When we examined the amount of total sequence space in human genes that is covered by 15mers in claims from current patents (Additional file 2), we found 58 patents whose claims covered at least 10% of the bases of all human genes. The top patent was US7795422, whose claims' sequences matched 91.5% of human genes. Interestingly, we also observed a patent for improving bovine traits (US7468248) with explicit claims for 15mers that matched 84% of human genes. This patent was not even aimed at any human sequence, yet covered a majority of human genes once we examined the claim's matches at the 15mer scale.
Discussion
These results have striking implications for the AMP v. Myriad Genetics case, gene patent litigation, and other patent legislation. The demonstrated non-specificity of sequence uniqueness across the genome suggests that the Supreme Court should use this case to clarify the law on gene patents. If patent claims that use these 15mer or other short k-mer sizes are enforced, it could potentially create a situation where a piece of every gene in the human genome is patented by a phalanx of competing patents, with potentially harmful consequences for genetic testing laboratories and research groups performing targeted sequencing on any gene, in virtually all species.
Our data show that currently claimed nucleotide sequences in US patents cover at least 41% of existing genes, as identified by BLAST alignment matches, and as many as 100% when allowing for 15mers. We also observed a large number of cross-kingdom exact matches of 15mers, indicating that not only human genetic sequences are in play: entire patent families from plant genomics can similarly claim the majority of the human genome. As both plant and animal patents claim short sequences, as well as those with low homology (45 to 55%), any claimed sequence will inevitably match many others. Yet, importantly, we observe that there is no ideal k-mer size that will preclude matches with another gene in the genome. Thus, the non-specificity needed for diagnostic patents to find any mutated sequence of one gene expands their property rights to hundreds or thousands of other genes.
Some have commented that whole-genome sequencing (WGS) might avoid some of these infringement problems [8], as no targeted fragment that contains a patented sequence is specifically isolated when shotgun-based approaches are used for sequencing. There is, however, no specific case precedent that yet supports this conclusion, and as such, legal ambiguities still affect WGS. These infringement risks are also still very clear for PCR-based or enrichment-capture strategies, which directly overlap with these patents by enriching for a molecule that contains BRCA1 or the targets of other patented genes. These targeted methods currently represent the vast majority of clinical sequencing for diagnostic medical decisions in molecular pathology, and they will likely be used for a long time as validation technology for any WGS approaches.
These claims' non-specificity highlight a large problem with gene patents, but there are at least four other potential dangers inherent to these patents. First, since almost all claimed genetic sequences from gene patents are simple DNA sequences that lack atomic-scale molecular structures, the patenting of specific gene sequences could prevent their use in other modalities of research. For example, patents could prevent work involving epigenetic and transcriptional studies of a gene in which some bases contain methyl groups (for example, a comparison of TACTGG and TACmTGG) or hydroxyl-methyl groups, both of which are important for cancer [16] and RNA regulation [17]. Second, patents on one gene can prevent research on any pseudogene of the original gene, such as BRCA1's pseudogene (BRCA1P1), even though pseudogenes can have their own independent function in a cell. Third, if these 15mer and other gene patent claims are allowed, new regions of the human genome that are still being discovered and annotated could be patented as soon as they are sequenced. Last, understanding pleiotropy for any gene depends on the ability to examine a gene in every context, and one patent on a gene's single known function limits any ability to discover the gene's many other possible functions. For example, beyond breast and ovarian cancer, there are 19 other cancers in The Cancer Genome Atlas [18] that are also associated with common mutations in BRCA1 (Figure 5), yet only Myriad Genetics has the right to an isolated molecule containing BRCA1 from any of these patients. Taken together, gene patents represent a sharp conflict between the public goods of medical knowledge and improved health and the private goods of rewarding innovation and entrepreneurial risk-taking.
Figure 5.
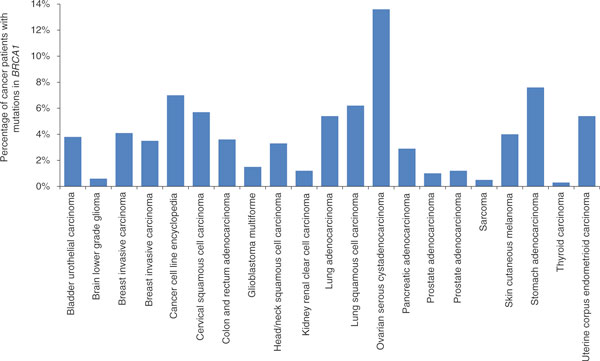
Patients with other cancer types who carry BRCA1 mutations. We used data from The Cancer Genome Atlas (TCGA) to examine the percentage of patients with other tumor types who show deleterious mutations (missense, nonsense, copy-number, and structural variations) in their BRCA1 gene. We found that ovarian and breast cancers are only 2 of 21 total cancer types likely to be associated with relevant mutations in BRCA1, as determined by TCGA.
Fortunately, there is precedence in US history for resolving such a medical-legal conflict. In 1992, a US patent was issued (#5,080,111) for a 'self-sealing episcleral incision', and this patent required a license of $4.00 per operation for any doctor to use the method. A lawsuit was filed against the patent by the American Medical Association (AMA), which strongly condemned 'the patenting of medical and surgical procedures' and began to work with Congress to outlaw the practice. The AMA argued that the best surgical techniques should not be denied to patients, simply because of legal reasons or fees. In 1995, the United States Code was changed accordingly, to add language that exempted any 'patient, physician, licensed healthcare practitioner, or a health care entity from infringement of a patented medical or surgical procedure, therapy, or diagnosis'. Thus, any surgeon could still patent a new method and stake claim as the inventor, but the patients' need to get access to the best medical care out-weighed the infringement of the intellectual property rights of surgeons.
Now that the era of genomic medicine is here, the US Supreme Court has the chance to shape the balance of the medical good and inventor protection. Given the falling price of genome sequencing and targeted re-sequencing, and the ubiquity of the genomics technologies, the urgency to resolve this uncertainly around gene patents has never been more salient. Failure to resolve these ambiguities perpetuates a direct threat to 'genomic liberty', or the right to examine one's own DNA. Our analysis and data provide strong evidence that the Supreme Court and Congress should limit the patenting of existing nucleotide sequences because of their broad scope and non-specificity in the human genome. Finally, we suggest that a research exemption or limited liability for patent infringement be implemented, as has been done for surgical techniques, which could craft a functional balance between the rights of inventors and the best application of personalized genomics for improved medical care.
Methods and data
Data sources
We used gene sequences from the CCDS database, which were downloaded from the NCBI web site [19]. This is a curated set of high-confidence genes prepared by an international consortium [11]. It contains 18,382 genes having a total of 26,355 isoforms. For this analysis, we used the longest isoform of each gene as a single, representative sequence for that gene.
Tumor data from The Cancer Genome Atlas
We used the public database from The Cancer Genome Atlas (TCGA) [18], and specifically the query and search tool at Memorial Sloan Kettering Cancer Center (MSKCC) [20]. We used the query 'BRCA1' for the database search. Data queried on 9 March 2013.
Claim-specific sequence identification
Patents in the inClaims subset of the Cambia [21] patent sequence database were downloaded from [22]. This database has been curated to include all of the sequences from US Patents that are specifically contained within the claims of the inventions, and we validated 25 of these manually (from nt-inClaims.fsa) to confirm that they were correct.
Analysis
Short-read alignments
For the comparisons of k-mers between genes, we used Bowtie (v0.12.5) to report all of the matches for each query. Each CCDS gene was split into all overlapping k-mers of the designated length and these sequences were searched against all of the other genes.
Long-read alignments
The comparison of patents to genes was performed using the criteria used by Jensen and Murray [10]; specifically using BLAST version 2.2.25+ [23] to match at least 150 nucleotides and an e-value of 0. This was performed using the BLAST command: blastn -evalue 1e-307 -word_size 150.
In addition, the criteria of e-value <0.05 was used, which is a typical set of parameters for BLAST. The command was: blastn -evalue 0.05. For the exact matching of 15 bp sequences, Bowtie was used. The bowtie command was: bowtie -f -a -best -v 0.
Mutational complexity estimate
For the calculations in our text about mutational complexity, we show that for a gene with N mutations, there are [(2N) - 1] combinations of mutated forms of that gene. For example, given a sequence with ten bases, where '=' represents an un-modified base pair, where three mutations (A,G,T) exist, there are [(2N) - 1] = [23 - 1] = [8 - 1] = 7 possible combinations.
These seven possible mutated forms that are different from the wild-type are shown here:
==========(wild-type)
===AG===T=(mutant #1)
===A======(mutant #2)
====G=====(mutant #3)
========T=(mutant #4)
====G===T=(mutant #5)
===A====T=(mutant #6)
===AG=====(mutant #7)
Abbreviations
AMA: American Medical Association; AMP: Association of Molecular Pathologists; BLAST: Basic Local Alignment Search Tool; CCDS: Consensus Coding Sequences; WGS: whole-genome sequencing.
Competing Interests
The authors declare that they have no competing interests.
Acknowledgements
The results published here are in whole or part based upon data generated by The Cancer Genome Atlas pilot project established by the NCI and NHGRI. Information about TCGA and the investigators and institutions who constitute the TCGA research network can be found at the project website [18].
Supplementary Material
Table S1. A list of the genes that are claimed by each patent.
Table S2. The percentage of the coding sequence in the human genome covered by each patent.
Contributor Information
Jeffrey A Rosenfeld, Email: rosenfj1@umdnj.edu.
Christopher E Mason, Email: chm2042@med.cornell.edu.
References
- Federal Register. 2001. No. 4, 5 January.
- Rogers EJ Can you patent genes - yes and no. J Pat & Trademark Off Soc 2011931923559147 [Google Scholar]
- Mason CE, Seringhaus MR, Brito CSS. Personalized genomic medicine with a patchwork, partially owned genome. Yale J Biol Med. 2007;80:145–151. [PMC free article] [PubMed] [Google Scholar]
- Ford D, Easton DF, Bishop DT, Narod SA, Goldgar DE. Risks of cancer in BRCA1-mutation carriers. Lancet. 1994;343:692–695. doi: 10.1016/S0140-6736(94)91578-4. [DOI] [PubMed] [Google Scholar]
- Cook-Deegan R, Heaney C. Patents in genomics and human genetics. Annu Rev Genomics Hum Genet. 2010;11:383–425. doi: 10.1146/annurev-genom-082509-141811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kepler TB, Crossman C, Cook-Deegan R. Metastasizing patent claims on BRCA1. Genomics. 2010;95:312–314. doi: 10.1016/j.ygeno.2010.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salzberg SL. The perils of gene patents. Clin Pharmacol Ther. 2012;91:969–971. doi: 10.1038/clpt.2012.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holman CM. Debunking the myth that whole-genome sequencing infringes thousands of gene patents. Nat Biotechnol. 2012;30:240–244. doi: 10.1038/nbt.2146. [DOI] [PubMed] [Google Scholar]
- Easton D, Bishop D, Ford D, Crockford G. Genetic linkage analysis in familial breast and ovarian cancer: results from 214 families. The Breast Cancer Linkage Consortium. Am J Hum Genet. 1993;52:678. [PMC free article] [PubMed] [Google Scholar]
- Jensen K, Murray F. Intellectual property landscape of the human genome. Science. 2005;310:239–240. doi: 10.1126/science.1120014. [DOI] [PubMed] [Google Scholar]
- Pruitt KD, Harrow J, Harte RA, Wallin C, Diekhans M, Maglott DR, Searle S, Farrell CM, Loveland JE, Ruef BJ. The consensus coding sequence (CCDS) project: identifying a common protein-coding gene set for the human and mouse genomes. Genome Res. 2009;19:1316–1323. doi: 10.1101/gr.080531.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg S. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurtz S, Narechania A, Stein J, Ware D. A new method to compute K-mer frequencies and its application to annotate large repetitive plant genomes. BMC Genomics. 2008;9:517. doi: 10.1186/1471-2164-9-517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salzberg SL, Phillippy AM, Zimin A, Puiu D, Magoc T, Koren S, Treangen TJ, Schatz MC, Delcher AL, Roberts M, Marçais G, Pop M, Yorke JA. GAGE: a critical evaluation of genome assemblies and assembly algorithms. Genome Res. 2012;22:557–567. doi: 10.1101/gr.131383.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herold J, Kurtz S, Giegerich R. Efficient computation of absent words in genomic sequences. BMC Bioinformatics. 2008;9:167. doi: 10.1186/1471-2105-9-167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akalin A, Garrett-Bakelman FE, Kormaksson M, Busuttil J, Zhang L, Khrebtukova I, Milne TA, Huang Y, Biswas D, Hess JL, Allis CD, Roeder RG, Valk PJ, Löwenberg B, Delwel R, Fernandez HF, Paietta E, Tallman MS, Schroth GP, Mason CE, Melnick A, Figueroa ME. Base-pair resolution DNA methylation sequencing reveals profoundly divergent epigenetic landscapes in acute myeloid leukemia. PLOS Genetics. 2012;8:e1002781. doi: 10.1371/journal.pgen.1002781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saletore Y, Meyer K, Korlach J, Vilfan I, Jaffrey S, Mason CE. The birth of the epitranscriptome: deciphering the function of RNA modifications. Genome Biol. 2012;13:175. doi: 10.1186/gb-2012-13-10-175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Cancer Genome Atlas. http://cancergenome.nih.gov/
- Consensus Coding Sequences database. ftp://ftp.ncbi.nlm.nih.gov/pub/CCDS/archive/Hs37.3/?
- Query and search tool at Memorial Sloan Kettering Cancer Center. http://www.cbioportal.org/public-portal/
- Bacon N, Ashton D, Jefferson RA, Connett MB. Biological sequences named and claimed in US patents and patent applications-Cambia Patent Lens OS4 Initiative. 2006. http://www.bios.net/daisy/bios/g2/2456/2462.html
- Cambia's Sequence Project. http://www.patentlens.net/sequence/US_B/nt-inClaims.fsa.gz
- Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, Madden T. BLAST+: architecture and applications. BMC Bioinformatics. 2009;10:421. doi: 10.1186/1471-2105-10-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. A list of the genes that are claimed by each patent.
Table S2. The percentage of the coding sequence in the human genome covered by each patent.